
Angiotensin-(1–7) Peptide Hormone Reduces Inflammation and Pathogen Burden during Mycoplasma pneumoniae Infection in Mice

 ,
,  and
and 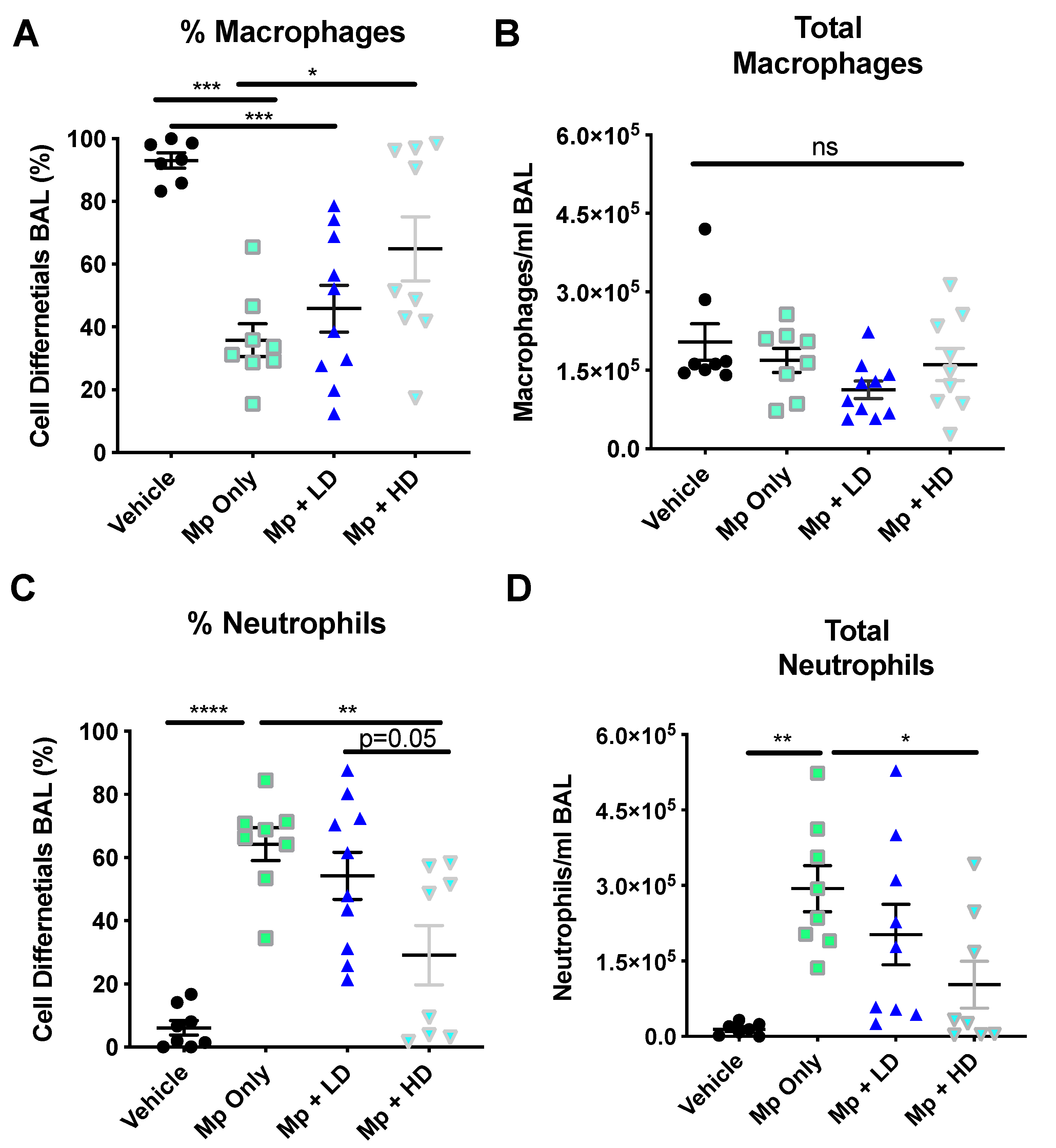{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Animal Models
2.2. Pathogen-Challenge and Drug Treatment
2.3. Bronchoalveolar Lavage (BAL) Fluid Cell Counts and Cell Differentiation
2.4. Lung Tissue Preparation for Histological Analysis
2.5. RT-PCR Analysis
2.6. Growth and Stimulation of RAW 264.7 Cells
2.7. ELISA Analysis
2.8. Statistical Analysis
3. Results
3.1. Effect of Ang-(1–7) on Inflammatory Cells in the BALF Post Mp-Infection
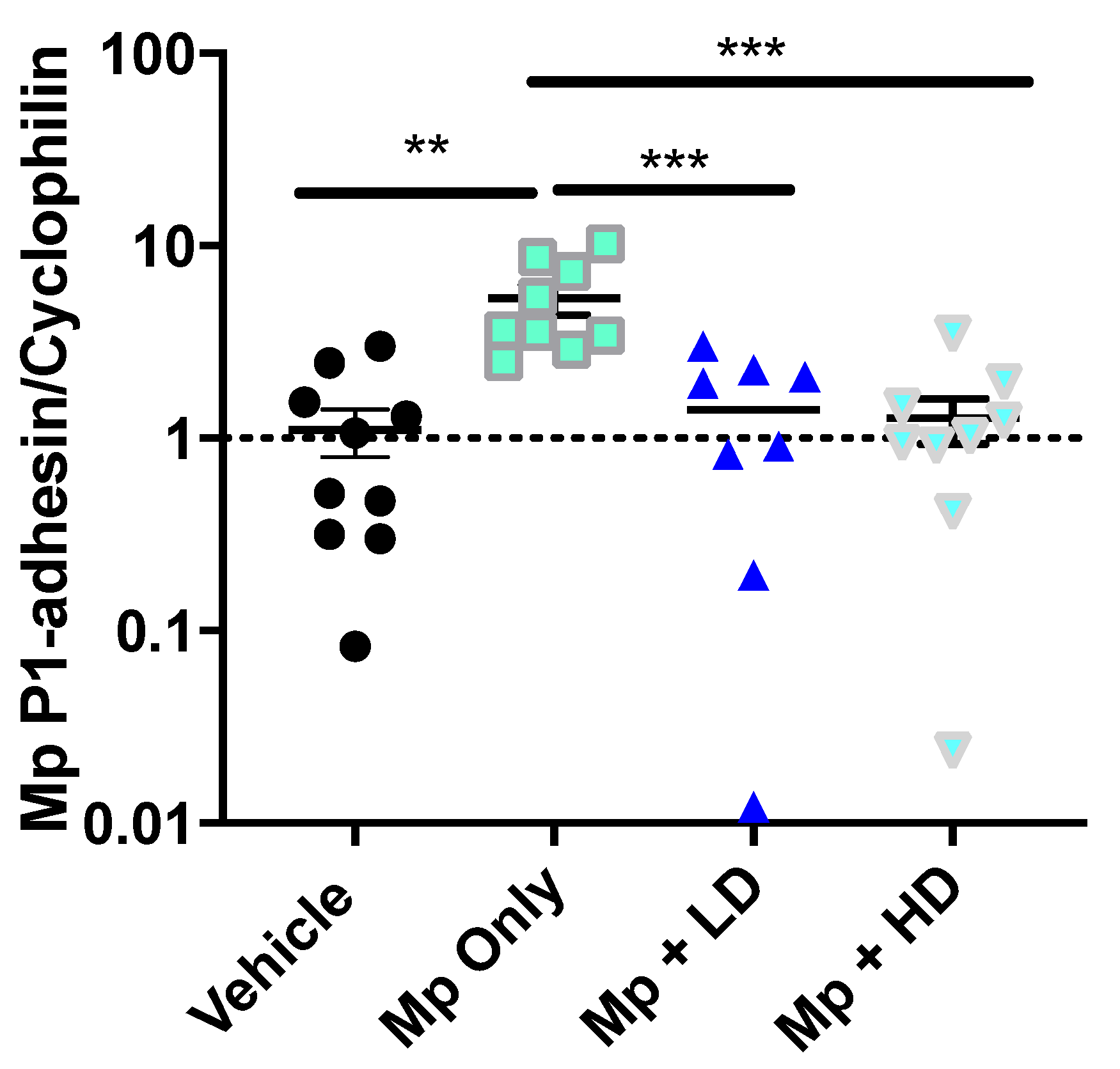
3.2. Effect of Ang-(1–7) on Mp Burden in Lung Tissue
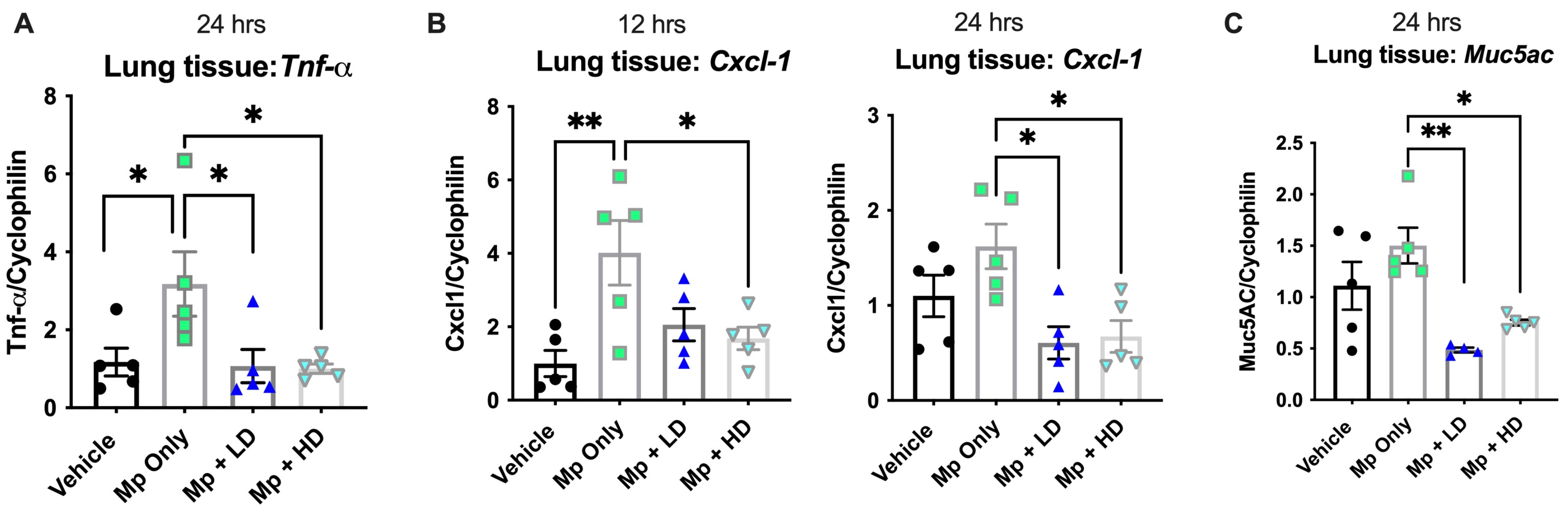
3.3. Effect of Ang-(1–7) on Muc5ac, Cytokines, and Chemokines during Mp Infection
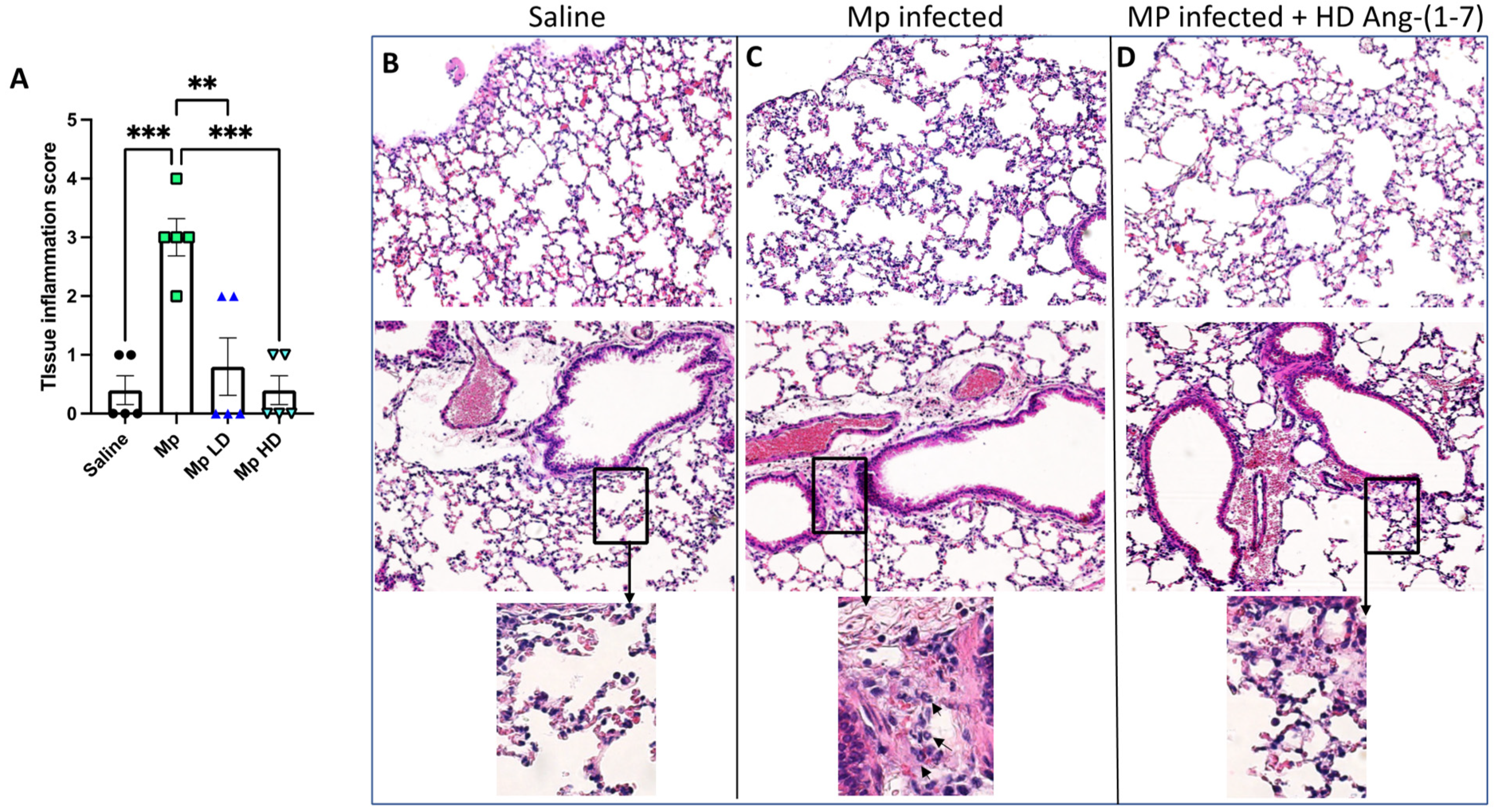
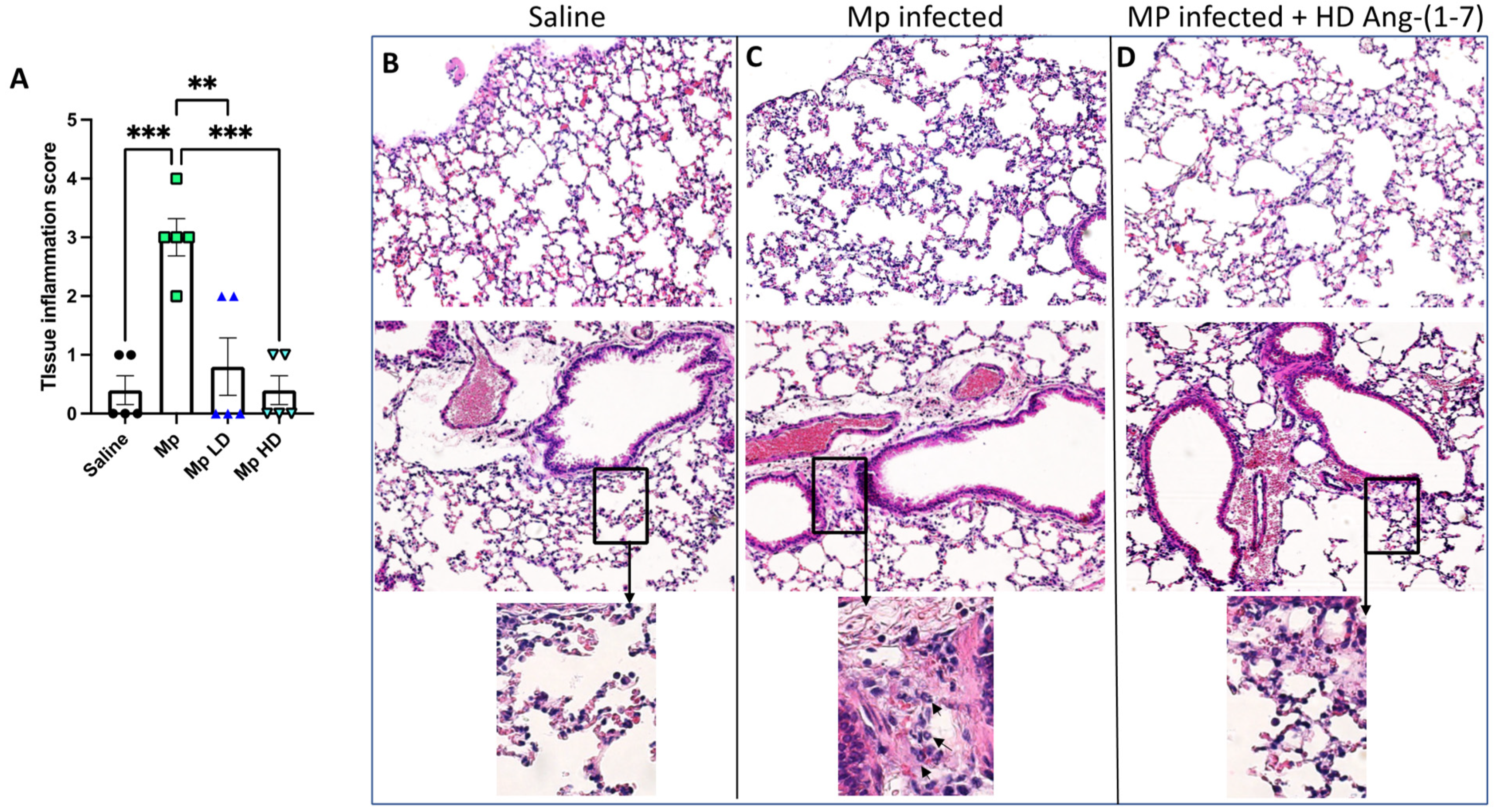
3.4. Effect of Ang-(1–7) Treatment on Lung Tissue Inflammation in Histological Sections
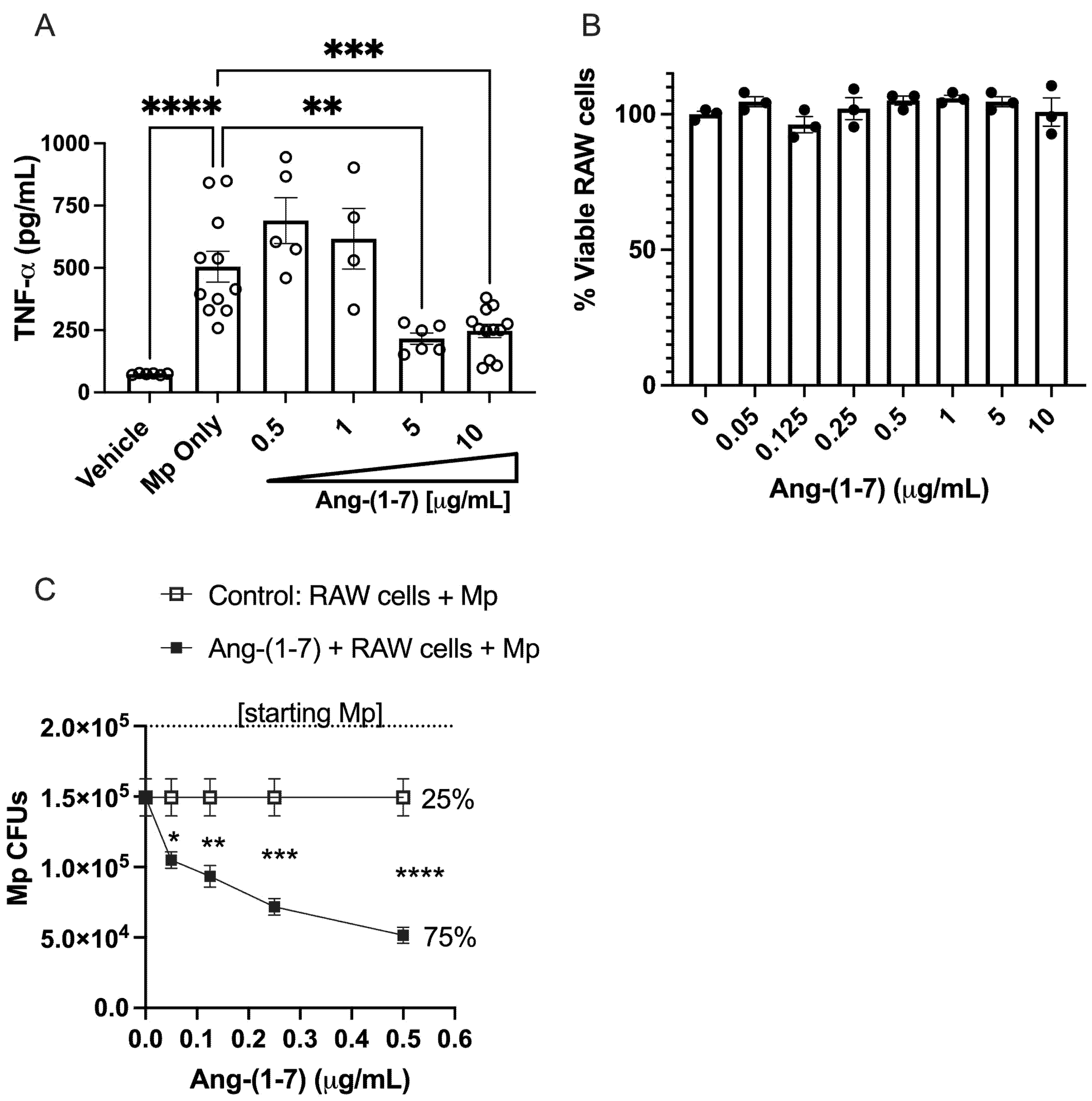
3.5. Ang-(1–7) Dose-Dependently Inhibits TNF-α Secretion from RAW 264.7 Cells Following Mp Challenge
3.6. Ang-(1–7) Dose-Dependently Promotes Killing of Mp by RAW Cells In Vitro
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kostenis, E.; Milligan, G.; Christopoulos, A.; Sanchez-Ferrer, C.F.; Heringer-Walther, S.; Sexton, P.; Gembardt, F.; Kellett, E.; Martini, L.; Vanderheyden, P.; et al. G-Protein–Coupled Receptor Mas Is a Physiological Antagonist of the Angiotensin II Type 1 Receptor. Circulation 2005, 111, 1806–1813. [Google Scholar] [CrossRef]
