
Induction of Apoptosis by Isoalantolactone in Human Hepatocellular Carcinoma Hep3B Cells through Activation of the ROS-Dependent JNK Signaling Pathway

 , , ,
, , ,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture and IALT Treatment
2.3. Cell Viability Assay
2.4. Detection of Apoptotic Morphological Changes
2.5. Determination of Apoptosis by Flow Cytometry
2.6. TUNEL Assay
2.7. Western Blot Analysis
2.8. Caspase Activity Assay
2.9. Measurement of Mitochondrial Membrane Potential (MMP)
2.10. Determination of ROS Generation
2.11. Spheroid Formation Assay
2.12. Determination of ROS Levels in the Spheroids
2.13. TUNEL Assay of the Spheroids
2.14. Western Blot Analysis of the Spheroids
2.15. Statistical Analysis
3. Results
3.1. IALT Inhibits Cell Viability and Induces Apoptosis in Hep3B Cells
3.2. IALT Activates Caspases in Hep3B Cells
3.3. IALT Modulates the Expression of Bcl-2 Family Proteins and Increases Mitochondrial Dysfunction in Hep3B Cells
3.4. IALT Increases Intracellular ROS Generation in Hep3B Cells
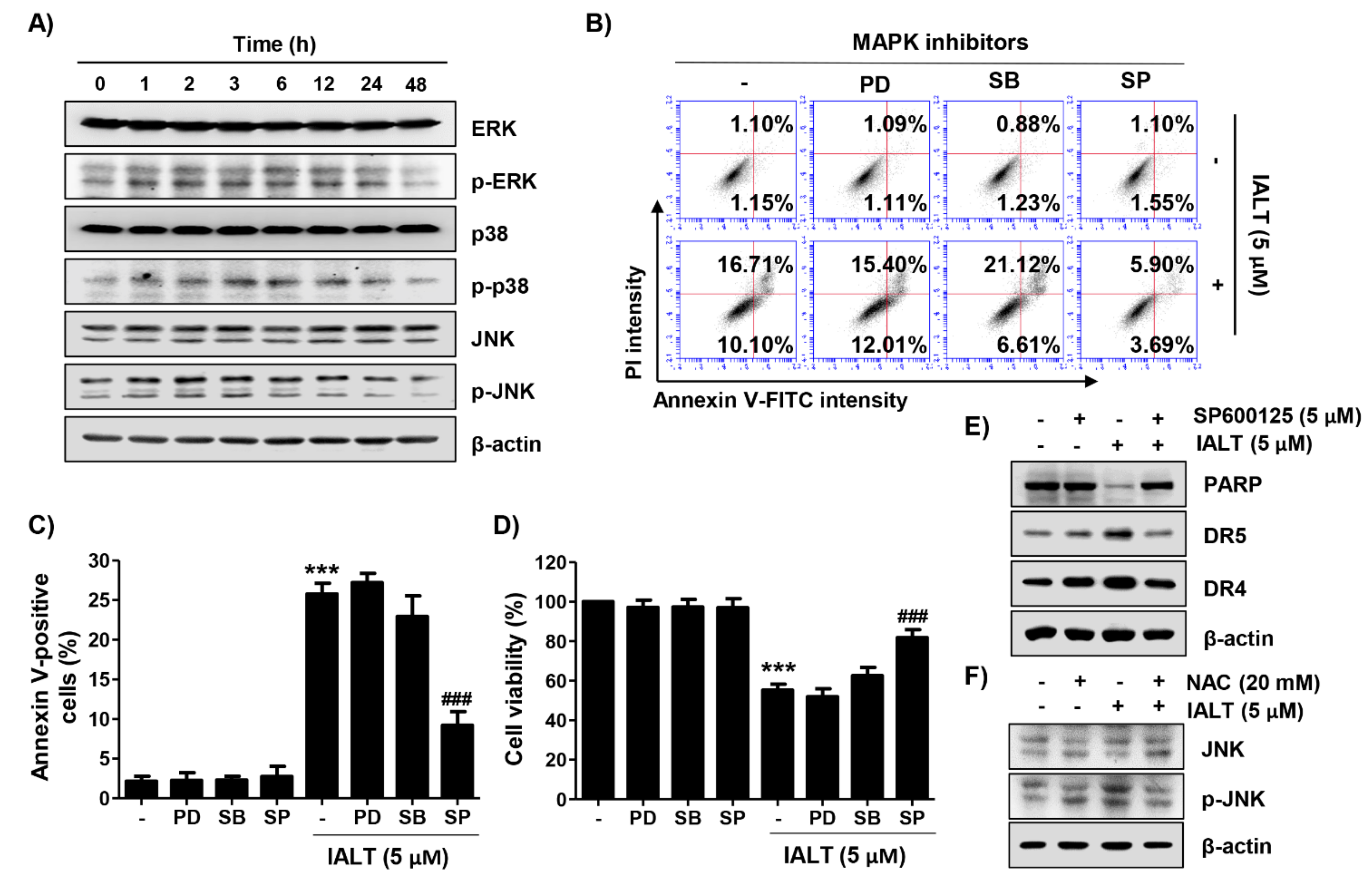
3.5. IALT Activates the MAPK Signaling Pathway in Hep3B Cells
3.6. IALT Suppresses the Growth of Hep3B MTSs
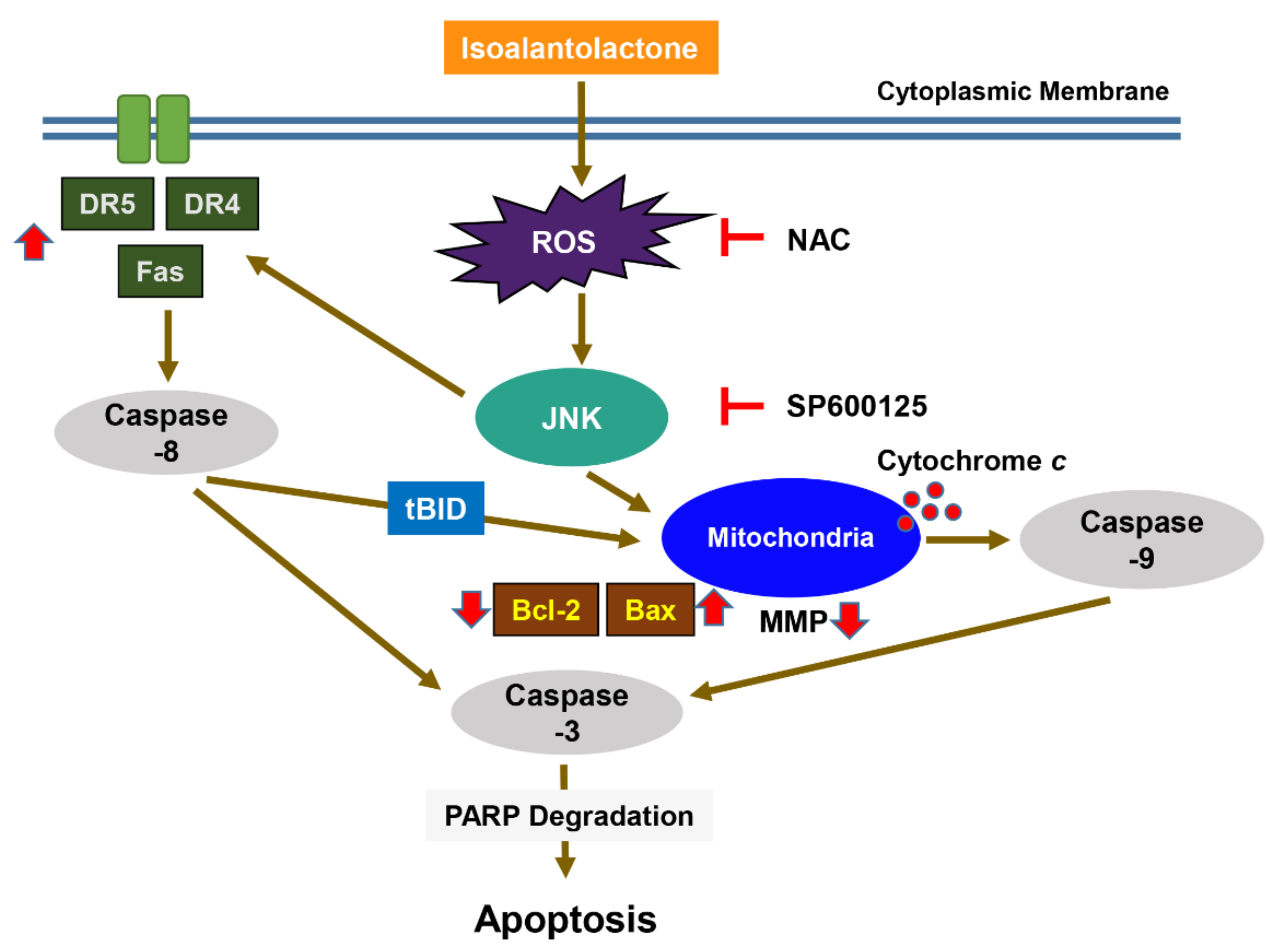
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular Therapies and Precision Medicine for Hepatocellular Carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Bruix, J.; Gores, G.J.; Mazzaferro, V. Hepatocellular Carcinoma: Clinical Frontiers and Perspectives. Gut 2014, 63, 844–855. [Google Scholar] [CrossRef]
- Moeini, A.; Cornellà, H.; Villanueva, A. Emerging Signaling Pathways in Hepatocellular Carcinoma. Liver Cancer 2012, 1, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced Hepatocellular Carcinoma. J. Hepatol. 2016, 64 (Suppl. 1), S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The Mechanisms of Sorafenib Resistance in Hepatocellular Carcinoma: Theoretical Basis and Therapeutic Aspects. Signal. Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Muñoz-Martínez, S.; Iserte, G.; Sanduzzi-Zamparelli, M.; Llarch, N.; Reig, M. Current Pharmacological Treatment of Hepatocellular Carcinoma. Curr. Opin. Pharmacol. 2021, 60, 141–148. [Google Scholar] [CrossRef]
