
D-Peptide-Based Probe for CXCR4-Targeted Molecular Imaging and Radionuclide Therapy

 ,
,  , , , ,
, , , ,
Abstract
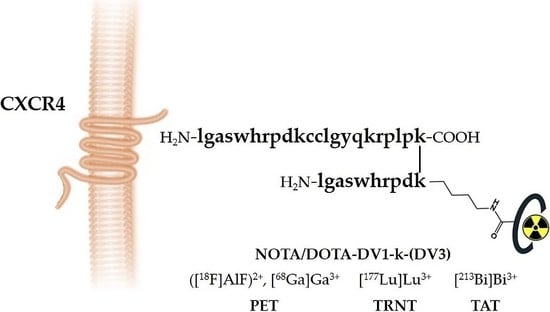
:
1. Introduction
2. Materials and Methods
2.1. Synthesis and Radiolabeling
2.2. Analysis
2.2.1. LC-HRMS
2.2.2. iTLC
2.2.3. RadioHPLC
2.2.4. Animals and Cell Lines
2.3. In Vitro Evaluation
2.3.1. Determination of hCXCR4/mCXCR4 Affinity
2.3.2. Stability Assessment (Supplementary Materials)
2.3.3. Binding and Internalization Assay
2.4. In Vivo Evaluation
Stability Assessment
2.5. Ex Vivo Autoradiography
2.6. Preparation of Tumor Xenograft Model
2.7. PET/CT Imaging and Biodistribution Study
2.8. Image Processing and Analysis
2.9. Statistical Analysis
3. Results & Discussion
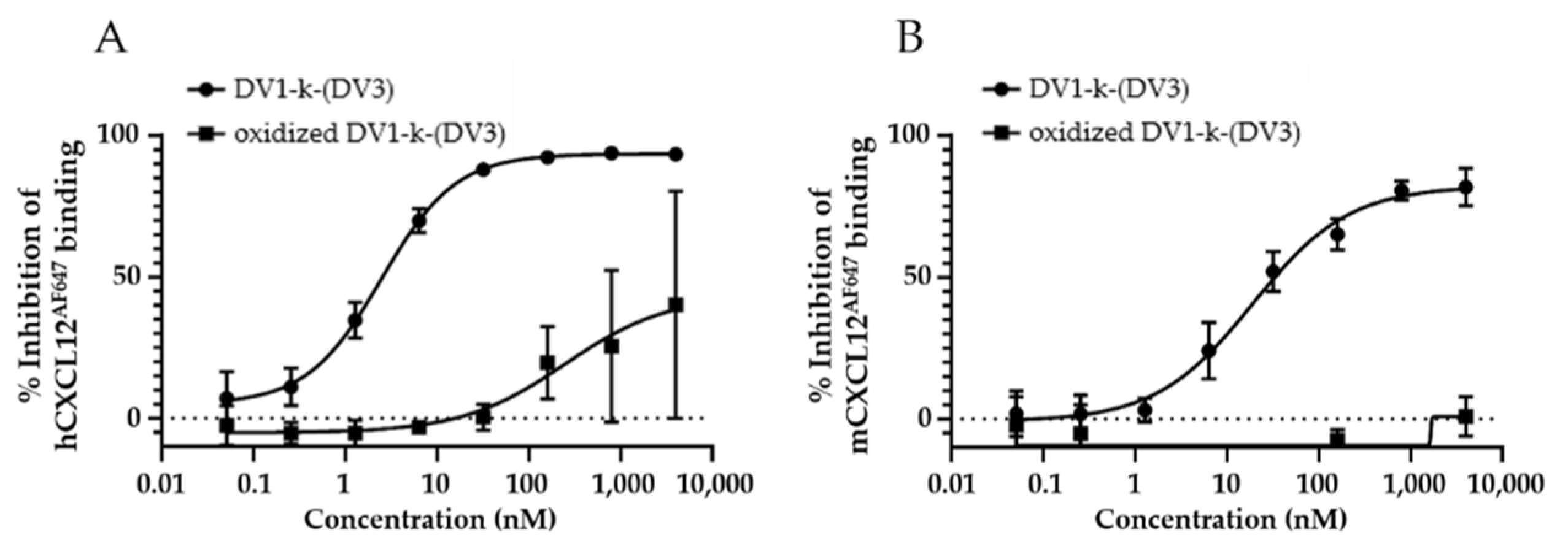
3.1. In Vitro Evaluation
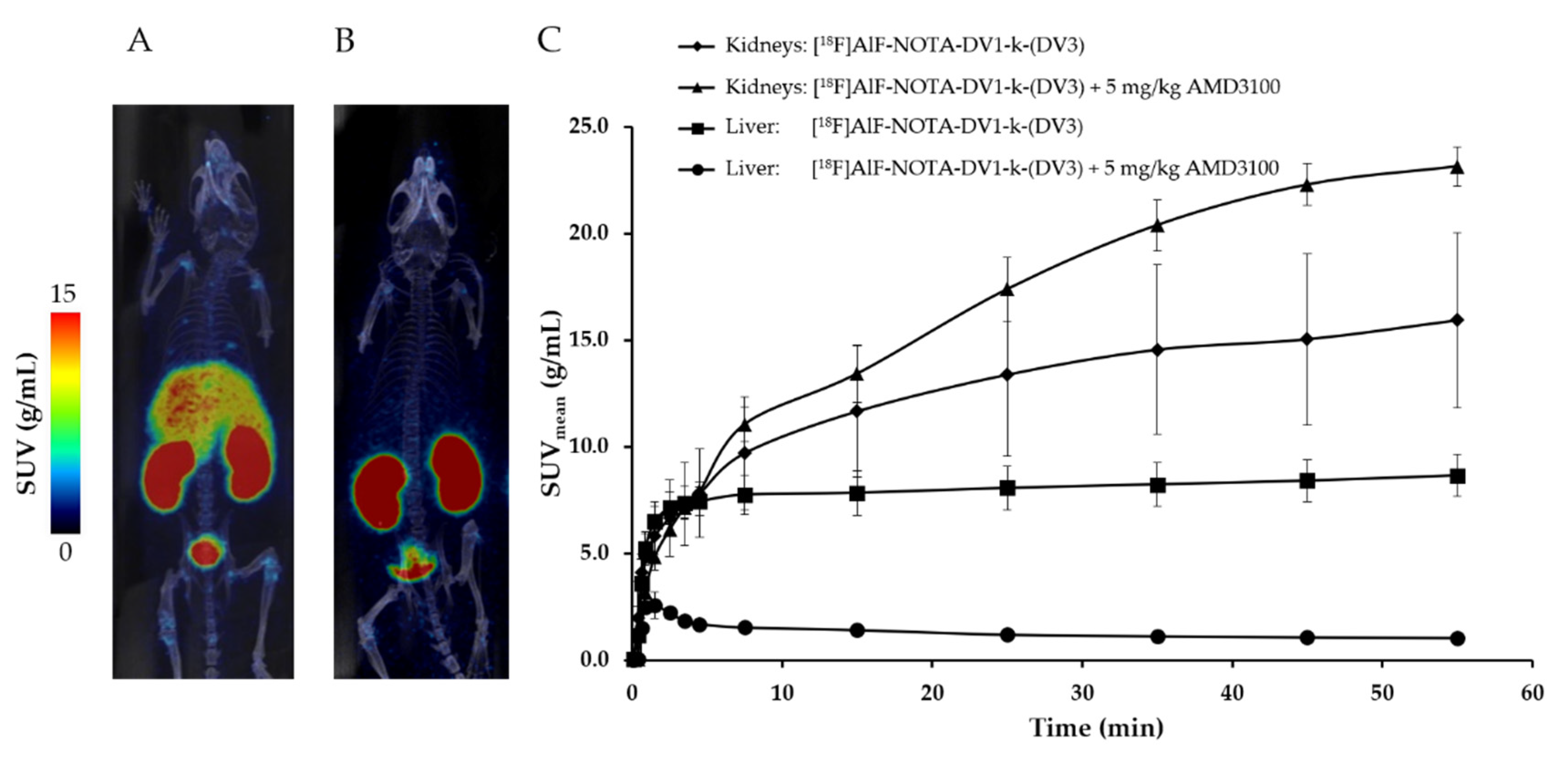
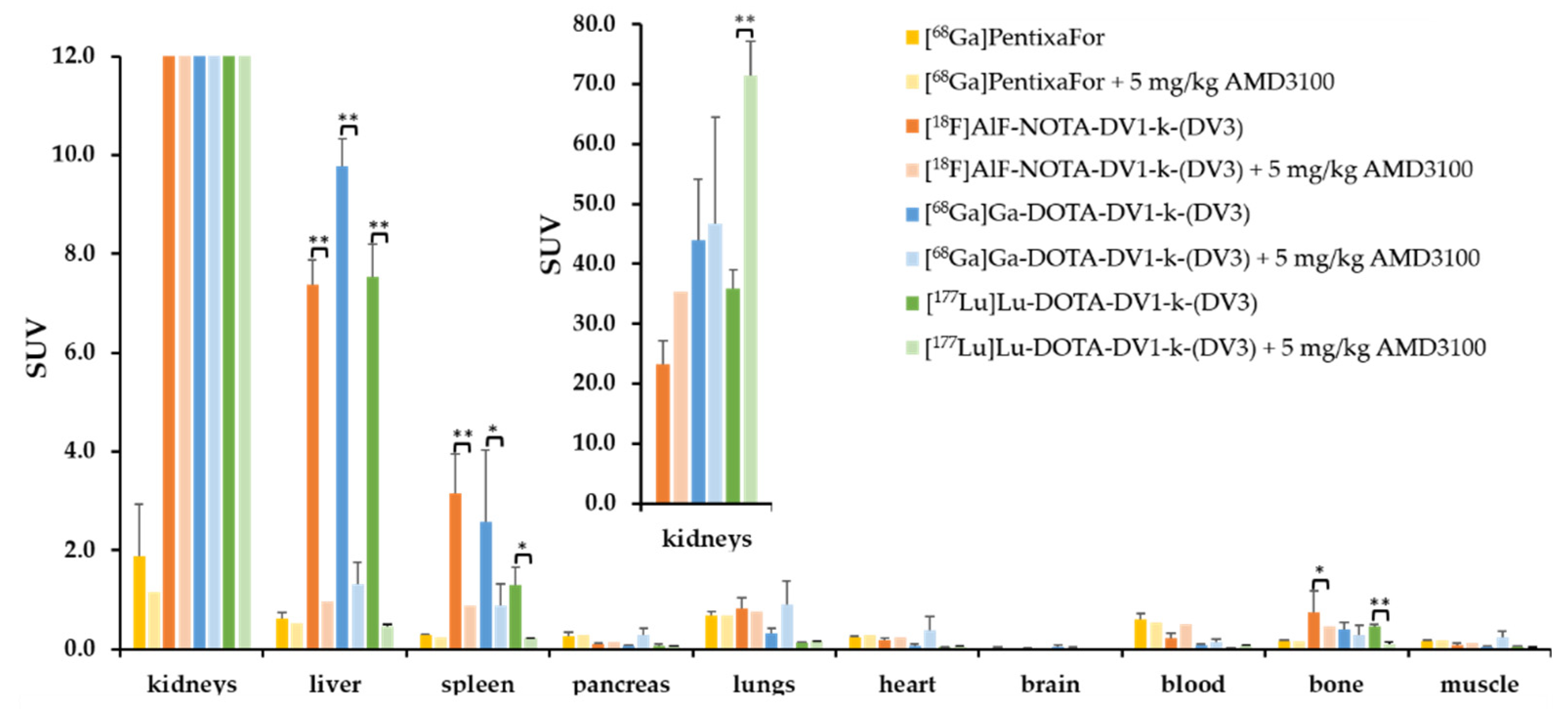
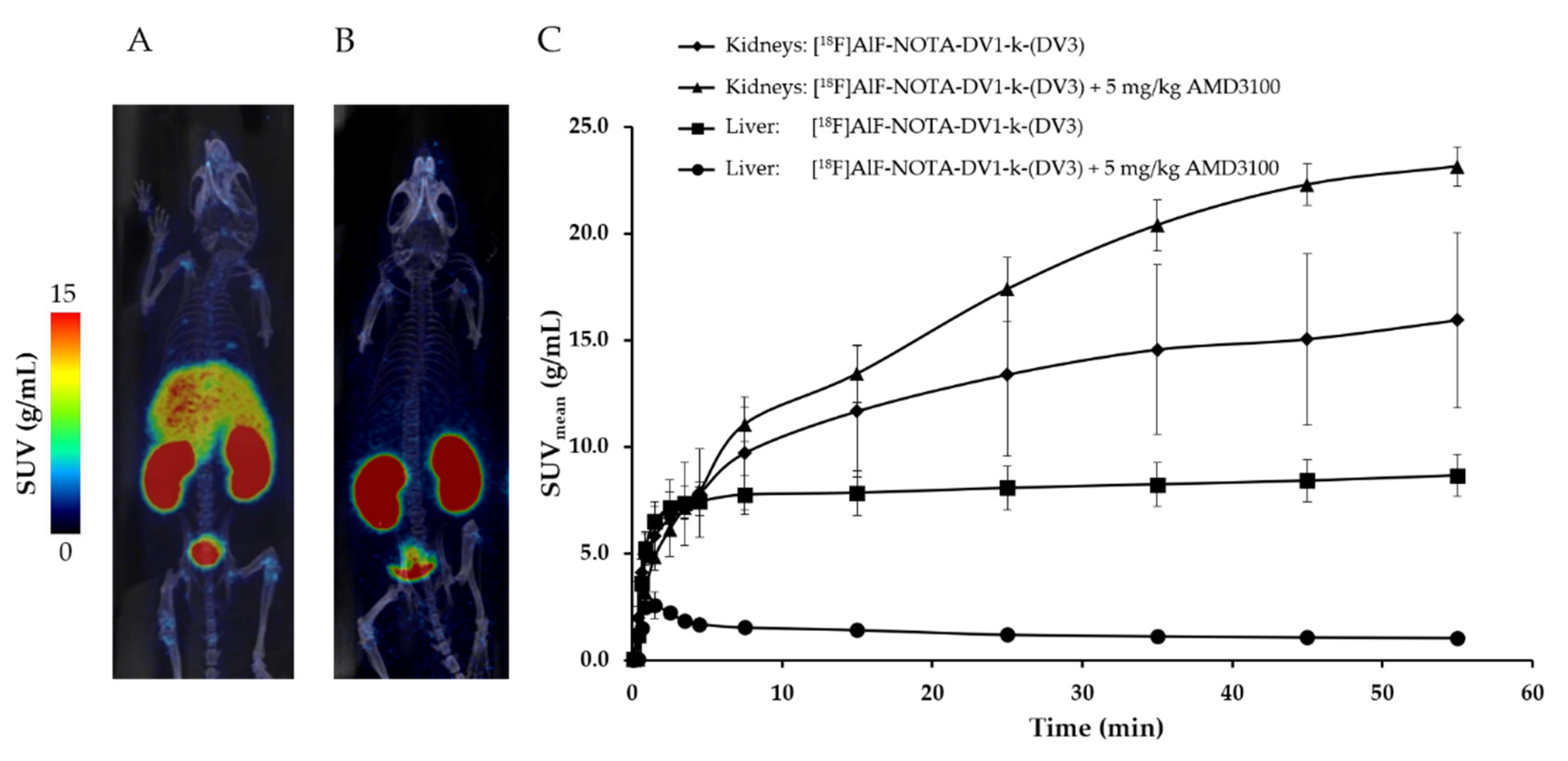
3.2. In Vivo Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kircher, M.; Herhaus, P.; Schottelius, M.; Buck, A.K.; Werner, R.A.; Wester, H.-J.; Keller, U.; Lapa, C. CXCR4-directed theranostics in oncology and inflammation. Ann. Nucl. Med. 2018, 32, 503–511. [Google Scholar] [CrossRef] [Green Version]
