In Vitro Release Study of the Polymeric Drug Nanoparticles: Development and Validation of a Novel Method


Abstract
1. Introduction
2. Material and Methods
2.1. Materials
2.2. Preparation of Polymeric Drug Nanoparticles
2.3. Analysis of Particle Size and Particle Size Distribution
2.4. Measurement of Zeta Potential
2.5. High Performance Liquid Chromatography (HPLC)
2.6. Measurement of Drug Recovery During Filtration
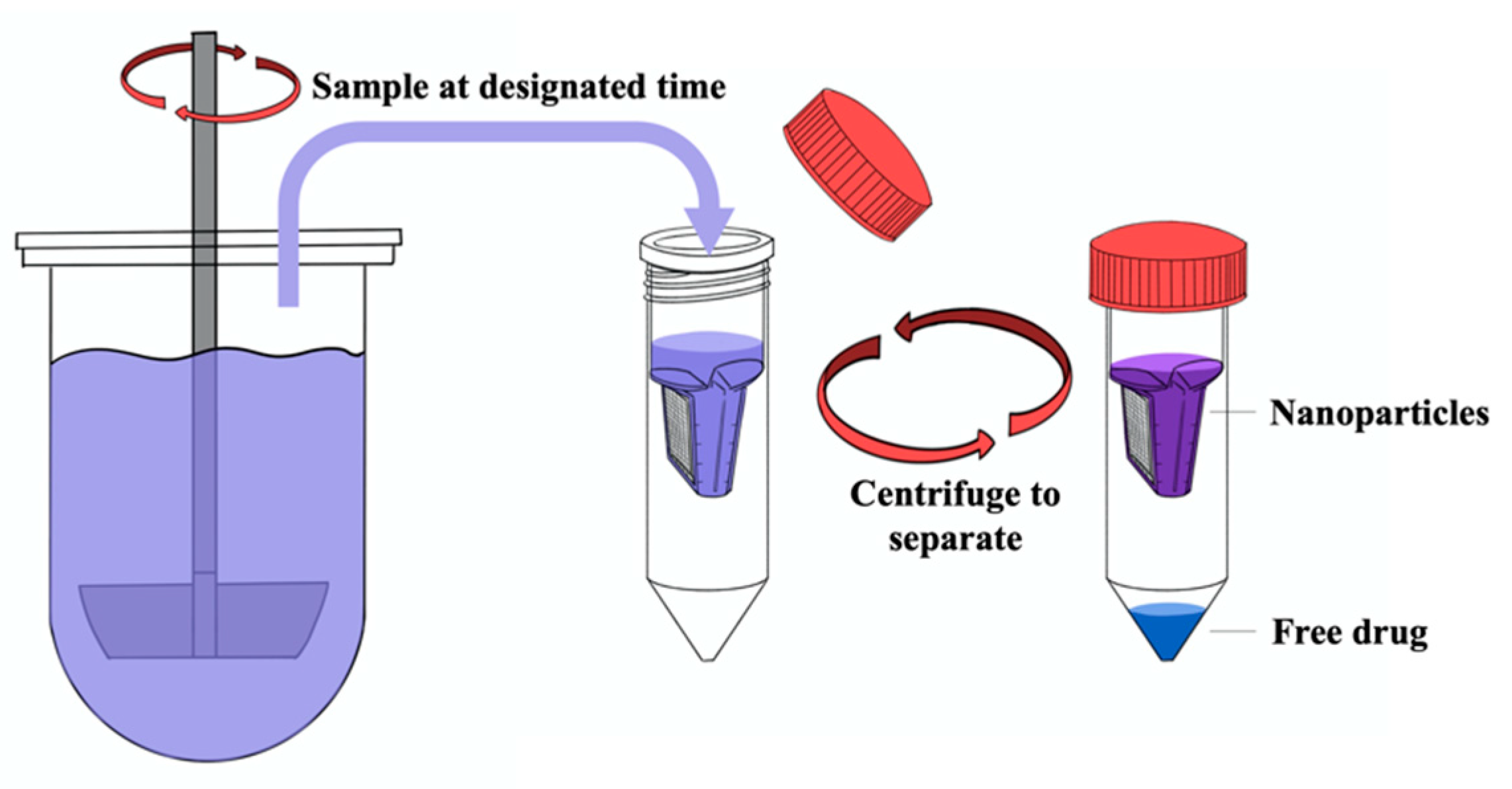
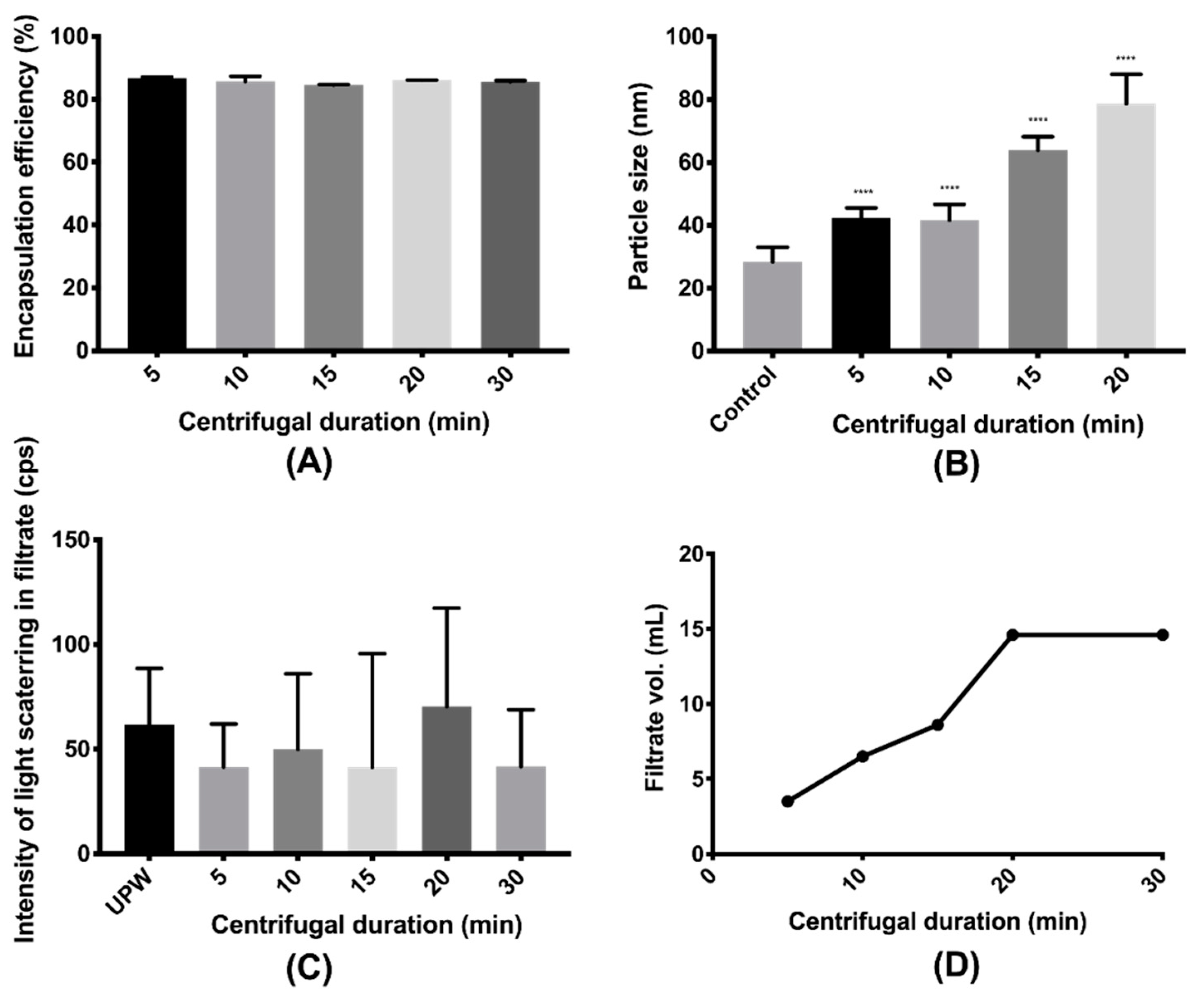
2.7. Validation of Centrifugal Ultrafiltration
2.8. Determination of Drug Loading and Encapsulation Efficiency
2.9. Measurement of Drug Solubilities in Water and Release Medium
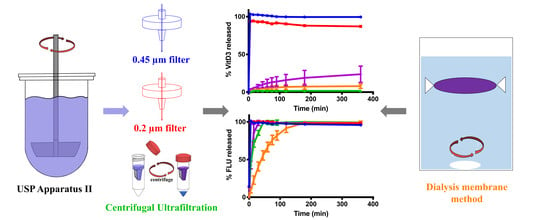
2.10. Release Study under Sink Condition
2.11. Mathematical Modeling of the Release Kinetics
2.12. Statistical Analysis
3. Results and Discussion
3.1. Preparation and Characterization of Nanoparticles
3.2. Validation of the Separation Methods
3.3. Release Profiles of Nanoparticles
3.4. Significance of the Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
List of Abbreviations
| Abbreviation | Meaning |
| ACE | Acetone |
| ACN | Acetonitrile |
| CLT | Cholesterol |
| CF | Continuous flow |
| CU | Centrifugal ultrafiltration |
| DL | Drug loading |
| DLS | Dynamic light scattering |
| DM | Dialysis membrane |
| DMF | Dimethylformamide |
| EE | Encapsulation efficiency |
| EtOH | Ethanol |
| FLU | Flurbiprofen |
| HPLC | High performance liquid chromatography |
| ITZ | Itraconazole |
| IVIVC | In vitro-in vivo correlation |
| LOD | Limit of detection |
| MeOH | Methanol |
| MIMV | Multi-inlet vortex mixer |
| PDI | Polydispersity index |
| RSD | Relative standard deviation |
| SDS | Sodium dodecyl sulfate |
| SS | Sample and separate |
| TPGS | D-α-tocopheryl polyethylene glycol 1000 succinate |
| UPW | Ultra-purified water |
| VitD3 | Cholecalciferol |
References
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef]