- Sarcognato, S.; García-Lezana, T.; Villanueva, A. Mechanisms of Action of Drugs Effective in Hepatocellular Carcinoma. Clin. Liver Dis. 2019, 14, 62–65. [Google Scholar] [CrossRef]
- Degroote, H.; Van Dierendonck, A.; Geerts, A.; Van Vlierberghe, H.; Devisscher, L. Preclinical and Clinical Therapeutic Strategies Affecting Tumor-associated Macrophages in Hepatocellular Carcinoma. J. Immunol. Res. 2018, 2018, 7819520. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, Y. Promising New Strategies for Hepatocellular Carcinoma. Hepatol. Res. 2017, 47, 251–265. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the Hallmarks of Cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major Apoptotic Mechanisms and Genes Involved in Apoptosis. Tumour Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling Pathway of MAPK/ERK in Cell Proliferation, Differentiation, Migration, Senescence and Apoptosis. J. Recept. Signal. Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK Pathways in the Regulation of Metabolic Reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef]
- Badrinath, N.; Yoo, S.Y. Mitochondria in Cancer: In the Aspects of Tumorigenesis and Targeted Therapy. Carcinogenesis 2018, 39, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS Signaling in the Biology of Cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Fakhri, S.; Tomas, M.; Capanoglu, E.; Hussain, Y.; Abbaszadeh, F.; Lu, B.; Hu, X.; Wu, J.; Zou, L.; Smeriglio, A.; et al. Antioxidant and Anticancer Potentials of Edible Flowers: Where Do We Stand? Crit. Rev. Food Sci. Nutr. 2021, 1–57. [Google Scholar] [CrossRef] [PubMed]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Surowiak, A.K.; Balcerzak, L.; Lochyński, S.; Strub, D.J. Biological Activity of Selected Natural and Synthetic Terpenoid Lactones. Int. J. Mol. Sci. 2021, 22, 5036. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Gómez, A.; Ontiveros-Rodríguez, J.C.; Pablo-Pérez, S.S.; Vargas-Díaz, M.E.; Garduño-Siciliano, L. The Potential Role of Sesquiterpene Lactones Isolated from Medicinal Plants in the Treatment of the Metabolic Syndrome—A Review. S. Afr. J. Bot. 2020, 135, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-P.; Jia, Z.-L.; Huo, X.-K.; Tian, X.-G.; Feng, L.; Wang, C.; Zhang, B.-J.; Zhao, W.-Y.; Ma, X.-C. Medicinal Inula Species: Phytochemistry, Biosynthesis, and Bioactivities. Am. J. Chin. Med. 2021, 49, 315–358. [Google Scholar] [CrossRef] [PubMed]