- Walenkamp, A.M.E.; Lapa, C.; Herrmann, K.; Wester, H.-J. CXCR4 Ligands: The Next Big Hit? J. Nucl. Med. 2017, 58, 77S–82S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasawa, T. CXC chemokine ligand 12 (CXCL12) and its receptor CXCR4. J. Mol. Med. 2014, 92, 433–439. [Google Scholar] [CrossRef]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.R.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Azad, B.B.; Nimmagadda, S. The Intricate Role of CXCR4 in Cancer. Adv. Cancer Res. 2014, 124, 31–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wester, H.J.; Keller, U.; Schottelius, M.; Beer, A.; Philipp-Abbrederis, K.; Hoffmann, F.; Šimeček, J.; Gerngross, C.; Lassmann, M.; Herrmann, K.; et al. Disclosing the CXCR4 Expression in Lymphoproliferative Diseases by Targeted Molecular Imaging. Theranostics 2015, 5, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, K.; Schottelius, M.; Lapa, C.; Osl, T.; Poschenrieder, A.; Hänscheid, H.; Lückerath, K.; Schreder, M.; Bluemel, C.; Knott, M.; et al. First-in-Human Experience of CXCR4-Directed Endoradiotherapy with 177Lu- and 90Y-Labeled Pentixather in Advanced-Stage Multiple Myeloma with Extensive Intra- and Ex-tramedullary Disease. J. Nucl. Med. 2016, 57, 248–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodnick, M.E.; Sollert, C.; Stark, D.; Clark, M.; Katsifis, A.; Hockley, B.G.; Parr, D.C.; Frigell, J.; Henderson, B.D.; Abghari-Gerst, M.; et al. Cyclotron-based production of 68Ga, [68Ga]GaCl3, and [68Ga]Ga-PSMA-11 from a liquid target. EJNMMI Radiopharm. Chem. 