- Couvreur, P. Nanoparticles in drug delivery: Past, present and future. Adv. Drug Deliv. Rev. 2013, 65, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in nanomedicine: Approved and investigational nanodrugs. Pharm. Ther. 2017, 42, 742. [Google Scholar]
- Ventola, C.L. The nanomedicine revolution: Part 3: Regulatory and safety challenges. Pharm. Ther. 2012, 37, 631. [Google Scholar]
- Cardot, J.; Beyssac, E.; Alric, M. In vitro-in vivo correlation: Importance of dissolution in IVIVC. Dissolution Technol. 2007, 14, 15. [Google Scholar] [CrossRef]
- Sjögren, E.; Abrahamsson, B.; Augustijns, P.; Becker, D.; Bolger, M.B.; Brewster, M.; Brouwers, J.; Flanagan, T.; Harwood, M.; Heinen, C. In vivo methods for drug absorption–comparative physiologies, model selection, correlations with in vitro methods (IVIVC), and applications for formulation/API/excipient characterization including food effects. Eur. J. Pharm. 2014, 57, 99–151. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.K.; Friedel, H.D.; Barker, A.R.; Buhse, L.F.; Keitel, S.; Cecil, T.L.; Kraemer, J.; Morris, J.M.; Reppas, C.; Stickelmeyer, M.P. FIP/AAPS joint workshop report: Dissolution/in vitro release testing of novel/special dosage forms. AAPS PharmSciTech 2011, 12, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Siewert, M.; Dressman, J.; Brown, C.K.; Shah, V.P.; Aiache, J.-M.; Aoyagi, N.; Bashaw, D.; Brown, C.; Brown, W.; Burgess, D. FIP/AAPS guidelines to dissolution/in vitro release testing of novel/special dosage forms. AAPS PharmSciTech 2003, 4, 43–52. [Google Scholar] [CrossRef]
- D’Souza, S. A review of in vitro drug release test methods for nano-sized dosage forms. Adv. Pharm. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- D’Souza, S.S.; DeLuca, P.P. Methods to assess in vitro drug release from injectable polymeric particulate systems. Pharm. Res. 2006, 23, 460–474. [Google Scholar] [CrossRef]
