
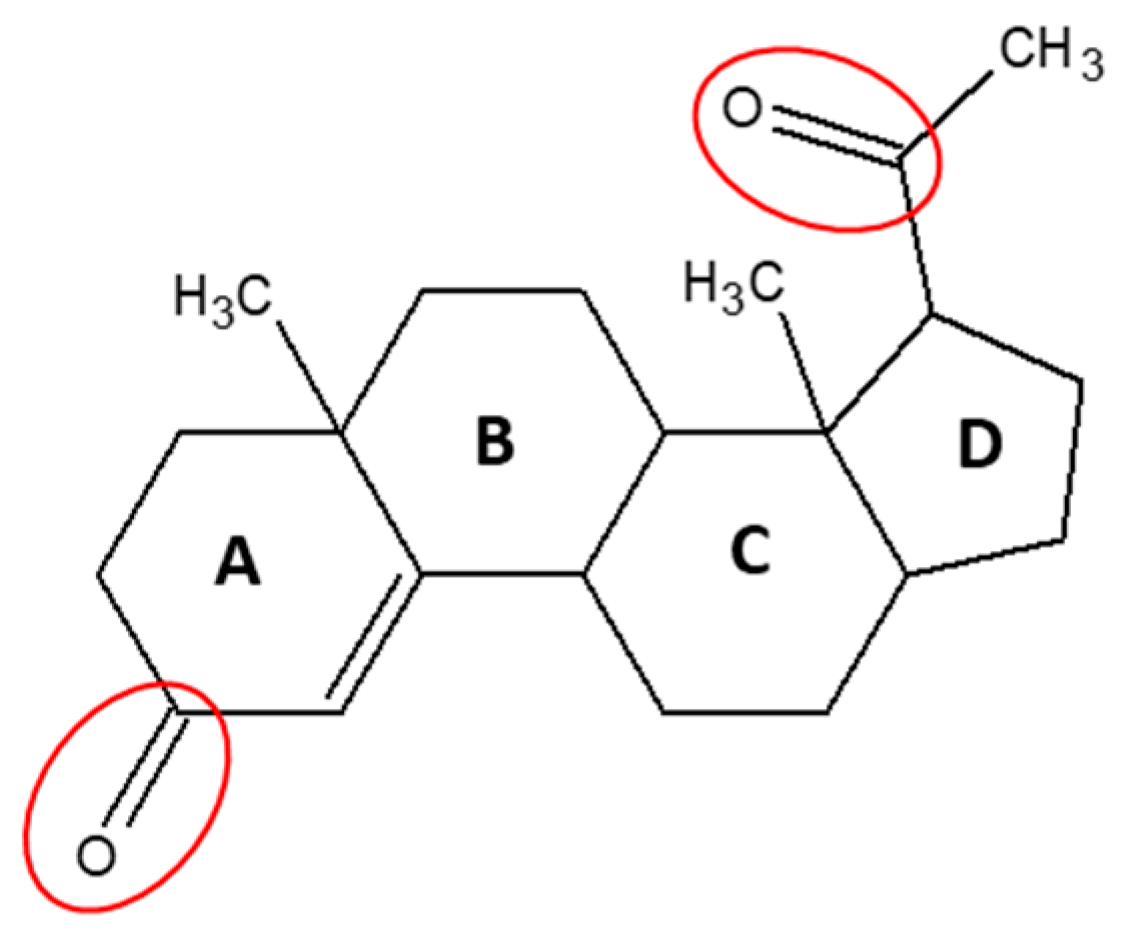
Progesterone Metabolism by Human and Rat Hepatic and Intestinal Tissue

 ,
,  ,
,  and
and 
Abstract
:
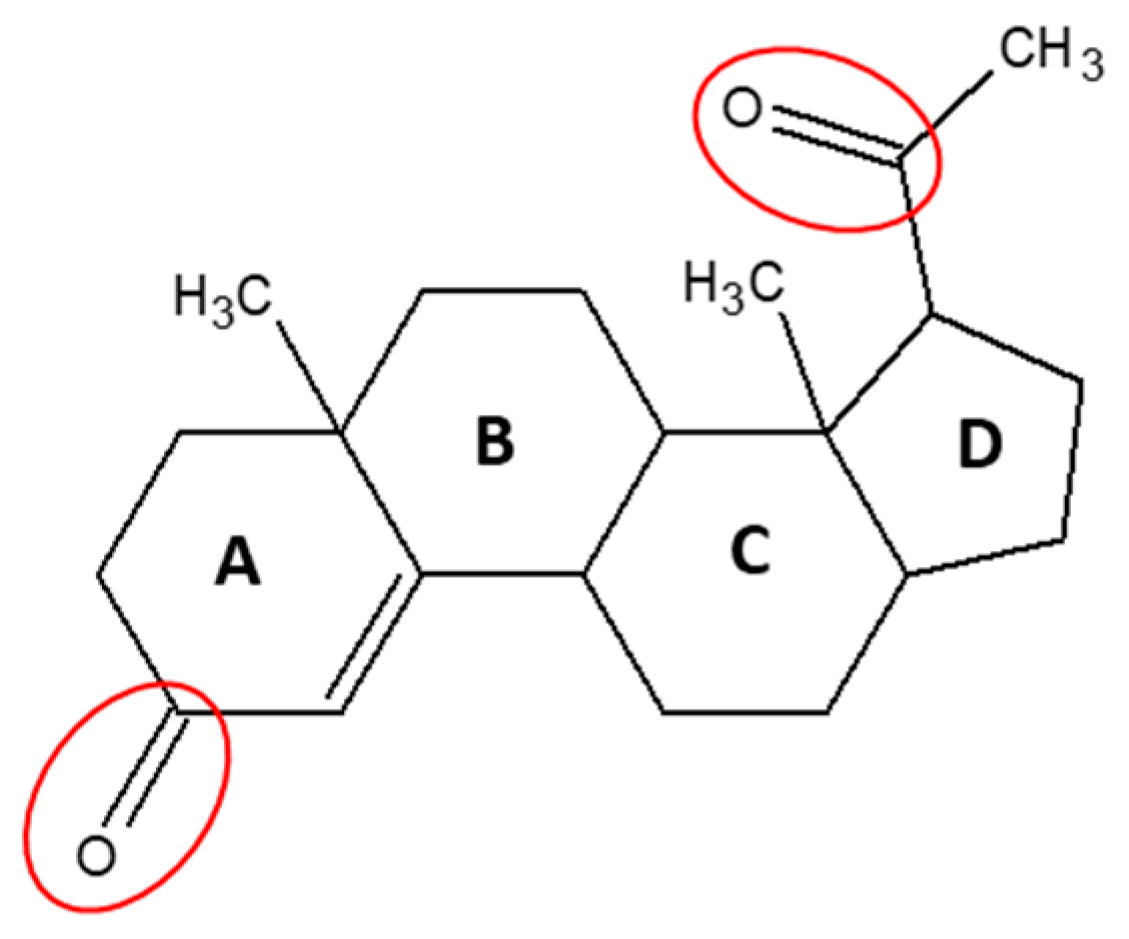
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Tissue Homogenate Stability Assay