- Papinska, A.M.; Soto, M.; Meeks, C.J.; Rodgers, K.E. Long-term administration of angiotensin (1-7) prevents heart and lung dysfunction in a mouse model of type 2 diabetes (db/db) by reducing oxidative stress, inflammation and pathological remodeling. Pharmacol. Res. 2016, 107, 372–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, E.J.; Chisolm, G.M.; Ferrario, C.M.; Tallant, E.A. Angiotensin-(1-7) Inhibits Vascular Smooth Muscle Cell Growth. Hypertension 1996, 28, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Yin, A.; Zhang, Q.; Zhong, T.; O’Rourke, S.T.; Sun, C. Angiotensin-(1–7) attenuates angiotensin II-induced cardiac hypertrophy via a Sirt3-dependent mechanism. Am. J. Physiol. Circ. Physiol. 2017, 312, H980–H991. [Google Scholar] [CrossRef] [PubMed]
- Kittana, N. Angiotensin-converting enzyme 2-Angiotensin 1-7/1-9 system: Novel promising targets for heart failure treatment. Fundam Clin. Pharmacol. 2018, 32, 14–25. [Google Scholar] [CrossRef] [PubMed]
- El-Hashim, A.Z.; Renno, W.M.; Raghupathy, R.; Abduo, H.T.; Akhtar, S.; Benter, I.F. Angiotensin-(1-7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-κB-dependent pathways. Br. J. Pharmacol. 2012, 166, 1964–1976. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cao, Y.; Zeng, Z.; Liang, M.; Xue, Y.; Xi, C.; Zhou, M.; Jiang, W. Angiotensin-converting enzyme 2/angiotensin-(1-7)/Mas axis prevents lipopolysaccharide-induced apoptosis of pulmonary microvascular endothelial cells by inhibiting JNK/NF-κB pathways. Sci. Rep. 2015, 5, 8209. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.A.S.; Ferreira, A.J.; Verano-Braga, T.; Bader, M. Angiotensin-converting enzyme 2, angiotensin-(1–7) and Mas: New players of the renin–angiotensin system. J. Endocrinol. 2012, 216, R1–R17. [Google Scholar] [CrossRef] [Green Version]