- Modi, S.; Anderson, B.D. Determination of drug release kinetics from nanoparticles: Overcoming pitfalls of the dynamic dialysis method. Mol. Pharm. 2013, 10, 3076–3089. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yuan, W.; Li, D.; Schwendeman, A.; Schwendeman, S.P. Predicting Drug Release Kinetics from Nanocarriers inside Dialysis Bags. J. Control. Release 2019, 315, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Zambito, Y.; Pedreschi, E.; Di Colo, G. Is dialysis a reliable method for studying drug release from nanoparticulate systems?—A case study. Int. J. Pharm. 2012, 434, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Cetin, M.; Atila, A.; Kadioglu, Y. Formulation and in vitro characterization of Eudragit® L100 and Eudragit® L100-PLGA nanoparticles containing diclofenac sodium. AAPS Pharmscitech. 2010, 11, 1250–1256. [Google Scholar] [CrossRef]
- Boyd, B.J. Characterisation of drug release from cubosomes using the pressure ultrafiltration method. Int. J. Pharm. 2003, 260, 239–247. [Google Scholar] [CrossRef]
- Sun, W.; Mao, S.; Shi, Y.; Li, L.C.; Fang, L. Nanonization of itraconazole by high pressure homogenization: Stabilizer optimization and effect of particle size on oral absorption. J. Pharm. Sci. 2011, 100, 3365–3373. [Google Scholar] [CrossRef]
- Ricci, M.; Giovagnoli, S.; Blasi, P.; Schoubben, A.; Perioli, L.; Rossi, C. Development of liposomal capreomycin sulfate formulations: Effects of formulation variables on peptide encapsulation. Int. J. Pharm. 2006, 311, 172–181. [Google Scholar] [CrossRef]
- Liu, J.; Sonshine, D.A.; Shervani, S.; Hurt, R.H. Controlled release of biologically active silver from nanosilver surfaces. ACS Nano 2010, 4, 6903–6913. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.F.; Sun, C.C.; Chow, A.H.L. Assessment of the relative performance of a confined impinging jets mixer and a multi-inlet vortex mixer for curcumin nanoparticle production. Eur. J. Pharm. Biopharm. 2014, 88, 462–471. [Google Scholar] [CrossRef]
- Prentice, A.G.; Glasmacher, A. Making sense of itraconazole pharmacokinetics. J. Antimicrob. Chemother 2005, 56, 17–22. [Google Scholar] [CrossRef]
