1. Introduction
New insights into tumor biology, gained from the perpetually growing field of oncology, have prompted researchers to contrive novel treatment strategies and regimens with the aim of reducing tumor burden and improving quality of life [
1]. Prodigious work has been done to explore the antibody architecture, which demonstrates remarkable success in the clinic as therapeutic agents [
2]. A plethora of antibody strategies is currently being investigated in clinical studies. Approaches include evaluating multiple specificities, valences, and combinations thereof for improved and more selective tumor targeting or for immune system co-engagement [
3]. The conjugation of cytotoxic drugs has been widely used to increase potency [
4], which has become even more relevant with strategies that increase the selectivity for tumor tissues, e.g., by using prodrug formats of antibodies [
5,
6].
In addition to monoclonal antibodies and antibody derivatives, several non-immunoglobulin scaffold affinity proteins have been developed [
7]. One such scaffold is the affibody molecule, a small (approximately 7 kDa) three-helical bundle consisting of 58 amino acids based on the staphylococcal protein A derived Z-domain [
8]. Target-specific affibody molecules are developed from combinatorial protein engineering by randomization of typically 13 surface exposed positions. The scaffold exhibits desirable biophysical properties with regards to small size for improved tissue penetration and extravasation, high stability, and fast refolding kinetics. Amenability to the modular extension of the molecule with additional functional domains provides a versatile framework for novel drug design. Some examples include the modulation of valency, specificity, linker lengths, and other functionalities, such as albumin-binding for a prolonged circulatory half-life. The scaffold is devoid of native cysteines, which enables straightforward cytoplasmic protein production in bacterial hosts and site-directed modifications by the introduction of terminal cysteines, including the incorporation of cytotoxic drugs for increased potency and chelators for labeling with radionuclides. Rapid renal clearance, as a consequence of its small size, makes the scaffold an excellent probe for companion diagnostics and other imaging applications [
8].
Receptor tyrosine kinases in cancer have for decades been recognized as excellent targets for therapeutic intervention [
9]. In particular, human epidermal growth factor receptors (HER) have been extensively scrutinized for their pivotal role in numerous malignancies. Perhaps the most studied of these receptors is the HER2, which is upregulated in 25–30% of breast cancer patients [
10]. Potent HER signaling complexes are formed by the homo- or heterodimerization of the HER family members, which activate downstream signaling pathways that mediate cellular proliferation, migration, and survival [
11]. The dysregulation of these receptors might convert otherwise innocuous cellular processes into transforming oncogenic signals. HER3 has attracted much attention in recent years for its important function as a driver of tumor progression and acquired treatment resistance [
12]. The overexpression of this receptor, which occurs at a high frequency in several types of cancer, including breast, pancreatic, and ovarian cancer, has been associated with the decreased survival of patients, in particular with concomitant HER2 overexpression [
13]. HER3 adopts a dimer-competent conformation upon the binding of its cognate ligands. Although the kinase activity of HER3 is impaired, the potent activation of downstream signaling pathways is achieved by heterodimerization with a kinase functional coreceptor, preferentially HER2, resulting in the transphosphorylation of six tyrosine residues situated on the cytosolic tail. The phosphotyrosines of HER3 act as docking sites for the initiation of signaling cascades, especially via the direct binding of the p85 subunit of PI-3K [
14]. Furthermore, the HER3/HER2 heterodimer is thought to be the most potent mitogenic unit within the HER family and has a crucial function for HER2-mediated transformation and the continued growth of breast cancer cell lines driven by HER2 overexpression, demonstrated by two different HER3 knockdown models [
15,
16]. In clinical studies, it was demonstrated that a combination of anti-HER2 and anti-HER3 immunotherapies resulted in an encouraging overall response rate of 55% [
17]. Thus, anti-HER3 therapies would be desirable for the treatment of patients with HER3-driven resistance.
There is currently no FDA-approved treatment specifically targeting HER3 and the two most prominent HER3-targeting antibody drug candidates, patritumab and seribantumab, were terminated in late-phase clinical trials for lack of efficacy (NCT02134015, NCT03241810). Nonetheless, numerous HER3-targeting strategies are being evaluated as a single or combination therapies in early stages of clinical development [
12]. As it stands, HER3-mediated resistance to HER-targeted treatments, which encompass a wide range of therapeutics [
18,
19,
20], poses major challenges for cancer therapy and calls for new and improved HER3-targeting therapeutics to be developed.