- Ferrario, C.M. Angiotensin-converting enzyme 2 and angiotensin-(1-7): An evolving story in cardiovascular regulation. Hypertension 2006, 47, 515–521. [Google Scholar] [CrossRef]
- Raizada, M.K.; Ferreira, A.J. ACE2: A New Target for Cardiovascular Disease Therapeutics. J. Cardiovasc. Pharmacol. 2007, 50, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of Biological Peptides by Human Angiotensin-converting Enzyme-related Carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [Green Version]
- Lazartigues, Y.F.A.J.L.L.E.; Feng, Y.; Lavoie, J. The Two fACEs of the Tissue Renin-Angiotensin Systems: Implication in Cardiovascular Diseases. Curr. Pharm. Des. 2007, 13, 1231–1245. [Google Scholar] [CrossRef]
- Hay, M.; Polt, R.; Heien, M.L.; Vanderah, T.W.; Largent-Milnes, T.M.; Rodgers, K.; Falk, T.; Bartlett, M.J.; Doyle, K.P.; Konhilas, J. A Novel Angiotensin-(1-7) Glycosylated Mas Receptor Agonist for Treating Vascular Cognitive Impairment and Inflammation-Related Memory Dysfunction. J. Pharmacol. Exp. Ther. 2019, 369, 9–25. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Qian, C.; Roks, A.J.; Westermann, D.; Schumacher, S.-M.; Escher, F.; Schoemaker, R.G.; Reudelhuber, T.L.; Van Gilst, W.H.; Schultheiss, H.-P.; et al. Circulating Rather Than Cardiac Angiotensin-(1-7) Stimulates Cardioprotection After Myocardial Infarction. Circ. Hear. Fail. 2010, 3, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Karnik, S.S.; Singh, K.D.; Tirupula, K.; Unal, H. Significance of angiotensin 1-7 coupling with MAS1 receptor and other GPCRs to the renin-angiotensin system: IUPHAR Review 22. Br. J. Pharmacol. 2017, 174, 737–753. [Google Scholar] [CrossRef]
- Gopallawa, I.; Uhal, B.D. Molecular and cellular mechanisms of the inhibitory effects of ACE-2/ANG1-7/Mas axis on lung injury. Curr. Top. Pharmacol. 2014, 18, 71–80. [Google Scholar]