- Miyama, T.; Takanaga, H.; Matsuo, H.; Yamano, K.; Yamamoto, K.; Iga, T.; Naito, M.; Tsuruo, T.; Ishizuka, H.; Kawahara, Y. P-glycoprotein-mediated transport of itraconazole across the blood-brain barrier. Antimicrob. Agents Chemother. 1998, 42, 1738–1744. [Google Scholar] [CrossRef]
- Norkus, C.L. Veterinary Technician’s Manual for Small Animal Emergency and Critical Care; John Wiley & Sons: Hoboken, NJ, USA, 2018. [Google Scholar]
- Domanska, U.; Pobudkowska, A.; Pelczarska, A.; Gierycz, P. PKa and solubility of drugs in water, ethanol, and 1-octanol. J. Phys. Chem. B 2009, 113, 8941–8947. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A.; Hoekman, D. Exploring QSAR: Fundamentals and Applications in Chemistry and Biology; American Chemical Society: Washington, DC, USA, 1995; Volume 557. [Google Scholar]
- United States Pharmacopeial Convention. The United States Pharmacopeia: The National Formulary; United States Pharmacopeial Convention: Rockville, MD, USA, 1980. [Google Scholar]
- Chow, S.F.; Wan, K.Y.; Cheng, K.K.; Wong, K.W.; Sun, C.C.; Baum, L.; Chow, A.H.L. Development of highly stabilized curcumin nanoparticles by flash nanoprecipitation and lyophilization. Eur. J. Pharm. Biopharm. 2015, 94, 436–449. [Google Scholar] [CrossRef]
- Wan, K.Y.; Wong, S.N.; Wong, K.W.; Chow, S.F.; Lum Chow, A.H. Interplay between amphiphilic stabilizers and cholesterol in the stabilization of itraconazole nanoparticles prepared by flash nanoprecipitation. Mol. Pharm. 2019, 16, 195–204. [Google Scholar] [CrossRef]
- Weng, J.; Wong, S.; Wan, K.; Chow, A.; Chow, S. Comparative Evaluation of Four Structurally-Related Profens for Nanoparticle Formulation. In Proceedings of the AAPS PharmSci 360, Washington, DC, USA, 4–7 November 2018. [Google Scholar]
- Weng, J.; Wong, S.N.; Xu, X.; Xuan, B.; Wang, C.; Chen, R.; Sun, C.C.; Lakerveld, R.; Kwok, P.C.L.; Chow, S.F. Cocrystal Engineering of Itraconazole with Suberic Acid via Rotary Evaporation and Spray Drying. Cryst. Growth Des. 2019, 19, 2736–2745. [Google Scholar] [CrossRef]
- FDA. Dissolution Methods Database; FDA: Washington, DC, USA, 2019.