- Tavares, W.R.; Seca, A.M.L.; Inula, L. Secondary Metabolites against Oxidative Stress-Related Human Diseases. Antioxidants 2019, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Kaval, I.; Behçet, L.; Cakilcioglu, U. Ethnobotanical Study on Medicinal Plants in Geçitli and Its Surrounding (Hakkari-Turkey). J. Ethnopharmacol. 2014, 155, 171–184. [Google Scholar] [CrossRef]
- Zhou, Y.; Guo, Y.; Wen, Z.; Ci, X.; Xia, L.; Wang, Y.; Deng, X.; Wang, J. Isoalantolactone Enhances the Antimicrobial Activity of Penicillin G against Staphylococcus aureus by Inactivating beta-lactamase during Protein Translation. Pathogens 2020, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.-S.; Lee, H.-S.; Cho, H.-R.; Kim, K.-J.; Kim, J.-H.; Safe, S.; Lee, S.-O. Dual Targeting of Nur77 and AMPKα by Isoalantolactone Inhibits Adipogenesis in vitro and Decreases Body Fat Mass in vivo. Int. J. Obes. 2019, 43, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Arha, D.; Ramakrishna, E.; Gupta, A.P.; Rai, A.K.; Sharma, A.; Ahmad, I.; Riyazuddin, M.; Gayen, J.R.; Maurya, R.; Tamrakar, A.K. Isoalantolactone Derivative Promotes Glucose Utilization in Skeletal Muscle Cells and Increases Energy Expenditure in db/db Mice via Activating AMPK-dependent Signaling. Mol. Cell Endocrinol. 2018, 460, 134–151. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, K.; Gao, X.; Zhao, K.; Chen, H.; Xu, M. Anti-inflammatory Effects of Isoalantolactone on LPS-stimulated BV2 Microglia Cells through Activating GSK-3beta-Nrf2 Signaling Pathway. Int. Immunopharmacol. 2018, 65, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.B.; Tian, L.; Yang, B.; Zhou, H.Y. Isoalantolactone Protects LPS-induced Acute Lung Injury through Nrf2 Activation. Microb. Pathog. 2018, 123, 213–218. [Google Scholar] [CrossRef]
- Seo, J.Y.; Lim, S.S.; Kim, J.; Lee, K.W.; Kim, J.S. Alantolactone and Isoalantolactone Prevent Amyloid β25-35-induced Toxicity in Mouse Cortical Neurons and Scopolamine-induced Cognitive Impairment in Mice. Phytother. Res. 2017, 31, 801–811. [Google Scholar] [CrossRef]