2020, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Cleeren, F.; Lecina, J.; Ahamed, M.; Raes, G.; Devoogdt, N.; Caveliers, V.; McQuade, P.; Rubins, D.J.; Li, W.; Verbruggen, A.; et al. Al18F-Labeling Of Heat-Sensitive Biomolecules for Positron Emission Tomography Imaging. Theranostics 2017, 7, 2924–2939. [Google Scholar] [CrossRef]
- Poschenrieder, A.; Osl, T.; Schottelius, M.; Hoffmann, F.; Wirtz, M.; Schwaiger, M.; Wester, H.-J. First 18F-Labeled Pentixafor-Based Imaging Agent for PET Imaging of CXCR4 Expression In Vivo. Tomography 2016, 2, 85–93. [Google Scholar] [CrossRef]
- Schottelius, M.; Osl, T.; Poschenrieder, A.; Hoffmann, F.; Beykan, S.; Hänscheid, H.; Schirbel, A.; Buck, A.K.; Kropf, S.; Schwaiger, M.; et al. [177Lu] pentixather: Comprehensive Preclinical Characterization of a First CXCR4-directed Endoradiotherapeutic Agent. Theranostics 2017, 7, 2350–2362. [Google Scholar] [CrossRef]
- Xu, Y.; Duggineni, S.; Espitia, S.; Richman, D.D.; An, J.; Huang, Z. A synthetic bivalent ligand of CXCR4 inhibits HIV infection. Biochem. Biophys. Res. Commun. 2013, 435, 646–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, N.; Luo, Z.; Luo, J.; Hall, J.W.; Huang, Z. A novel peptide antagonist of CXCR4 derived from the N-terminus of viral chemokine vMIP-II. Biochemistry 2000, 39, 3782–3787. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Luo, Z.; Luo, J.; Fan, X.; Cayabyab, M.; Hiraoka, M.; Liu, D.; Han, X.; Pesavento, J.; Dong, C.-Z.; et al. Exploring the Stereochemistry of CXCR4-Peptide Recognition and Inhibiting HIV-1 Entry with d-Peptides Derived from Chemokines. J. Biol. Chem. 2002, 277, 17476–17485. [Google Scholar] [CrossRef] [Green Version]