2.3. Human Cytosol Stability Assay
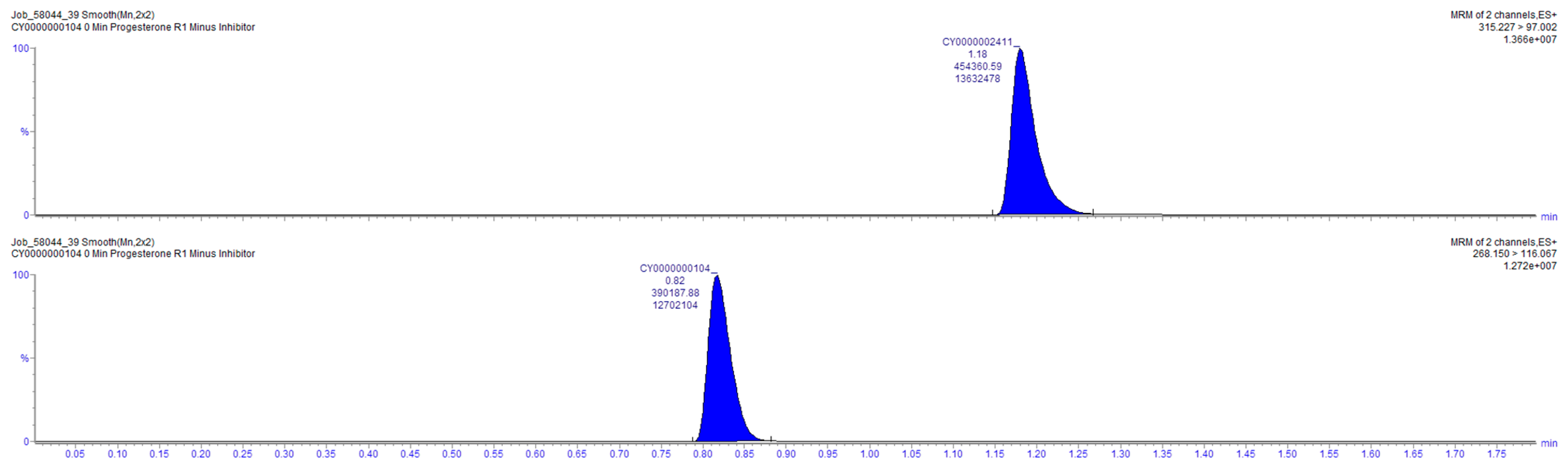
2.4. Sample Analysis
2.5. Data Analysis
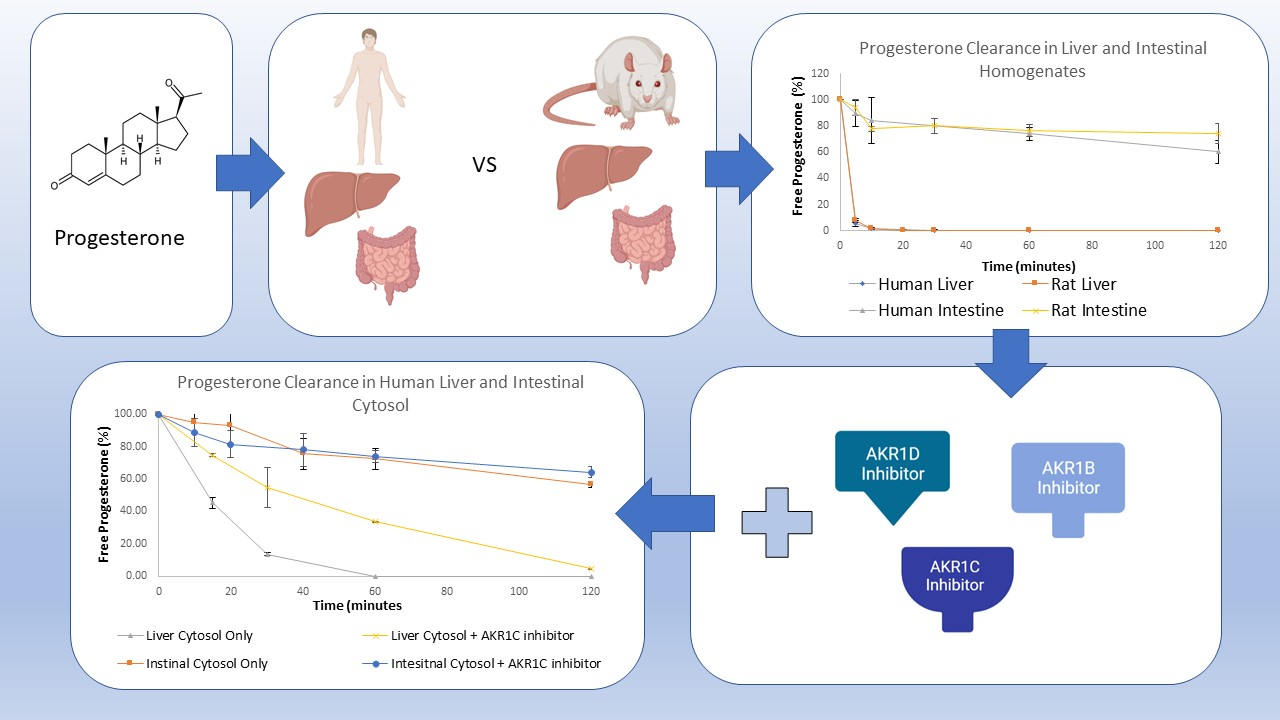
3. Results and Discussion
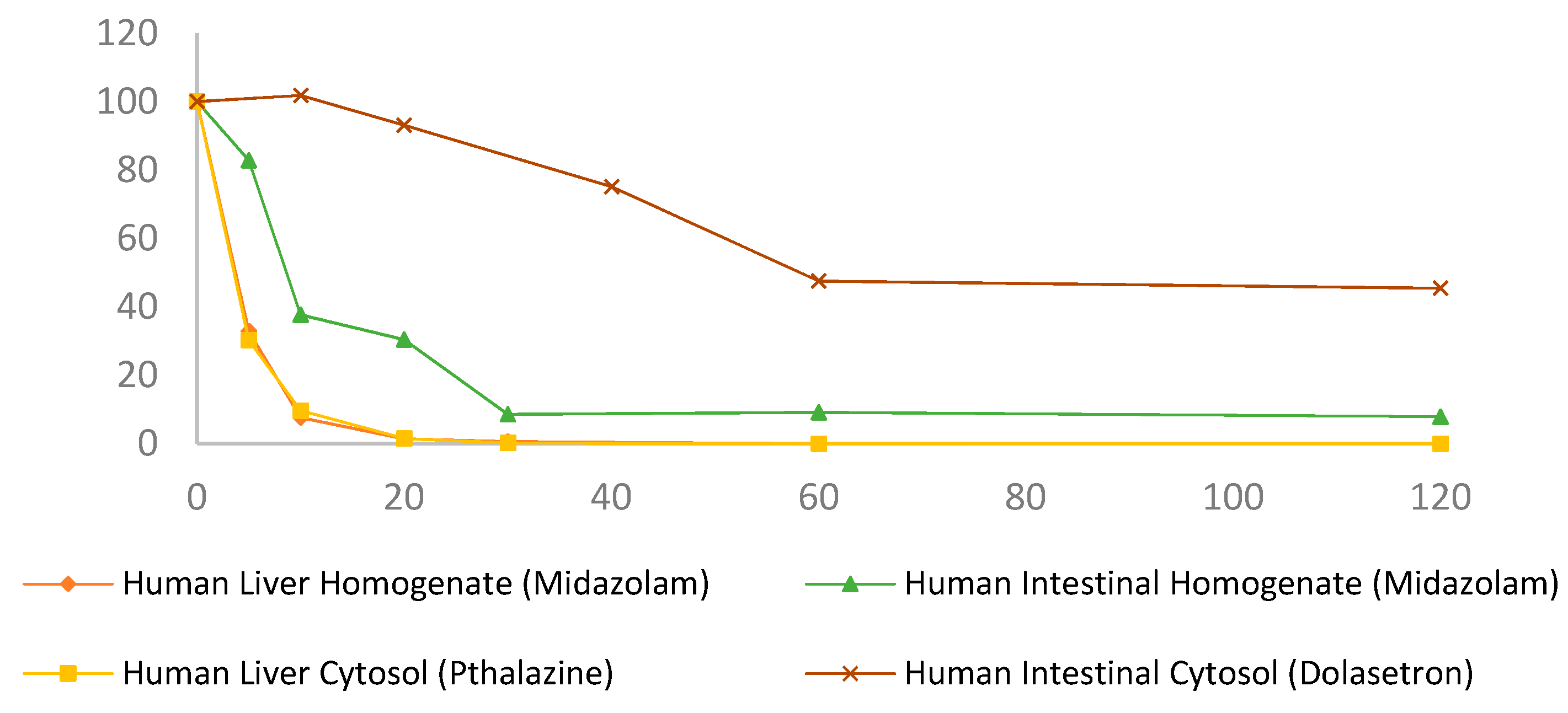