- Liu, Y.; Meng, Q.; Jing, L.; Feng, L.; Zhou, Z.; Ni, Z. 11, 13-Dehydro Lactone Moiety in Gynecologic Cancer Cells. Iran. J. Public Health 2020, 49, 2103–2110. [Google Scholar]
- Xu. R.; Peng, Y.; Wang, M.; Li, X. Intestinal Absorption of Isoalantolactone and Alantolactone, Two Sesquiterpene Lactones from Radix Inulae, using Caco-2 Cells. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 295–303. [Google Scholar] [CrossRef]
- Weng, Z.; Gao, H.; Hu, J.; Fan, Y.; Wang, H.; Li, L. Isoalantolactone Induces Autophagic Cell Death in SKOV3 Human Ovarian Carcinoma Cells via Upregulation of PEA-15. Oncol. Rep. 2016, 35, 833–840. [Google Scholar] [CrossRef]
- Xu, R.; Zhou, G.; Peng, Y.; Wang, M.; Li, X. Pharmacokinetics, Tissue Distribution and Excretion of Isoalantolactone and Alantolactone in Rats after Oral Administration of Radix Inulae Extract. Molecules 2015, 20, 7719–7736. [Google Scholar] [CrossRef]
- Zhang, C.; Huang, L.; Xiong, J.; Xie, L.; Ying, S.; Jia, Y.; Yao, Y.; Song, X.; Zeng, Z.; Yuan, J. Isoalantolactone Inhibits Pancreatic Cancer Proliferation by Regulation of PI3K and Wnt Signal Pathway. PLoS ONE 2021, 16, e0247752. [Google Scholar]
- Xing, J.S.; Wang, X.; Lan, Y.L.; Lou, J.C.; Ma, B.; Zhu, T.; Zhang, H.; Wang, D.; Yu, Z.; Yuan, Z.; et al. Isoalantolactone Inhibits IKKbeta Kinase Activity to Interrupt the NF-kappaB/COX-2-mediated Signaling Cascade and Induces Apoptosis Regulated by the Mitochondrial Translocation of Cofilin in Glioblastoma. Cancer Med. 2019, 8, 1655–1670. [Google Scholar] [CrossRef]
- Chen, W.; Li, P.; Liu, Y.; Yang, Y.; Ye, X.; Zhang, F.; Huang, H. Isoalantolactone Induces Apoptosis through ROS-mediated ER Stress and Inhibition of STAT3 in Prostate Cancer Cells. J. Exp. Clin. Cancer Res. 2018, 37, 309. [Google Scholar] [CrossRef]
- Lu, Z.; Zhang, G.; Zhang, Y.; Hua, P.; Fang, M.; Wu, M.; Liu, T. Isoalantolactone Induces Apoptosis through Reactive Oxygen Species-dependent Upregulation of Death Receptor 5 in Human Esophageal Cancer Cells. Toxicol. Appl. Pharmacol. 2018, 352, 46–58. [Google Scholar] [CrossRef]