- McBride, W.J.; Sharkey, R.M.; Goldenberg, D.M. Radiofluorination using aluminum-fluoride (Al18F). EJNMMI Res. 2013, 3, 36. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Ui, T.; Nagano, A.; Hino, A.; Arano, Y. C-terminal–modified LY2510924: A versatile scaffold for targeting C-X-C chemokine receptor type. Sci. Rep. 2019, 9, 15284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tshibangu, T.; Cawthorne, C.; Serdons, K.; Pauwels, E.; Gsell, W.; Bormans, G.; Deroose, C.M.; Cleeren, F. Automated GMP compliant production of [18F] AlF-NOTA-octreotide. EJNMMI Radiopharm. Chem. 2020, 5, 4. [Google Scholar] [CrossRef]
- Schoofs, G.; Van Hout, A.; D’huys, T.; Schols, D.; Van Loy, T. A Flow Cytometry-based Assay to Identify Compounds That Disrupt Binding of Fluorescently-labeled CXC Chemokine Ligand 12 to CXC Chemokine Receptor 4. J. Vis. Exp. 2018, 133, 57271. [Google Scholar] [CrossRef]
- Du Sert, N.P.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. Br. J. Pharmacol. 2020, 177, 3617–3624. [Google Scholar] [CrossRef]
- Burke, B.P.; Miranda, C.S.; Lee, R.E.; Renard, I.; Nigam, S.; Clemente, G.S.; D’Huys, T.; Ruest, T.; Domarkas, J.; Thompson, J.A.; et al. 64Cu PET Imaging of the CXCR4 Chemokine Receptor Using a Cross-Bridged Cyclam Bis-Tetraazamacrocyclic Antagonist. J. Nucl. Med. 2020, 61, 123–128. [Google Scholar] [CrossRef]