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Baker, R.; Lonsdale, H. Controlled Release: Mechanisms and Release; Springer: Berlin/Heidelberg, Germany, 1974. [Google Scholar]
- Wan, K.Y.; Wong, K.W.; Chow, A.H.L.; Chow, S.F. Impact of molecular rearrangement of amphiphilic stabilizers on physical stability of itraconazole nanoparticles prepared by flash nanoprecipitation. Int. J. Pharm. 2018, 542, 221–231. [Google Scholar] [CrossRef]
- Bi, L.; Yang, L.; Narsimhan, G.; Bhunia, A.K.; Yao, Y. Designing carbohydrate nanoparticles for prolonged efficacy of antimicrobial peptide. J. Control. Release 2011, 150, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Kisich, K.; Gelperina, S.; Higgins, M.; Wilson, S.; Shipulo, E.; Oganesyan, E.; Heifets, L. Encapsulation of moxifloxacin within poly (butyl cyanoacrylate) nanoparticles enhances efficacy against intracellular Mycobacterium tuberculosis. Int. J. Pharm. 2007, 345, 154–162. [Google Scholar] [CrossRef]
- Wallace, S.J.; Li, J.; Nation, R.L.; Boyd, B. Drug release from nanomedicines: Selection of appropriate encapsulation and release methodology. Drug Deliv. Transl. Res. 2012, 2, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Abouelmagd, S.A.; Sun, B.; Chang, A.C.; Ku, Y.J.; Yeo, Y. Release kinetics study of poorly water-soluble drugs from nanoparticles: Are we doing it right? Mol. Pharm. 2015, 12, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Wen, Y.; Wang, L.; An, X.; Zhu, X.; Ni, Y. Adsorption of polyethylene glycol (PEG) onto cellulose nano-crystals to improve its dispersity. Carbohydr. Polym. 2015, 123, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Wu, J.; Tian, H.; Zhang, L.; Xu, J. High-Strength Cellulose/Poly (ethylene glycol) Gels. ChemSusChem 2008, 1, 558–563. [Google Scholar] [CrossRef]
- Jain, R.; Sukla, S.K.; Nema, N.; Panday, A. Drug nanoparticle: A release kinetics. J. Nanomed. Nanotechnol. 2015, 6, 1. [Google Scholar]
- Azadi, S.; Ashrafi, H.; Azadi, A. Mathematical modeling of drug release from swellable polymeric nanoparticles. J. Appl. Pharm. Sci. 2017, 7, 125–133. [Google Scholar]
- Siepmann, J.; Peppas, N.A. Higuchi equation: Derivation, applications, use and misuse. Int. J. Pharm. 2011, 418, 6–12. [Google Scholar] [CrossRef]
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Merck KgaA. User Guide- Amicon® Ultra-15 Centrifugal Filter Devices for Volume up to 15 mL; Merck KgaA: Darmstadt, Germany, 2018. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | pKa | Log P | Aqueous Solubility (µg/mL) | Solubility in Release Medium (µg/mL) |
|---|---|---|---|---|