- Jin, C.; Zhang, G.; Zhang, Y.; Hua, P.; Song, G.; Sun, M.; Li, X.; Tong, T.; Li, B.; Zhang, X. Isoalantolactone Induces Intrinsic Apoptosis through p53 Signaling Pathway in Human Lung Squamous Carcinoma Cells. PLoS ONE 2017, 12, e0181731. [Google Scholar] [CrossRef]
- Li, Z.; Qin, B.; Qi, X.; Mao, J.; Wu, D. Isoalantolactone Induces Apoptosis in Human Breast Cancer Cells via ROS-mediated Mitochondrial Pathway and Downregulation of SIRT1. Arch. Pharm. Res. 2016, 39, 1441–1453. [Google Scholar] [CrossRef]
- Wang, J.; Cui, L.; Feng, L.; Zhang, Z.; Song, J.; Liu, D.; Jia, X. Isoalantolactone Inhibits the Migration and Invasion of Human Breast Cancer MDA-MB-231 Cells via Suppression of the p38 MAPK/NF-κB Signaling Pathway. Oncol. Rep. 2016, 36, 1269–1276. [Google Scholar] [CrossRef]
- Fan, Y.; Weng, Z.; Gao, H.; Hu, J.; Wang, H.; Li, L.; Liu, H. Isoalantolactone Enhances the Radiosensitivity of UMSCC-10A Cells via Specific Inhibition of Erk1/2 Phosphorylation. PLoS ONE 2015, 10, e0145790. [Google Scholar] [CrossRef] [PubMed]
- Di, W.; Khan, M.; Rasul, A.; Sun, M.; Sui, Y.; Zhong, L.; Yang, L.; Zhu, Q.; Feng, L.; Ma, T. Isoalantolactone Inhibits Constitutive NF-κB Activation and Induces Reactive Oxygen Species-mediated Apoptosis in Osteosarcoma U2OS Cells through Mitochondrial Dysfunction. Oncol. Rep. 2014, 32, 1585–1593. [Google Scholar] [CrossRef] [PubMed]
- Rasul, A.; Khan, M.; Yu, B.; Ali, M.; Bo, Y.J.; Yang, H.; Ma, T. Isoalantolactone, a Sesquiterpene Lactone, Induces Apoptosis in SGC-7901 Cells via Mitochondrial and Phosphatidylinositol 3-kinase/Akt Signaling Pathways. Arch. Pharm. Res. 2013, 36, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Ding, C.; Rasul, A.; Yi, F.; Li, T.; Gao, H.; Gao, R.; Zhong, L.; Zhang, K.; Fang, X.; et al. Isoalantolactone Induces Reactive Oxygen Species Mediated Apoptosis in Pancreatic Carcinoma PANC-1 Cells. Int. J. Biol. Sci. 2012, 8, 533–547. [Google Scholar] [CrossRef]
- Bordanaba-Florit, G.; Madarieta, I.; Olalde, B.; Falcón-Pérez, J.M.; Royo, F. 3D Cell Cultures as Prospective Models to Study Extracellular Vesicles in Cancer. Cancers 2021, 13, 307. [Google Scholar] [CrossRef]
- Heredia-Soto, V.; Redondo, A.; Kreilinger, J.J.P.; Martínez-Marín, V.; Berjón, A.; Mendiola, M. 3D Culture Modelling: An Emerging Approach for Translational Cancer Research in Sarcomas. Curr. Med. Chem. 2020, 27, 4778–4788. [Google Scholar] [CrossRef]