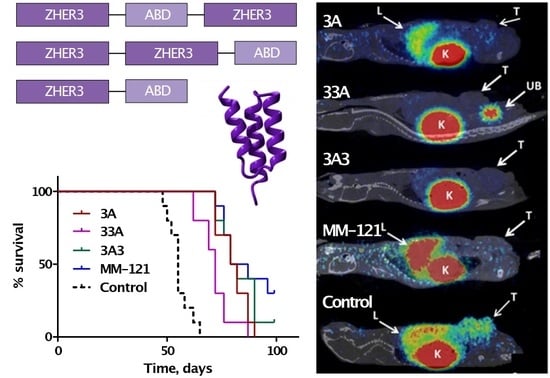
We have previously investigated the potential for affibody-based therapeutics targeting HER3 by evaluating the bivalent affibody-based construct TAM-HER3 in a preclinical in vivo model [
21]. The molecule consists of two identical HER3-targeting affibody domains flanking an albumin-binding domain (ABD). The affibody domain Z
HER3:08698 has previously been selected by our group and binds HER3 antagonistically in the picomolar range with cross-reactivity to the mouse ortholog [
22]. The engineered 46-residue (approximately 5.2 kDa) ABD binds human serum albumin with femtomolar affinity and was included for the extension of construct half-life in circulation [
23]. In order to elucidate the influence of domain orientation and valency on in vivo tumor targeting and biodistribution properties, five ABD-fused HER3-targeting variants were designed in either a mono- or bivalent format [
24]. The monovalent (Z
HER3:08698-G
3S-ABD
035, ABD
035-G
3S-Z
HER3:08698), denoted by 3A and A3 respectively, and bivalent variants (Z
HER3:08698-G
3S-ABD
035-G
3S-Z
HER3:08698, Z
HER3:08698-G
3S-Z
HER3:08698-G
3S-ABD
035, ABD
035-G
3S-Z
HER3:08698-G
3S-Z
HER3:08698), denoted by 3A3, 33A, and A33, respectively, could all inhibit heregulin-induced phosphorylation in HER3-expressing BxPC-3 and DU-145 cell lines [
24]. Biodistribution studies conducted in mice bearing BxPC-3 xenografts revealed disparities in blood retention and organ uptake, most notably in excretory organs and in the tumors. Formats with an ABD located at the C-terminus (3A, 33A), as well as 3A3, provided the most favorable biodistribution profiles for tumor targeting in vivo. Additionally, differences in the internalization rate were observed for the studied variants in a panel of cell lines with varying levels of HER3 expression. The ability to induce rapid internalization is of relevance when designing payload delivery, which could be a viable strategy for HER3-targeted therapy [
25]. Based on our results, we hypothesized that the molecular design of affibody-based therapeutics targeting HER3 in terms of the relative position of functional domains and valency has an impact on tumor targeting, biodistribution properties, and internalization rates.
The aim of the present study is to expand on previous findings that revealed a significant influence of molecular design on the in vivo targeting properties and pharmacokinetics of HER3-targeting affibody constructs [
24]. Based on biodistribution data, the most promising formats (3A3, 33A, and 3A) were selected for the evaluation of therapeutic potential in a preclinical therapy efficacy study using a pancreatic adenocarcinoma xenograft model (BxPC-3). HER3 is often overexpressed in pancreatic cancer and is critical for mediating tumorigenesis via transactivation by epidermal growth factor receptor (EGFR) [
26]. Both 3A3 and 3A demonstrated similar efficacy to MM-121 in terms of tumor growth inhibition and the prolonged survival of treated mice, whereas moderate efficacy was observed for 33A. No apparent side effects were observed for the affibody variants.
2. Materials and Methods
2.1. General
In vitro and in vivo studies were performed on BxPC-3 cells, primary pancreatic adenocarcinoma, obtained from the American Type Culture Collection (ATCC) via LGC Promochem, Borås, Sweden. Cells were cultured according to ATCC recommendations. In vivo experiments were planned and performed in accordance with national legislation on laboratory animal protection and were approved by the Ethics Committee for Animal Research in Uppsala, Sweden (animal permission C143/14, approved 16 September 2014).