- Fridman, R.; Benton, G.; Aranoutova, I.; Kleinman, H.K.; Bonfil, R.D. Increased initiation and growth of tumor cell lines, cancer stem cells and biopsy material in mice using basement membrane matrix protein (Cultrex or Matrigel) co-injection. Nat. Protoc. 2012, 7, 1138–1144. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Q.; Gao, M.; Yang, X.; Huang, Z.; An, J. A Novel CXCR4-Selective High-Affinity Fluorescent Probe and Its Application in Competitive Binding Assays. Biochemistry 2014, 53, 4881–4883. [Google Scholar] [CrossRef]
- Arimont, M.; Sun, S.-L.; Leurs, R.; Smit, M.; De Esch, I.J.P.; De Graaf, C. Structural Analysis of Chemokine Receptor–Ligand Interactions. J. Med. Chem. 2017, 60, 4735–4779. [Google Scholar] [CrossRef] [PubMed]
- Ory, D.; Brande, J.V.D.; de Groot, T.; Serdons, K.; Bex, M.; Declercq, L.; Cleeren, F.; Ooms, M.; Van Laere, K.; Verbruggen, A.; et al. Retention of [18F] fluoride on reversed phase HPLC columns. J. Pharm. Biomed. Anal. 2015, 111, 209–214. [Google Scholar] [CrossRef]
- De Jong, M.; Barone, R.; Krenning, E.; Bernard, B.; Melis, M.; Visser, T.; Gekle, M.; Willnow, T.E.; Walrand, S.; Jamar, F.; et al. Megalin is essential for renal proximal tubule reabsorption of 111In-DTPA-octreotide. J. Nucl. Med. 2005, 46, 1696–1700. [Google Scholar] [PubMed]