- Magalhaes, G.; Barroso, L.C.; Reis, A.; Rodrigues-Machado, M.G.; Gregório, J.; Motta-Santos, D.; Oliveira, A.C.; Perez, D.A.; Barcelos, L.S.; Teixeira, M.M.; et al. Angiotensin-(1–7) Promotes Resolution of Eosinophilic Inflammation in an Experimental Model of Asthma. Front. Immunol. 2018, 9, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhaes, G.; Rodrigues-Machado, M.D.G.; Motta-Santos, D.; Alenina, N.; Bader, M.; Santos, R.A.S.; Barcelos, L.S.; Campagnole-Santos, M.J. Chronic allergic pulmonary inflammation is aggravated in angiotensin-(1–7) Mas receptor knockout mice. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L1141–L1148. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Liu, J.; Wang, W.; Liu, S.; Yang, X.; Chen, M.; Cheng, L.; Lu, J.; Guo, T.; Huang, F. Sini decoction ameliorates sepsis-induced acute lung injury via regulating ACE2-Ang (1-7)-Mas axis and inhibiting the MAPK signaling pathway. Biomed. Pharmacother. 2019, 115, 108971. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.F.; Kuang, X.D.; Yuan, Q.F.; Hao, H.; Zhang, T.; Huang, Y.H.; Zhou, X.Y. Lipoxin A attenuates LPS-induced acute lung injury via activation of the ACE2-Ang-(1-7)-Mas axis. Innate Immun. 2018, 24, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, E.M.; Del Sarto, J.; Vago, J.P.; Tavares, L.P.; Rago, F.; Gonçalves, A.P.F.; Machado, M.G.; Aranda-Pardos, I.; Valiate, B.V.; Cassali, G.D.; et al. Relevance of angiotensin-(1-7) and its receptor Mas in pneumonia caused by influenza virus and post-influenza pneumococcal infection. Pharmacol. Res. 2021, 163, 105292. [Google Scholar] [CrossRef] [PubMed]