Data were evaluated either by an unpaired, two-tailed t-test or by one-way ANOVA with Bonferroni correction for multiple comparisons using GraphPad Prism (version 6 for Windows GraphPad Software) in order to determine statistically significant differences (p < 0.05). Obtained values are presented as an average with standard deviation if not stated otherwise.
2.2. Production and HSA Purification
Genes for 3A3, 33A, and 3A, identical to previously investigated constructs [
24] but lacking C-terminal cysteine, were subcloned into a pET45b(+) vector (Thermo Scientific, Chicago, IL, USA). The plasmids were transformed into BL21*(DE3) Escherichia coli (
E. coli) (Thermo Scientific, Chicago, IL, USA) using a standard heat-shock protocol. Protein production was performed using fed-batch cultivation in a 6 × 1 L multifermenter system (GRETA4, Belach Bioteknik AB, Stockholm, Sweden). Shake flasks containing TSB + Y medium and 100 μg/mL carbenicillin were inoculated with single colonies and placed in a shake incubator at 37 °C and 180 rpm for 7 h. The inocula for each construct were added to separate fermenters and induced with a final concentration of 0.5 mM isopropyl β-
d-1-thiogalactopyranoside (IPTG). Protein expression proceeded for 5 h before harvest. The harvested cells were lysed using a French press and the proteins were recovered with affinity chromatography on an ÄKTApure system (GE Healthcare, Uppsala, Sweden) using human serum albumin (HSA) immobilized on a Sepharose matrix. The column was equilibrated with TST buffer (25 mM Tris-HCl, 1 mM EDTA, 200 mM NaCl, 0.05% Tween, pH 8.0), which was used as a running buffer. The loaded sample was washed with ammonium acetate (5 mM, pH 5.5), followed by the isocratic elution of bound proteins with acetic acid (0.5 M, pH 2.8).
2.3. Reversed-Phase Chromatography
The eluate from the HSA purifications was adjusted to an acetonitrile (ACN) concentration of 10%, sterile filtered, and applied on a HR16/10 column (GE Healthcare, Uppsala, Sweden) packed with Source 15RPC (GE Healthcare, Uppsala, Sweden) previously equilibrated with RPC A-buffer (0.1% TFA, 10% ACN, 90% MQ water). RPC purification was performed on an ÄKTAexplore 100 (GE Healthcare, Uppsala, Sweden) with a flow rate of 6 mL/min. After sample loading, the column was washed with RPC A-buffer. After the column wash, bound protein was eluted with a linear gradient 0–50% RPC B-buffer (0.1% TFA, 80% ACN, 20% MQ water).
2.4. Buffer Exchange, Concentration, and Endotoxin Level Determination
The RPC pool was buffer exchanged to 1×DPBS (Corning Inc., New York, NY, USA; endotoxin tested) on two HiPrep 26/10 Desalting columns (GE Healthcare, Uppsala, Sweden) connected in series. The buffer exchange was performed by three consecutive injections of the RPC pool by using an ÄKTAexplore 100 system with a flow rate of 5–8 mL/min. Eluted protein from each injection was collected and pooled.
The buffer-exchanged pool of 3A3 and 3A was concentrated to 4.0 mg/mL and 2.6 mg/mL, respectively, by using an Amicon Ultra-15 centrifugal filter with 3kDa NMWL (Merck Millipore, Burlington, MA, USA). The concentration of 33A was diluted to 0.766 mg/mL in PBS containing 50 mM L-arginine to prevent aggregation. The endotoxin levels were determined to be <0.2 EU/mL for 3A3 and 3A and <0.5 EU/mL for 33A using an Endoafe PTS unit (Charles River, MA, USA) with a 1–0.01 EU/mL cartridge (Charles River, MA, USA).
2.5. Purity and Mass Identity Determination
The purity was determined with reverse-phase high performance liquid chromatography (RP-HPLC) on a 1200 series HPLC system (Agilent Technologies, Santa Clara, CA, USA) using an analytical Zorbax 300SB-C18 column (Agilent Technologies, Santa Clara, CA, USA) with a 20–50% acetonitrile elution gradient over 20 min with a flow rate of 1 mL/min. Electrospray ionization mass spectrometry (ESI-MS) with a 6520 Accurate-Mass Q-TOF LC/MS (Agilent Technologies, Santa Clara, CA, USA) was used to confirm the mass identity of the purified constructs.