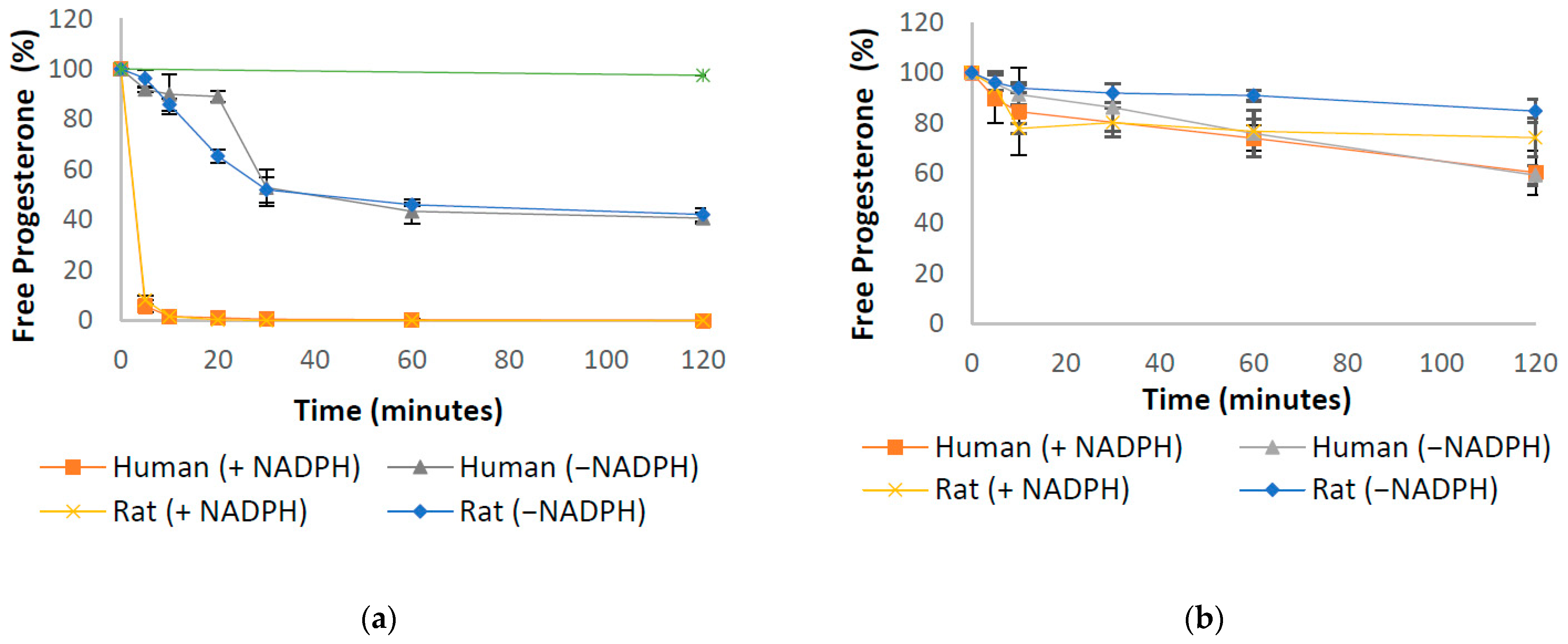
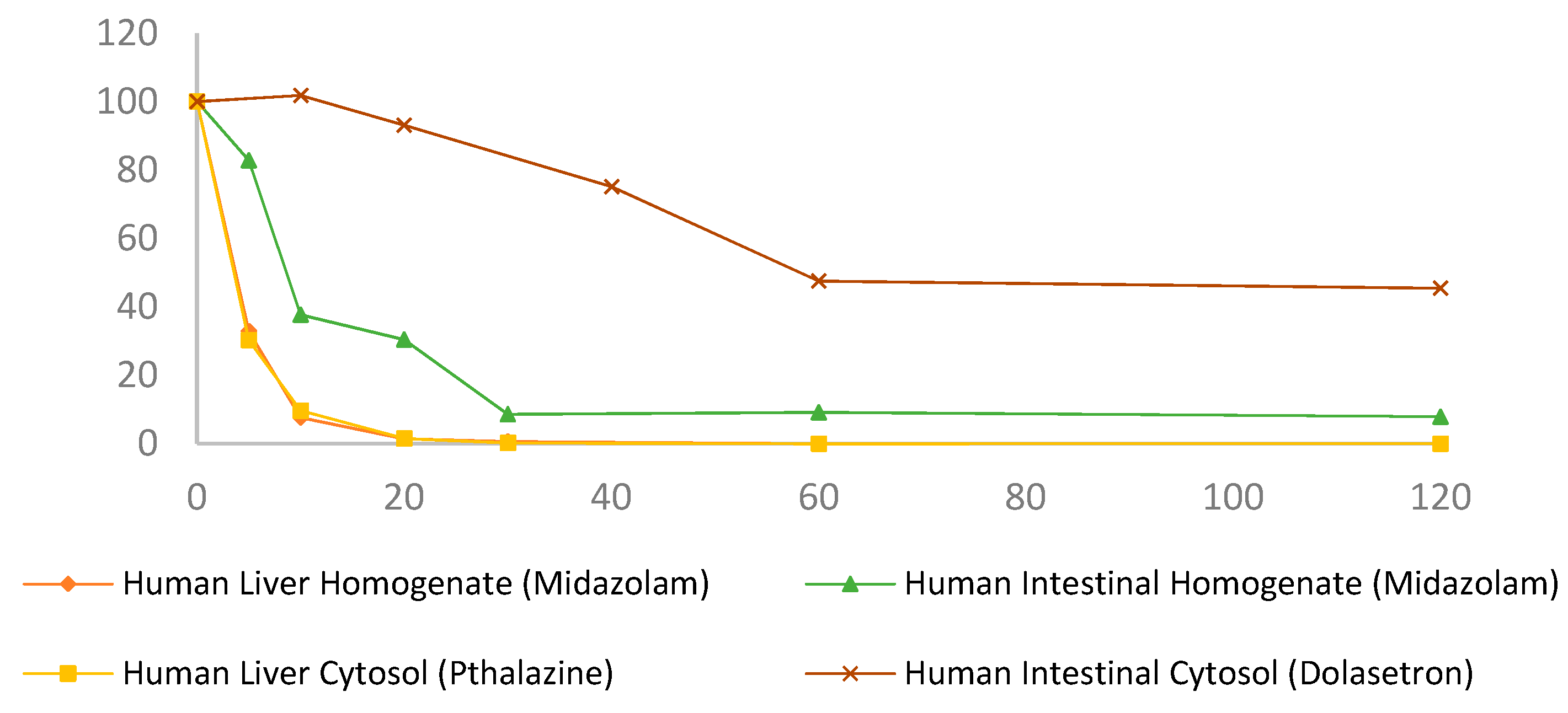
3.1. Progesterone Metabolism by Human and Rat Liver and Intestinal Homogenates
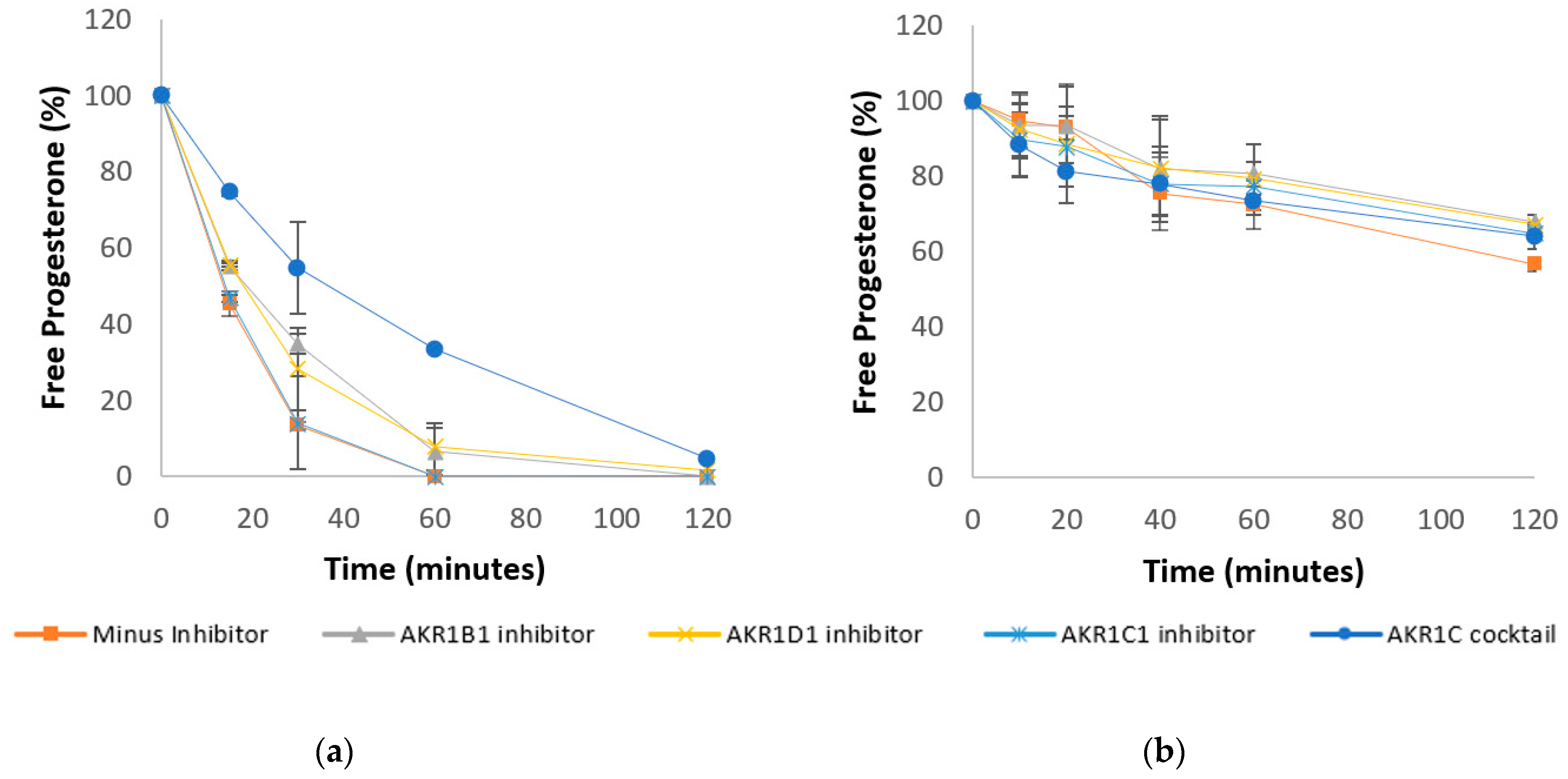
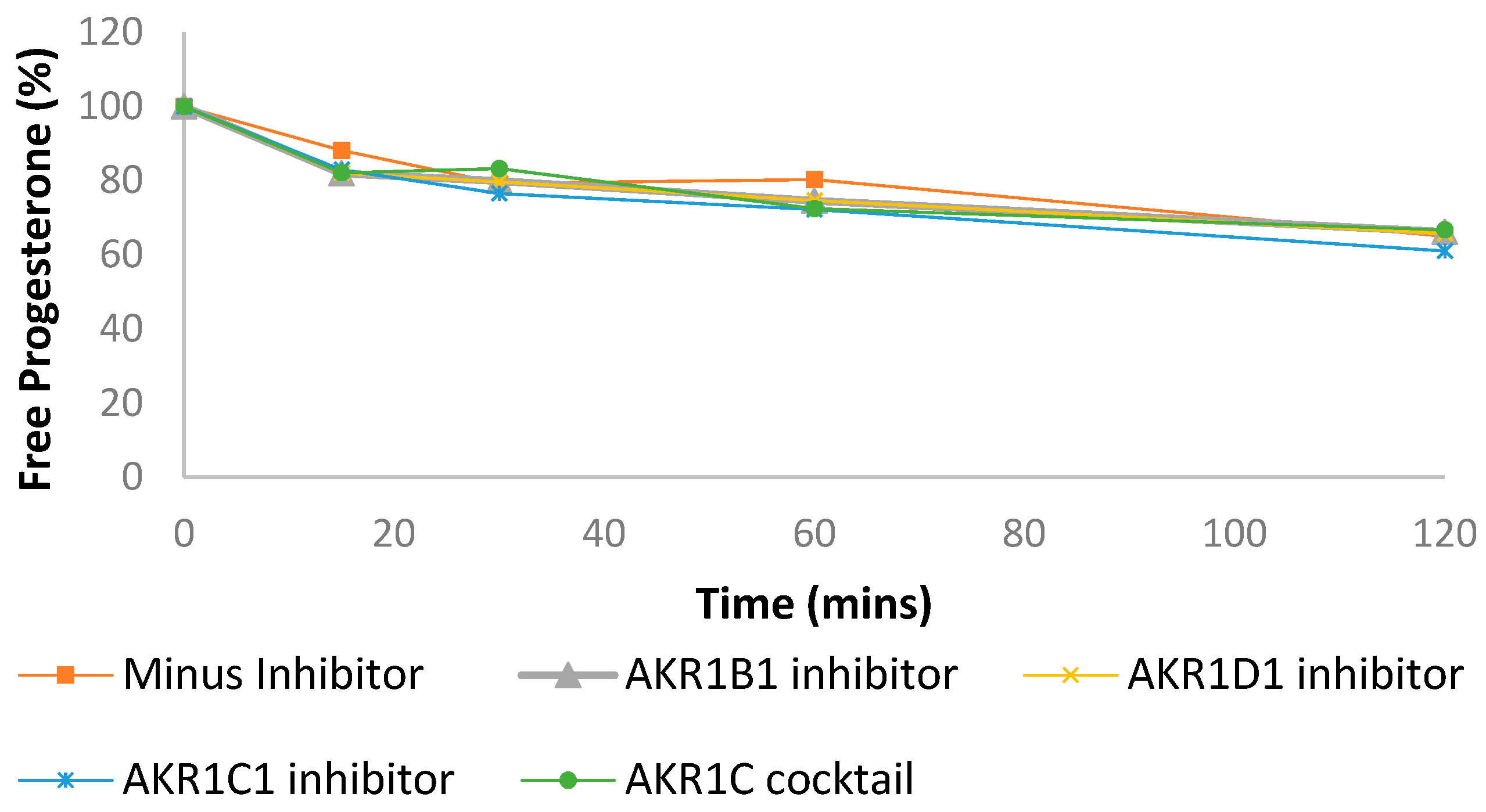