- McKinney, C.A.; Fattah, C.; Loughrey, C.; Milligan, G.; Nicklin, S.A. Angiotensin-(1–7) and angiotensin-(1–9): Function in cardiac and vascular remodelling. Clin. Sci. 2014, 126, 815–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraga-Silva, R.A.; Costa-Fraga, F.P.; De Sousa, F.; Alenina, N.; Bader, M.; Sinisterra, R.; Santos, R.A.S. An orally active formulation of angiotensin-(1-7) produces an antithrombotic effect. Clinics 2011, 66, 837–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, P.E.; Tallant, E.A. Inhibition of human lung cancer cell growth by angiotensin-(1-7). Carcinogenesis 2004, 25, 2045–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, H.; Schwartz, B.M.; Delmore, J.E.; Reed, E.; Cruickshank, S.; Drummond, L.; Rodgers, K.E.; Peterson, K.J.; Dizerega, G.S. Pharmacodynamic stimulation of thrombogenesis by angiotensin (1–7) in recurrent ovarian cancer patients receiving gemcitabine and platinum-based chemotherapy. Cancer Chemother. Pharmacol. 2013, 71, 965–972. [Google Scholar] [CrossRef]
- Stanhewicz, A.; Alexander, L.M. Local angiotensin-(1–7) administration improves microvascular endothelial function in women who have had preeclampsia. Am. J. Physiol. Integr. Comp. Physiol. 2020, 318, R148–R155. [Google Scholar] [CrossRef]
- Hay, M.; Vanderah, T.W.; Samareh-Jahani, F.; Constantopoulos, E.; Uprety, A.R.; Barnes, C.A.; Konhilas, J. Cognitive impairment in heart failure: A protective role for angiotensin-(1-7). Behav. Neurosci. 2017, 131, 99–114. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Kotch, C.; Barrett, D.; Teachey, D.T. Tocilizumab for the treatment of chimeric antigen receptor T cell-induced cytokine release syndrome. Expert Rev. Clin. Immunol. 2019, 15, 813–822. [Google Scholar] [CrossRef]