2.6. Secondary Structure and Thermal Stability Analysis
Alpha-helical content, thermal stability, and refolding capacity were analyzed using circular dichroism spectroscopy on a Chirascan spectropolarimeter (Applied Photophysics, Surrey, UK) with an optical path length of 1 mm and a protein concentration of 0.25 mg/mL. The evaluation of thermal stability was performed using variable temperature measurement (VTM). The change in ellipticity at 221 nm was monitored while heating (5 °C/min) the sample from 20 to 90 °C. The approximations of the melting temperatures were made from non-linear regressions of the generated curves using a Boltzmann Sigmoidal model (GraphPad Prism, version 7, GraphPad Software, La Jolla, CA, USA). Spectra obtained from measurements at wavelengths in the range 195–260 nm at 20 °C before and after VTM were compared to assess the refoldability of the affibody molecules after thermal denaturation.
2.7. Affinity Determination
The binding affinity of the constructs to human HER3 (Sino Biological, Wayne, PA, USA) and murine mErbB3 (Sino Biological, Wayne, PA, USA) was evaluated with surface plasmon resonance (SPR) on a BIAcore T200 system (GE Healthcare, Uppsala, Sweden) using a CM5 sensor chip immobilized with HSA. The constructs were captured on the HSA surface, followed by multi-cycle injections of five HER3 or mErbB3 concentrations (3.125, 6.25, 12.5, 25, and 50 nM). In addition, the binding affinity to HSA and mouse serum albumin (MSA) was evaluated using the same sensor chip and a multi-cycle setup with three concentrations (3.125, 6.25 and 12.5 nM) of the constructs. All experiments were performed in duplicate. The acquired sensorgrams were analyzed using a Langmuir 1:1 kinetic model.
2.8. Inhibition of In Vitro Phosphorylation
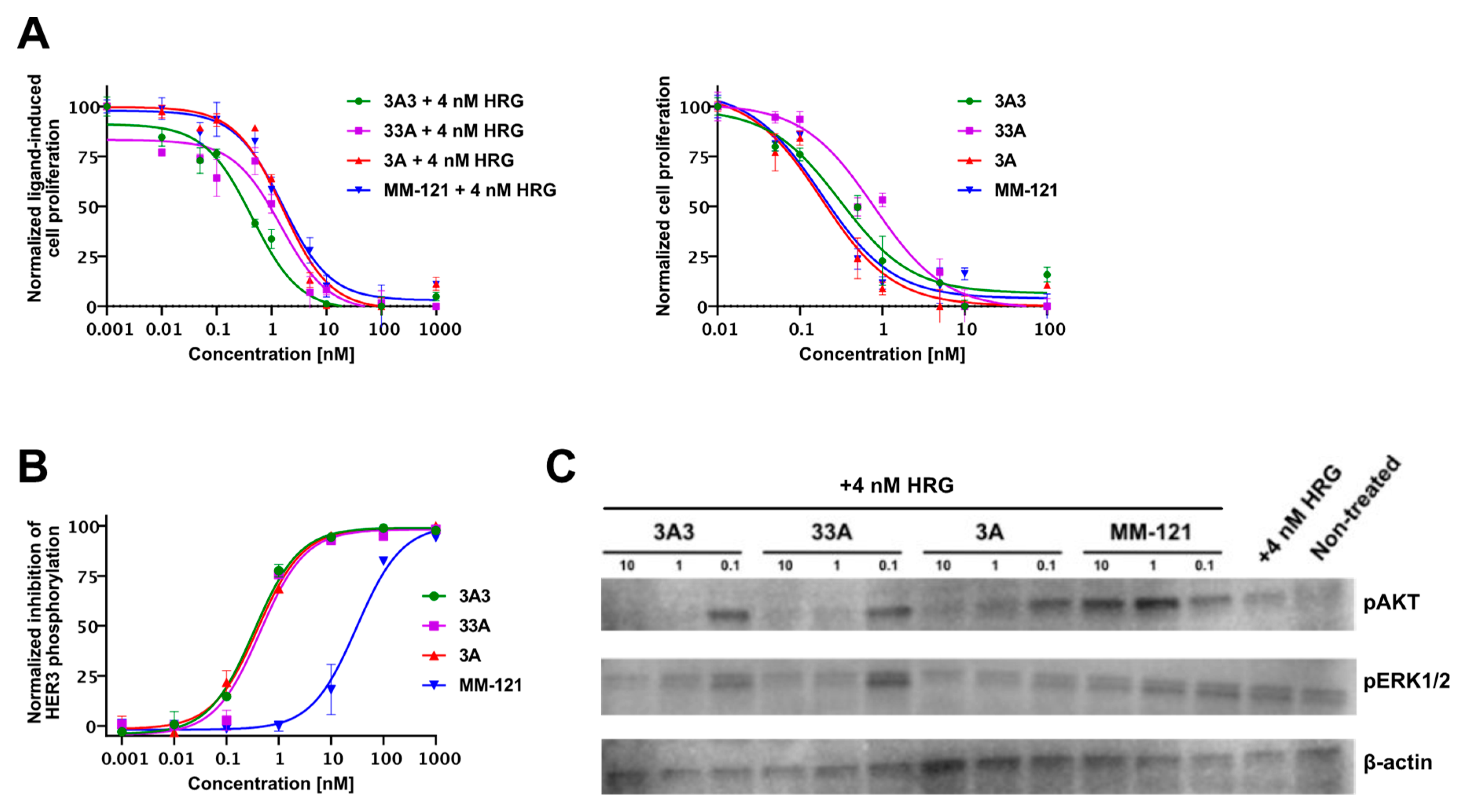
The inhibition of HER3 phosphorylation on BxPC-3 cells following treatment with affibody molecules was evaluated using a Phospho-HER3 ELISA assay (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s protocol. Western blot was used to evaluate the phosphorylation status of the downstream signaling molecules AKT and ERK1/2 on the same treated samples. The pAKT antibody (15306974; rabbit monoclonal, 1/500 dilution), pERK1/2 antibody (11372362; rabbit monoclonal, 1/1000 dilution), ß-actin antibody (15284898; rabbit monoclonal, 1/2000 dilution), and anti-rabbit HRP-conjugated antibody (11859140, goat polyclonal, 1/2000 dilution) were purchased from Fisher Scientific (Hampton, NH, USA).