- Han, S.J.; Kwon, S.; Kim, K.S. Challenges of Applying Multicellular Tumor Spheroids in Preclinical Phase. Cancer Cell Int. 2021, 21, 152. [Google Scholar] [CrossRef]
- Hamilton, G.; Rath, B. Applicability of Tumor Spheroids for in vitro Chemosensitivity Assays. Expert. Opin. Drug Metab. Toxicol. 2019, 15, 15–23. [Google Scholar] [CrossRef]
- Castro, F.; Pereira, C.L.; Macedo, M.H.; Almeida, A.; Silveira, M.J.; Dias, S.; Cardoso, A.P.; Oliveira, M.J.; Sarmento, B. Advances on Colorectal Cancer 3D Models: The Needed Translational Technology for Nanomedicine Screening. Adv. Drug Deliv. Rev. 2021, 175, 113824. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ouyang, G.; Lu, W.; Zhang, H. Long non-coding RNA HOTAIR Promotes Hepatocellular Carcinoma Progression by Regulating miR-526b-3p/DHX33 Axis. Genes Genom. 2021, 43, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H. Trans-cinnamaldehyde Protects C2C12 Myoblasts from DNA Damage, Mitochondrial Dysfunction and Apoptosis Caused by Oxidative Stress through Inhibiting ROS Production. Genes Genom. 2021, 43, 303–312. [Google Scholar] [CrossRef]
- Hwangbo, H.; Kim, S.Y.; Lee, H.; Park, S.-H.; Hong, S.H.; Park, C.; Kim, G.-Y.; Leem, S.-H.; Hyun, J.W.; Cheong, J.; et al. Auranofin Enhances Sulforaphane-Mediated Apoptosis in Hepatocellular Carcinoma Hep3B Cells through Inactivation of the PI3K/Akt Signaling Pathway. Biomol. Ther. (Seoul) 2020, 28, 443–455. [Google Scholar] [CrossRef]
- Park, S.; Kim, M.; Hong, Y.; Lee, H.; Tran, Q.; Kim, C.; Kwon, S.H.; Park, J.; Park, J.; Kim, S.-H. Myristoylated TMEM39AS41, a Cell-permeable Peptide, Causes Lung Cancer Cell Death. Toxicol. Res. 2020, 36, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.N.A.; Le, B.; Yang, S.H. Anticancer Activity of the Potential Pyropia yezoensis Galactan Fractionated in Human Prostate Cancer Cells. Biotechnol. Bioprocess Eng. 2021, 26, 63–70. [Google Scholar] [CrossRef]
- Liang, Y.; Kong, D.; Zhang, Y.; Li, S.; Li, Y.; Ramamoorthy, A.; Ma, J. Fisetin Inhibits Cell Proliferation and Induces Apoptosis via JAK/STAT3 Signaling Pathways in Human Thyroid TPC 1 Cancer Cells. Biotechnol. Bioprocess. Eng. 2020, 25, 197–205. [Google Scholar] [CrossRef]