- Murthy, H.; Iqbal, M.; Chavez, J.C.; A Kharfan-Dabaja, M. Cytokine Release Syndrome: Current Perspectives. ImmunoTargets Ther. 2019, 8, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, T.P.; Balish, M.F.; Waites, K.B. Epidemiology, clinical manifestations, pathogenesis and laboratory detection of Mycoplasma pneumoniae infections. FEMS Microbiol. Rev. 2008, 32, 956–973. [Google Scholar] [CrossRef] [Green Version]
- Carr, T.F.; Kraft, M. Chronic Infection and Severe Asthma. Immunol Allergy Clin. North. Am. 2016, 36, 483–502. [Google Scholar] [CrossRef]
- Kraft, M.; Hamid, Q. Mycoplasma in severe asthma. J. Allergy Clin. Immunol. 2006, 117, 1197–1198. [Google Scholar] [CrossRef]
- Kraft, M.; Cassell, G.H.; Pak, J.; Martin, R.J. Mycoplasma pneumoniae and Chlamydia pneumoniae in asthma: Effect of clarithromycin. Chest 2002, 121, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.L.; Younis, U.S.; Menghani, S.V.; Addison, K.J.; Whalen, M.; Pilon, A.L.; Cress, A.E.; Polverino, F.; Romanoki, C.E.; Kraft, M.; et al. CC16 Binding to α4β1 integrin protects against Mycoplasma pneumoniae infection. Am. J. Respir Crit Care Med. 2021, 203, 1410–1418. [Google Scholar] [CrossRef] [PubMed]
- Ledford, J.G.; Voelker, D.R.; Addison, K.J.; Wang, Y.; Nikam, V.S.; Degan, S.; Kadasamy, P.; Tanyaratsrisakul, S.; Fischer, B.M.; Kraft, M.; et al. Genetic variation in SP-A2 leads to differential binding to Mycoplasma pneumoniae membranes and regulation of host responses. J. Immunol. 2015, 194, 6123–6132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashyap, S.; Sarkar, M. Mycoplasma pneumonia: Clinical features and management. Lung India. 