- Mendt, M.; Cardier, J.E. Stromal-Derived Factor-1 and Its Receptor, CXCR4, Are Constitutively Expressed by Mouse Liver Sinusoidal Endothelial Cells: Implications for the Regulation of Hematopoietic Cell Migration to the Liver During Extramedullary Hematopoiesis. Stem Cells Dev. 2012, 21, 2142–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osl, T.; Schmidt, A.; Schwaiger, M.; Schottelius, M.; Wester, H.J. A new class of PentixaFor and PentixaTher-based theranostic agents with enhanced CXCR4-targeting efficiency. Theranostics 2020, 10, 8264–8280. [Google Scholar] [CrossRef]
- Nicolas, G.P.; Mansi, R.; McDougall, L.; Kaufmann, J.; Bouterfa, H.; Wild, D.; Fani, M. Biodistribution, Pharmacokinetics, and Dosimetry of 177Lu-, 90Y-, and 111In-Labeled Somatostatin Receptor Antagonist OPS201 in Comparison to the Agonist 177Lu-DOTATATE: The Mass Effect. J. Nucl. Med. 2017, 58, 1435–1441. [Google Scholar] [CrossRef] [Green Version]
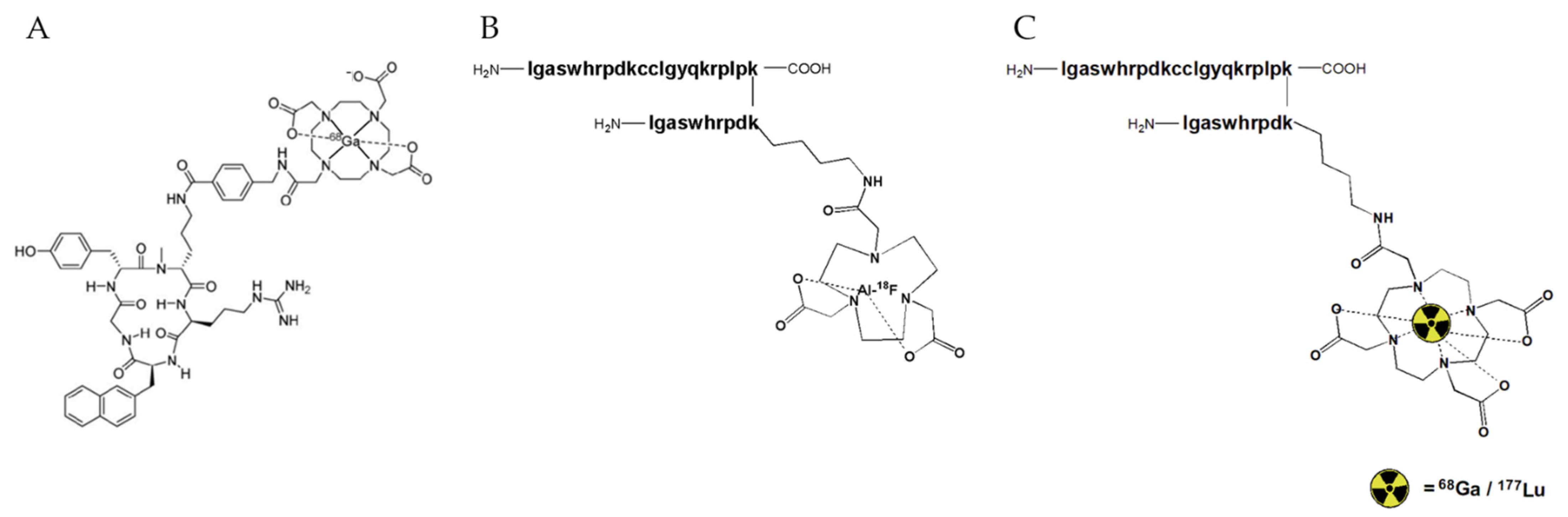



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | IC50 [nM] to hCXCR4 |
|---|---|
| [natGa]PentixaFor | 8.6 ± 1.1 |
| AMD3100 | 40.4 ± 3.5 |
| DV1 | 241.7 ± 18.1 |
| DV3 | >1000 |
| DV1-k-(DV3) | 2.9 ± 0.4 |
| NOTA-DV1-k-(DV3) | 14.2 ± 1.4 |
| DOTA-DV1-k-(DV3) | 27.3 ± 2.8 |
| AlF-NOTA-DV1-k-(DV3) | 5.3 ± 0.9 |
| [natGa]Ga-DOTA-DV1-k-(DV3) | 4.3 ± 0.3 |
| [natLu]Lu-DOTA-DV1-k-(DV3) | 2.9 ± 0.6 |