3.2. Progesterone Metabolism in Liver and Intestinal Cytosol and the Role of Aldo-Keto Reductases
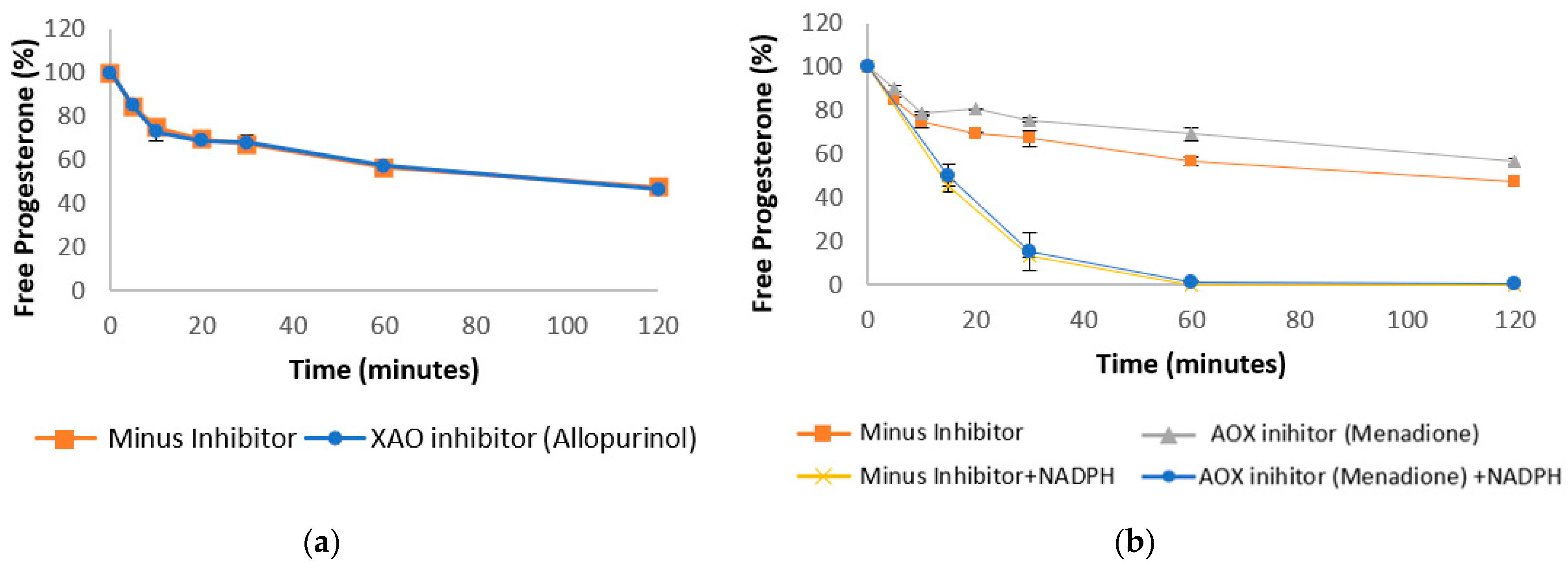
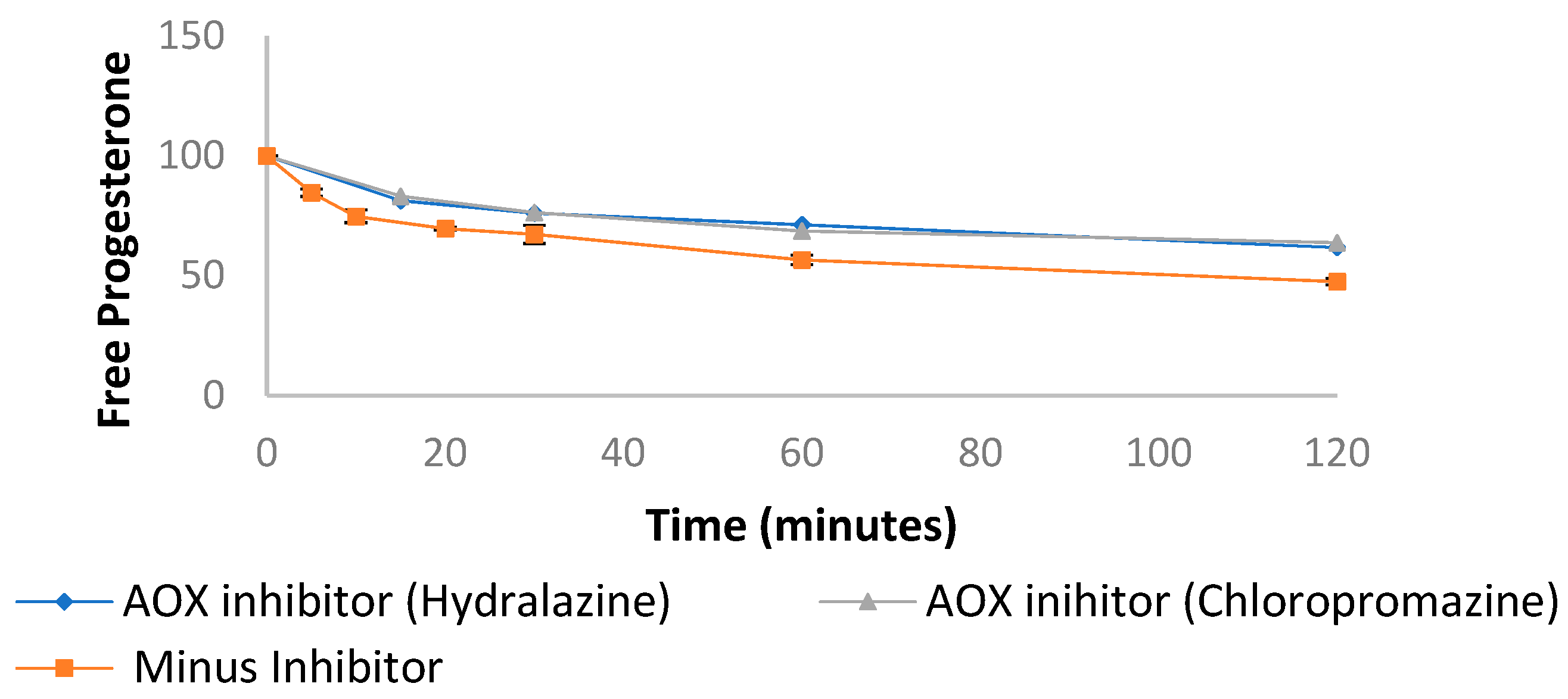
3.3. Progesterone Metabolism by Non-NADPH Dependent Enzymes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A


References
- Oettel, M.; Mukhopadhyay, A. Progesterone: The forgotten hormone in men? Aging Male 2004, 7, 236–257. [Google Scholar] [CrossRef] [PubMed]
- Progesterone. 2020. Available online: https://bnf.nice.org.uk/drug/progesterone.html#indicationsAndDoses (accessed on 5 August 2021).
- Utrogestan 100 mg Capsules Summary of Product Characteristics. 2019. Available online: https://www.medicines.org.uk/emc/product/352/smpc#PHARMACOKINETIC_PROPS (accessed on 5 August 2021).