- Prates Mori, M.; de Souza-Pinto, N.C. Role of Mitochondrial Dysfunction in the Pathophysiology of DNA Repair Disorders. Cell Biol. Int. 2018, 42, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Saki, M.; Prakash, A. DNA Damage Related Crosstalk Between the Nucleus and Mitochondria. Free Radic. Biol. Med. 2017, 107, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Birkinshaw, R.W.; Czabotar, P.E. The BCL-2 Family of Proteins and Mitochondrial Outer Membrane Permeabilisation. Semin. Cell Dev. Biol. 2017, 72, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Edlich, F. BCL-2 Proteins and Apoptosis: Recent Insights and Unknowns. Biochem. Biophys. Res. Commun. 2018, 500, 26–34. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.Y.; Lee, H.; Ji, S.Y.; Kim, S.Y.; Hwangbo, H.; Park, S.-H.; Kim, G.-Y.; Park, C.; Leem, S.-H.; Hong, S.H.; et al. Induction of Apoptosis by Isoalantolactone in Human Hepatocellular Carcinoma Hep3B Cells through Activation of the ROS-Dependent JNK Signaling Pathway. Pharmaceutics 2021, 13, 1627. https://doi.org/10.3390/pharmaceutics13101627
Kim MY, Lee H, Ji SY, Kim SY, Hwangbo H, Park S-H, Kim G-Y, Park C, Leem S-H, Hong SH, et al. Induction of Apoptosis by Isoalantolactone in Human Hepatocellular Carcinoma Hep3B Cells through Activation of the ROS-Dependent JNK Signaling Pathway. Pharmaceutics. 2021; 13(10):1627. https://doi.org/10.3390/pharmaceutics13101627
Chicago/Turabian StyleKim, Min Yeong, Hyesook Lee, Seon Yeong Ji, So Young Kim, Hyun Hwangbo, Shin-Hyung Park, Gi-Young Kim, Cheol Park, Sun-Hee Leem, Su Hyun Hong, and et al. 2021. "Induction of Apoptosis by Isoalantolactone in Human Hepatocellular Carcinoma Hep3B Cells through Activation of the ROS-Dependent JNK Signaling Pathway" Pharmaceutics 13, no. 10: 1627. https://doi.org/10.3390/pharmaceutics13101627
APA StyleKim, M. Y., Lee, H., Ji, S. Y., Kim, S. Y., Hwangbo, H., Park, S.-H., Kim, G.-Y., Park, C., Leem, S.-H., Hong, S. H., & Choi, Y. H. (2021). Induction of Apoptosis by Isoalantolactone in Human Hepatocellular Carcinoma Hep3B Cells through Activation of the ROS-Dependent JNK Signaling Pathway. Pharmaceutics, 13(10), 1627. https://doi.org/10.3390/pharmaceutics13101627