2010, 27, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Alabsi, W.; Acosta, M.F.; Al-Obeidi, F.A.; Hay, M.; Polt, R.; Mansour, H.M. Synthesis, Physicochemical Characterization, In Vitro 2D/3D Human Cell Culture, and In Vitro Aerosol Dispersion Performance of Advanced Spray Dried and Co-Spray Dried Angiotensin (1-7) Peptide and PNA5 with Trehalose as Microparticles/Nanoparticles for Targeted Respiratory Delivery as Dry Powder Inhalers. Pharmaceutics 2021, 13, 1278. [Google Scholar] [PubMed]
- Razin, S.; Yogev, D.; Naot, Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol Mol. Biol Rev. 1998, 62, 1094–1156. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.M.; Kim, K.; Tuvim, M.J.; Dickey, B.F. Mucus hypersecretion in asthma: Causes and effects. Curr. Opin. Pulm. Med. 2009, 15, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Darveaux, J.I.; Lemanske, R.F. Infection-Related Asthma. J. Allergy Clin. Immunol. Pr. 2014, 2, 658–663. [Google Scholar] [CrossRef] [Green Version]
- Benter, I.F.; Yousif, M.; Dhaunsi, G.S.; Kaur, J.; Chappell, M.C.; Diz, D.I. Angiotensin-(1–7) Prevents Activation of NADPH Oxidase and Renal Vascular Dysfunction in Diabetic Hypertensive Rats. Am. J. Nephrol. 2007, 28, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes e-Silva, A.C.; Oliveira, A.C.; Carvalho Bittencourt de Oliveira, F.; Gonçalves, R. The Anti-Inflammatory Potential of ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Evidence from Basic and Clinical Research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho Santuchi, M.; Dutra, M.F.; Vago, J.P.; Lima, K.M.; Galvao, I.; de Souza-Neto, F.P.; Morais e Silva, M.; Oliveira, A.C.; de Oliveira, F.C.; Gonçalves, R.; et al. Angiotensin-(1-7) and Alamandine Promote Anti-inflammatory Response in Macrophages In Vitro and In Vivo. Mediators Inflamm. 2019, 2019, 2401081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, K.E.; Oliver, J.; diZerega, G.S. Phase I/II dose escalation study of angiotensin 1-7 [A(1-7)] administered before and after chemotherapy in patients with newly diagnosed breast cancer. Cancer Chemother. Pharmacol. 2006, 57, 559–568. [Google Scholar] [CrossRef]