- Cacace, J.L.; Periscaner, P.H. Progesterone Formulations. U.S. Patent 20,150,148,323, 27 May 2015. [Google Scholar]
- Gadalla, H.H.; Soliman, G.M.; Mohammed, F.A.; El-Sayed, A.M. Development and in vitro/in vivo evaluation of Zn-pectinate microparticles reinforced with chitosan for the colonic delivery of progesterone. Drug Deliv. 2016, 23, 2541–2554. [Google Scholar] [CrossRef] [PubMed]
- Besins, A.; Besse, J. Pharmaceutical Composition Based on Micronized Progesterone, Preparation Method and Uses Thereof. Google Patents WO/2003/041720, 22 May 2003. [Google Scholar]
- Sitruk-Ware, R.; Bricaire, C.; De Lignieres, B.; Yaneva, H.; Mauvais-Jarvis, P. Oral micronized progesterone: Bioavailability pharmacokinetics, pharmacological and therapeutic implications—A review. Contraception 1987, 36, 373–402. [Google Scholar] [CrossRef]
- Penning, T.M.; Burczynski, M.E.; Jez, J.M.; Hung, C.F.; Lin, H.K.; Ma, H.; Moore, M.; Palackal, N.; Ratnam, K. Human 3α-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: Functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J. 2000, 351, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.B. Drug metabolism by cytochromes P450 in the liver and small bowel. Gastroenterol. Clin. N. Am. 1992, 21, 511–526. [Google Scholar] [CrossRef]
- De Sousa, I.P.; Bernkop-Schnürch, A. Pre-systemic metabolism of orally administered drugs and strategies to overcome it. J. Control. Release 2014, 192, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Tubic-Grozdanis, M.; Hilfinger, J.M.; Amidon, G.L.; Kim, J.S.; Kijek, P.; Staubach, P.; Langguth, P. Pharmacokinetics of the CYP 3A substrate simvastatin following administration of delayed versus immediate release oral dosage forms. Pharm. Res. 2008, 25, 1591–1600. [Google Scholar] [CrossRef]
- Mai, Y.; Dou, L.; Yao, Z.; Madla, C.M.; Gavins, F.K.; Taherali, F.; Yin, H.; Orlu, M.; Murdan, S.; Basit, A.W. Quantification of P-Glycoprotein in the Gastrointestinal Tract of Humans and Rodents: Methodology, Gut Region, Sex, and Species Matter. Mol. Pharm. 2021, 18, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Ohland, C.L.; Jobin, C. Microbial activities and intestinal homeostasis: A delicate balance between health and disease. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 28–40. [Google Scholar] [CrossRef] [Green Version]
- McCoubrey, L.E.; Gaisford, S.; Orlu, M.; Basit, A.W. Predicting drug-microbiome interactions with machine learning. Biotechnol. Adv. 2021, 107797. [Google Scholar] [CrossRef] [PubMed]
- Coombes, Z.; Yadav, V.; McCoubrey, L.E.; Freire, C.; Basit, A.W.; Conlan, R.S.; Gonzalez, D. Progestogens Are Metabolized by the Gut Microbiota: Implications for Colonic Drug Delivery. Pharmaceutics 2020, 12, 760. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Uehara, S.; Uno, Y.; Inoue, T.; Sasaki, E.; Yamazaki, H. Progesterone hydroxylation by cytochromes P450 2C and 3A enzymes in marmoset liver microsomes. Xenobiotica 2018, 48, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Mesaros, A.C.; Blair, I.A.; Penning, T.M. Stereospecific reduction of 5β-reduced steroids by human ketosteroid reductases of the AKR (aldo-keto reductase) superfamily: Role of AKR1C1–AKR1C4 in the metabolism of testosterone and progesterone via the 5β-reductase pathway. Biochem. J. 2011, 437, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Rižner, T.L.; Penning, T.M. Role of aldo-keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids 2014, 79, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Taylor, W. The metabolism of progesterone by animal tissues in vitro. 1. Factors influencing the metabolism of progesterone by rat liver and the investigation of the products of metabolism. Biochem. J. 1955, 60, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Liu, M.; Xie, T.; Zhang, B.-F.; Gao, X.-L. Chitosan-modified cholesterol-free liposomes for improving the oral bioavailability of progesterone. Colloids Surf. B Biointerfaces 2017, 159, 580–585. [Google Scholar] [CrossRef]
- Hatton, G.B.; Yadav, V.; Basit, A.W.; Merchant, H.A. Animal farm: Considerations in animal gastrointestinal physiology and relevance to drug delivery in humans. J. Pharm. Sci. 2015, 104, 2747–2776. [Google Scholar] [CrossRef]
- Düsterberg, B.; Hümpel, M.; Speck, U. Terminal half-lives in plasma and bioavailability of norethisterone, levonorgestrel, cyproterone acetate and gestodene in rats, beagles and rhesus monkeys. Contraception 1981, 24, 673–683. [Google Scholar] [CrossRef]
- Musther, H.; Olivares-Morales, A.; Hatley, O.J.; Liu, B.; Hodjegan, A.R. Animal versus human oral drug bioavailability: Do they correlate? Eur. J. Pharm. Sci. 2014, 57, 280–291. [Google Scholar] [CrossRef]
- Bussy, U.; Boujtita, M. Advances in the electrochemical simulation of oxidation reactions mediated by cytochrome P450. Chem. Res. Toxicol. 2014, 27, 1652–1668. [Google Scholar] [CrossRef] [PubMed]
- Desgrés, J.; Fay, L.; Jo, D.-H.; Guiguet, M.; Padieu, P. Oxidoreductive and hydroxylating metabolism of progesterone in rat liver epithelial cell lines. J. Steroid Biochem. 1984, 21, 391–403. [Google Scholar] [CrossRef]
- Blacker, T.S.; Duchen, M.R. Investigating mitochondrial redox state using NADH and NADPH autofluorescence. Free Radic. Biol. Med. 2016, 100, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Merchant, H.A.; Liu, F.; Gul, M.O.; Basit, A.W. Age-mediated changes in the gastrointestinal tract. Int. J. Pharm. 2016, 512, 382–395. [Google Scholar] [CrossRef]
- Luch, J.R. Metoprolol tartrate. Anal. Profiles Drug Subst. 1983, 12, 325–356. [Google Scholar]