| [natBi]Bi-DOTA-DV1-k-(DV3) | 8.8 ± 1.1 |
| La-DOTA-DV1-k-(DV3) | 14.7 ± 2.1 |
| Peptide | IC50 [nM] to mCXCR4 |
| [natGa]PentixaFor | >1000 |
| AMD3100 | 123.4 ± 17.0 |
| DV1-k-(DV3) | 41.5 ± 12.3 |
| AlF-NOTA-DV1-k-(DV3) | 33.4 ± 13.5 |
| Total Cell-Bound Fraction [% of Applied Activity] | Internalized Fraction [% of Total Cell-Bound Fraction] | ||
|---|---|---|---|
| AMD3100 | 100 µM | ||
| [68Ga]PentixaFor | 8.9 ± 0.3 | 0.1 ± 0.003 ** | 10.0 ± 0.1 |
| [18F]AlF-NOTA-DV1-k-(DV3) | 6.0 ± 0.1 | 1.1 ± 0.1 ** | 20.1 ± 2.0 |
| [68Ga]Ga-DOTA-DV1-k-(DV3) | 6.4 ± 0.1 | 1.9 ± 0.2 ** | 31.2 ± 2.4 |
| [177Lu]Lu-DOTA-DV1-k-(DV3) | 4.6 ± 0.5 | 0.5 ± 0.04 * | 29.4 ± 0.8 |
| [18F]AlF-NOTA-DV1-k-(DV3) | [18F]AlF-NOTA-DV1-k-(DV3) + 5 mg/kg AMD3100 | |
|---|---|---|
| liver | 8.2 ± 1.0 | 0.9 ± 0.3 ** |
| spleen | 2.5 ± 1.0 | 0.5 ± 0.1 * |
| bone | 0.4 ± 0.1 | 0.1 ± 0.04 * |
| tumor | 0.6 ± 0.2 | 0.4 ± 0.2 |
| muscle | 0.05 ± 0.02 | 0.1 ± 0.1 |
| blood | 0.1 ± 0.1 | 0.3 ± 0.2 |
| t/muscle | 12.4 ± 2.9 | 4.2 ± 1.1 * |
| t/blood | 4.6 ± 0.9 | 1.7 ± 0.5 ** |
| [18F]AlF-NOTA-DV1-k-(DV3) | |||||
|---|---|---|---|---|---|
| AMD3100 | 0 nmol n = 4 | 157 nmol n = 4 | |||
| DV1-k-(DV3) | 7 nmol n = 3 | 14 nmol n = 4 | 24 nmol n = 4 | ||
| liver | 8.2 ± 1.0 | 1.2 ± 0.1 *** | 1.2 ± 0.4 *** | 0.7 ± 0.1 *** | 0.9 ± 0.3 *** |
| spleen | 2.5 ± 1.0 | 0.5 ± 0.1 ** | 0.6 ± 0.2 * | 0.4 ± 0.1 ** | 0.5 ± 0.1 ** |