- Savage, P.D.; Lovato, J.; Brosnihan, K.B.; Miller, A.A.; Petty, W.J. Phase II Trial of Angiotensin-(1-7) for the Treatment of Patients with Metastatic Sarcoma. Sarcoma 2016, 2016, 1–7. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collins, K.L.; Younis, U.S.; Tanyaratsrisakul, S.; Polt, R.; Hay, M.; Mansour, H.M.; Ledford, J.G. Angiotensin-(1–7) Peptide Hormone Reduces Inflammation and Pathogen Burden during Mycoplasma pneumoniae Infection in Mice. Pharmaceutics 2021, 13, 1614. https://doi.org/10.3390/pharmaceutics13101614
Collins KL, Younis US, Tanyaratsrisakul S, Polt R, Hay M, Mansour HM, Ledford JG. Angiotensin-(1–7) Peptide Hormone Reduces Inflammation and Pathogen Burden during Mycoplasma pneumoniae Infection in Mice. Pharmaceutics. 2021; 13(10):1614. https://doi.org/10.3390/pharmaceutics13101614
Chicago/Turabian StyleCollins, Katie L., Usir S. Younis, Sasipa Tanyaratsrisakul, Robin Polt, Meredith Hay, Heidi M. Mansour, and Julie G. Ledford. 2021. "Angiotensin-(1–7) Peptide Hormone Reduces Inflammation and Pathogen Burden during Mycoplasma pneumoniae Infection in Mice" Pharmaceutics 13, no. 10: 1614. https://doi.org/10.3390/pharmaceutics13101614
APA StyleCollins, K. L., Younis, U. S., Tanyaratsrisakul, S., Polt, R., Hay, M., Mansour, H. M., & Ledford, J. G. (2021). Angiotensin-(1–7) Peptide Hormone Reduces Inflammation and Pathogen Burden during Mycoplasma pneumoniae Infection in Mice. Pharmaceutics, 13(10), 1614. https://doi.org/10.3390/pharmaceutics13101614