- Rao, L.; Taylor, W. The Metabolism of Progesterone by Animal Tissues In Vitro. Sex and Species Differences in Conjugate Formation during the Metabolism of (4-14C) Progesterone by Liver Homogenates. Biochem. J. 1965, 96, 172–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhl, H. Pharmacology of estrogens and progestogens: Influence of different routes of administration. Climacteric 2005, 8, 3–63. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.C.; Kim, H.R.; Cho, H.J.; Yi, H.; Cho, S.M.; Lee, D.G.; Abd El-Aty, A.M.; Kim, J.-S.; Sun, D.; Amidon, G.L. Comparative gene expression of intestinal metabolizing enzymes. Biopharm. Drug Dispos. 2009, 30, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Lemley, C.; Wilson, M. Effect of cytochrome P450 and aldo-keto reductase inhibitors on progesterone inactivation in primary bovine hepatic cell cultures. J. Dairy Sci. 2010, 93, 4613–4624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrns, M.C.; Jin, Y.; Penning, T.M. Inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3): Overview and structural insights. J. Steroid Biochem. Mol. Biol. 2011, 125, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt-Hyatt, M.; Lickteig, A.J.; Klaassen, C.D. Tissue distribution, ontogeny, and chemical induction of aldo-keto reductases in mice. Drug Metab. Dispos. 2013, 41, 1480–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penning, T.M.; Wangtrakuldee, P.; Auchus, R.J. Structural and Functional Biology of Aldo-Keto Reductase Steroid-Transforming Enzymes. Endocr. Rev. 2018, 40, 447–475. [Google Scholar] [CrossRef]
- Hatton, G.B.; Madla, C.M.; Rabbie, S.C.; Basit, A.W. All disease begins in the gut: Influence of gastrointestinal disorders and surgery on oral drug performance. Int. J. Pharm. 2018, 548, 408–422. [Google Scholar] [CrossRef]
- Madla, C.M.; Gavins, F.K.H.; Merchant, H.A.; Orlu, M.; Murdan, S.; Basit, A.W. Let’s talk about sex: Differences in drug therapy in males and females. Adv. Drug Deliv. Rev. 2021, 175, 113804. [Google Scholar] [CrossRef]
- Guerciolini, R.; Szumlanski, C.; Weinshilboum, R.M. Human liver xanthine oxidase: Nature and extent of individual variation. Clin. Pharmacol. Ther. 1991, 50, 663–672. [Google Scholar] [CrossRef]
- Montefiori, M.; Jørgensen, F.S.; Olsen, L. Aldehyde oxidase: Reaction mechanism and prediction of site of metabolism. Acs Omega 2017, 2, 4237–4244. [Google Scholar] [CrossRef]
- Gatzuli, E.; Aten, R.; Behrman, H. Inhibition of Gonadotropin Action and Progesterone Synthesis by Xanthine Oxidase in Rat Luteal Cells*. Endocrinology 1991, 128, 2253–2258. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Igarashi, S.; Tanaka, T. Xanthine oxidase in eutopic and ectopic endometrium in endometriosis and adenomyosis. Fertil. Steril. 2001, 75, 785–790. [Google Scholar] [CrossRef]
- Obach, R.S.; Huynh, P.; Allen, M.C.; Beedham, C. Human Liver Aldehyde Oxidase: Inhibition by 239 Drugs. J. Clin. Pharmacol. 2004, 44, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Floreani, M.; Carpenedo, F. Inhibition of rat liver monooxygenase activities by 2-methyl-1,4-naphthoquinone (menadione). Toxicol. Appl. Pharmacol. 1990, 105, 333–339. [Google Scholar] [CrossRef]
- Bentham, Z. A Novel Progesterone Oral Formulation for the Treatment of Endometrial Hyperplasia with Reduced Adverse Signalling Compared to Synthetic Progestins. Ph.D. Thesis, Swansea University, Swansea, UK, 2015. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solution | t1/2 (min) | % Drug Remaining After 60 min |
|---|---|---|
| Progesterone in liver homogenates (Figure 2a) | ||
| Human liver homogenate + NADPH | 2.7 ± 0.05 | 0.27 ± 0.26 |
| Human liver homogenate | 38.95 ± 11.68 | 43.32 ± 4.84 |
| Rat liver homogenate + NADPH | 2.72 ± 0.05 | 0.03 ± 0.0005 |
| Rat liver homogenate | 39.77 ± 10.16 | 46.04 ± 0.69 |
| Progesterone in intestinal homogenates (Figure 2b) | ||
| Human intestinal homogenate + NADPH | 157.22 ± 27.41 | 73.92 ± 5.11 |
| Human intestinal homogenate | 155.84 ± 13.75 | 75.82 ± 9.35 |
| Rat intestinal homogenate + NADPH | 197.63 ± 2.91 | 76.67 ± 4.72 |
| Rat intestinal homogenate | 352.06 ± 36.87 | 90.93 ± 2.19 |
| Progesterone in liver cytosol + NADPH (Figure 3a) | ||
| Human liver Cytosol | 13.73 ± 0.81 | 0.04 ± 0.01 |
| Human liver Cytosol + AKR1B1 inhibitor | 18.81 ± 0.96 | 6.6 ± 6.24 |
| Human liver Cytosol + AKR1D1 inhibitor | 18.46 ± 1.7 | 7.85 ± 6.31 |
| Human liver Cytosol + AKR1C1 inhibitor | 14.08 ± 0.24 | 0.11 ± 0.04 |
| Human liver Cytosol + AKR1C1 cocktail | 35.56 ± 6.67 | 33.5 ± 1.72 |
| Progesterone in intestinal cytosol + NADPH (Figure 3b) | ||
| Human intestinal Cytosol | 129.52 ± 2.75 | 72.51 ± 6.48 |
| Human intestinal Cytosol + AKR1B inhibitor | 184.12 ± 18.95 | 80.87 ± 7.84 |
| Human intestinal Cytosol + AKR1D1 inhibitor | 181.74 ± 4.39 | 79.56 ± 4.44 |
| Human intestinal Cytosol + AKR1C1 inhibitor | 167.39 ± 3.03 | 77.38 ± 6.37 |
| Human intestinal Cytosol + AKR1C cocktail | 161.82 ± 14.46 | 73.56 ± 3.69 |
| Progesterone in liver cytosol (Figure 4a) | ||
| Human intestinal Cytosol | 103.1 ± 9.38 | 56.6 ± 2.07 |
| Human liver Cytosol + XAO inhibitor | 99.63 ± 9.14 | 57.09 ± 2.01 |
| Progesterone in liver cytosol (Figure 4b) | ||
| Human intestinal Cytosol | 103.1 ± 9.38 | 56.6 ± 2.07 |
| Human liver Cytosol + AOX inhibitor | 132.6 ± 4.69 | 69.14 ± 2.91 |
| Human liver Cytosol + NADPH | 13.73 ± 0.81 | 0.04 ± 0.01 |
| Human liver Cytosol + NADPH + AOX inhibitor | 15.49 ± 2.03 | 0.97 ± 0.88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coombes, Z.; Plant, K.; Freire, C.; Basit, A.W.; Butler, P.; Conlan, R.S.; Gonzalez, D. Progesterone Metabolism by Human and Rat Hepatic and Intestinal Tissue. Pharmaceutics 2021, 13, 1707. https://doi.org/10.3390/pharmaceutics13101707
Coombes Z, Plant K, Freire C, Basit AW, Butler P, Conlan RS, Gonzalez D. Progesterone Metabolism by Human and Rat Hepatic and Intestinal Tissue. Pharmaceutics. 2021; 13(10):1707. https://doi.org/10.3390/pharmaceutics13101707
Chicago/Turabian StyleCoombes, Zoe, Katie Plant, Cristina Freire, Abdul W. Basit, Philip Butler, R. Steven Conlan, and Deyarina Gonzalez. 2021. "Progesterone Metabolism by Human and Rat Hepatic and Intestinal Tissue" Pharmaceutics 13, no. 10: 1707. https://doi.org/10.3390/pharmaceutics13101707
APA StyleCoombes, Z., Plant, K., Freire, C., Basit, A. W., Butler, P., Conlan, R. S., & Gonzalez, D. (2021). Progesterone Metabolism by Human and Rat Hepatic and Intestinal Tissue. Pharmaceutics, 13(10), 1707. https://doi.org/10.3390/pharmaceutics13101707