| ITZ | 3.7 [21] | 5.7 [22] | <0.09 * | 8.6 ± 0.3 |
| VitD3 | - | 7.5 [23] | <0.05 * | 90.3 ± 6.7 |
| FLU | 4.5 [24] | 4.2 [25] | 22.0 ± 5.7 | 674.5 ± 4.9 |
| Formulation | Drug | Initial Drug Conc. * (mg/mL) | Initial TPGS Conc. * (mg/mL) | Co-Stabilizer | Initial Co-Stabilizer Conc. (mg/mL) | Organic Solvent |
|---|---|---|---|---|---|---|
| ITZ nanoparticle | ITZ | 5 | 5 | CLT | 1 * | DMF |
| VitD3 nanoparticle | VitD3 | 10 | 5 | CLT | 1 * | EtOH |
| FLU nanoparticle | FLU | 10 | 20 | PVA | 0.5 | ACE |
| Formulation | Size (nm) | PDI (Polydispersity Index) | Zeta Potential (mV) | Drug Loading (%) | Encapsulation Efficiency (%) | Physical Stability (h) |
|---|---|---|---|---|---|---|
| ITZ nanoparticle | 93.75 ± 4.33 | 0.19 ± 0.02 | −0.87 ± 0.1 | 42.88 ± 0.78 | >99.96 * | 6.00 ± 0.00 |
| VitD3 nanoparticle | 41.62 ± 3.49 | 0.23 ± 0.05 | −0.31 ± 0.22 | 65.72 ± 1.23 | >99.98 * | 7.00 ± 4.58 |
| FLU nanoparticle | 28.18 ± 4.22 | 0.26 ± 0.01 | −61.23 ± 3.09 | 23.45 ± 0.99 | 86.06 ± 1.22 | 0.67 ± 0.29 |
| Drug | 0.45 μm Filter | 0.2 μm Filter | Amicon® Ultra-15 |
|---|---|---|---|
| ITZ | 99.79 ± 0.14 | 99.86 ± 0.07 | 100.25 ± 1.01 |
| VitD3 | 99.96 ± 0.04 | 99.90 ± 0.02 | 99.82 ± 0.37 |
| FLU | 100.44 ± 0.56 | 99.9 ± 30.47 | 99.78 ± 0.23 |
| Formulation | Korsmeyer-Peppas Model | Baker-Lonsdale Model | |
|---|---|---|---|
| R2 | n | R2 | |
| ITZ nanoparticles | |||
| DM | 0.987 | 0.680 | 0.994 |
| SS + CU | 0.939 | 0.510 | 0.902 |
| VitD3 nanoparticles | |||
| DM | 0.985 | 0.290 | 0.995 |
| SS + CU | 0.966 | 0 | 0.989 |
| FLU nanoparticles | |||
| DM | 0.999 | 0.816 | 0.995 |
| SS + CU | 0.994 | 0.465 | 0.991 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weng, J.; Tong, H.H.Y.; Chow, S.F. In Vitro Release Study of the Polymeric Drug Nanoparticles: Development and Validation of a Novel Method. Pharmaceutics 2020, 12, 732. https://doi.org/10.3390/pharmaceutics12080732
Weng J, Tong HHY, Chow SF. In Vitro Release Study of the Polymeric Drug Nanoparticles: Development and Validation of a Novel Method. Pharmaceutics. 2020; 12(8):732. https://doi.org/10.3390/pharmaceutics12080732
Chicago/Turabian StyleWeng, Jingwen, Henry H. Y. Tong, and Shing Fung Chow. 2020. "In Vitro Release Study of the Polymeric Drug Nanoparticles: Development and Validation of a Novel Method" Pharmaceutics 12, no. 8: 732. https://doi.org/10.3390/pharmaceutics12080732
APA StyleWeng, J., Tong, H. H. Y., & Chow, S. F. (2020). In Vitro Release Study of the Polymeric Drug Nanoparticles: Development and Validation of a Novel Method. Pharmaceutics, 12(8), 732. https://doi.org/10.3390/pharmaceutics12080732