| bone | 0.4 ± 0.1 | 0.2 ± 0.04 ** | 0.3 ± 0.1 * | 0.2 ± 0.04 ** | 0.1 ± 0.04 ** |
| tumor | 0.6 ± 0.2 | 0.7 ± 0.1 | 0.9 ± 0.3 | 0.6 ± 0.2 | 0.4 ± 0.2 |
| t/liver | 0.1 ± 0.02 | 0.6 ± 0.1 ** | 0.8 ± 0.2 ** | 0.9 ± 0.2 *** | 0.5 ± 0.2 * |
| t/muscle | 12.4 ± 2.9 | 5.4 ± 2.2 ** | 4.8 ± 2.1 ** | 3.0 ± 0.4 ** | 4.2 ± 1.1 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luyten, K.; Van Loy, T.; Cawthorne, C.; Deroose, C.M.; Schols, D.; Bormans, G.; Cleeren, F. D-Peptide-Based Probe for CXCR4-Targeted Molecular Imaging and Radionuclide Therapy. Pharmaceutics 2021, 13, 1619. https://doi.org/10.3390/pharmaceutics13101619
Luyten K, Van Loy T, Cawthorne C, Deroose CM, Schols D, Bormans G, Cleeren F. D-Peptide-Based Probe for CXCR4-Targeted Molecular Imaging and Radionuclide Therapy. Pharmaceutics. 2021; 13(10):1619. https://doi.org/10.3390/pharmaceutics13101619
Chicago/Turabian StyleLuyten, Kaat, Tom Van Loy, Christopher Cawthorne, Christophe M. Deroose, Dominique Schols, Guy Bormans, and Frederik Cleeren. 2021. "D-Peptide-Based Probe for CXCR4-Targeted Molecular Imaging and Radionuclide Therapy" Pharmaceutics 13, no. 10: 1619. https://doi.org/10.3390/pharmaceutics13101619
APA StyleLuyten, K., Van Loy, T., Cawthorne, C., Deroose, C. M., Schols, D., Bormans, G., & Cleeren, F. (2021). D-Peptide-Based Probe for CXCR4-Targeted Molecular Imaging and Radionuclide Therapy. Pharmaceutics, 13(10), 1619. https://doi.org/10.3390/pharmaceutics13101619