Treated cell samples were prepared as previously described [
24]. Briefly, BxPC-3 cells were expanded in RPMI-1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin–streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). Cells were seeded at 10
6 cells per well in six-well plates and allowed to adhere overnight. The medium was subsequently changed to a starvation medium (RPMI-1640 supplemented with 0.5% FBS) and incubated overnight at 37 °C. Cells were treated with seven different concentrations of either affibody construct (3A3, 33A and 3A) or MM-121 for 10 min at 37 °C. The volume in wells with untreated cells were equalized using the starvation medium. Cells were then treated with 4 nM heregulin and incubated for an additional 10 min at 37 °C. The plates were transferred to ice, lysed using Cell Lysis Buffer (Cell Signaling Technology, Danvers, MA, USA), containing 100 mM orthovanadate (BioNordika, Stockholm, Sweden), 100 mM PMSF (Sigma-Aldrich, St. Louis, MO, USA), and PhosSTOP (Sigma-Aldrich, St. Louis, MO, USA), and collected.
2.9. Inhibition of In Vitro Proliferation
The HER3-expressing cancer cell line BxPC-3 was cultured in RPMI-1640 medium supplemented with 10% FBS and 1% penicillin–streptomycin. The cells were treated with TrypLE Express Enzyme (Thermo Fisher Scientific, MA, USA), diluted using RPMI-1640 supplemented with 4% FBS, and seeded in 96-well plates at a density of 2000 cells per well and allowed to adhere overnight. On the subsequent day, cells were treated with ten concentrations of the affibody constructs and MM-121 (the production and characterization of the antibody is as previously described [
21]) in the presence or absence of 4 nM heregulin and incubated at 37 °C for 5 days. Cell viability was determined by measuring fluorescence after incubating with alamarBlue Cell Viability Reagent (Invitrogen, Waltham, MA, USA) for 2 h.
2.10. Therapy Study
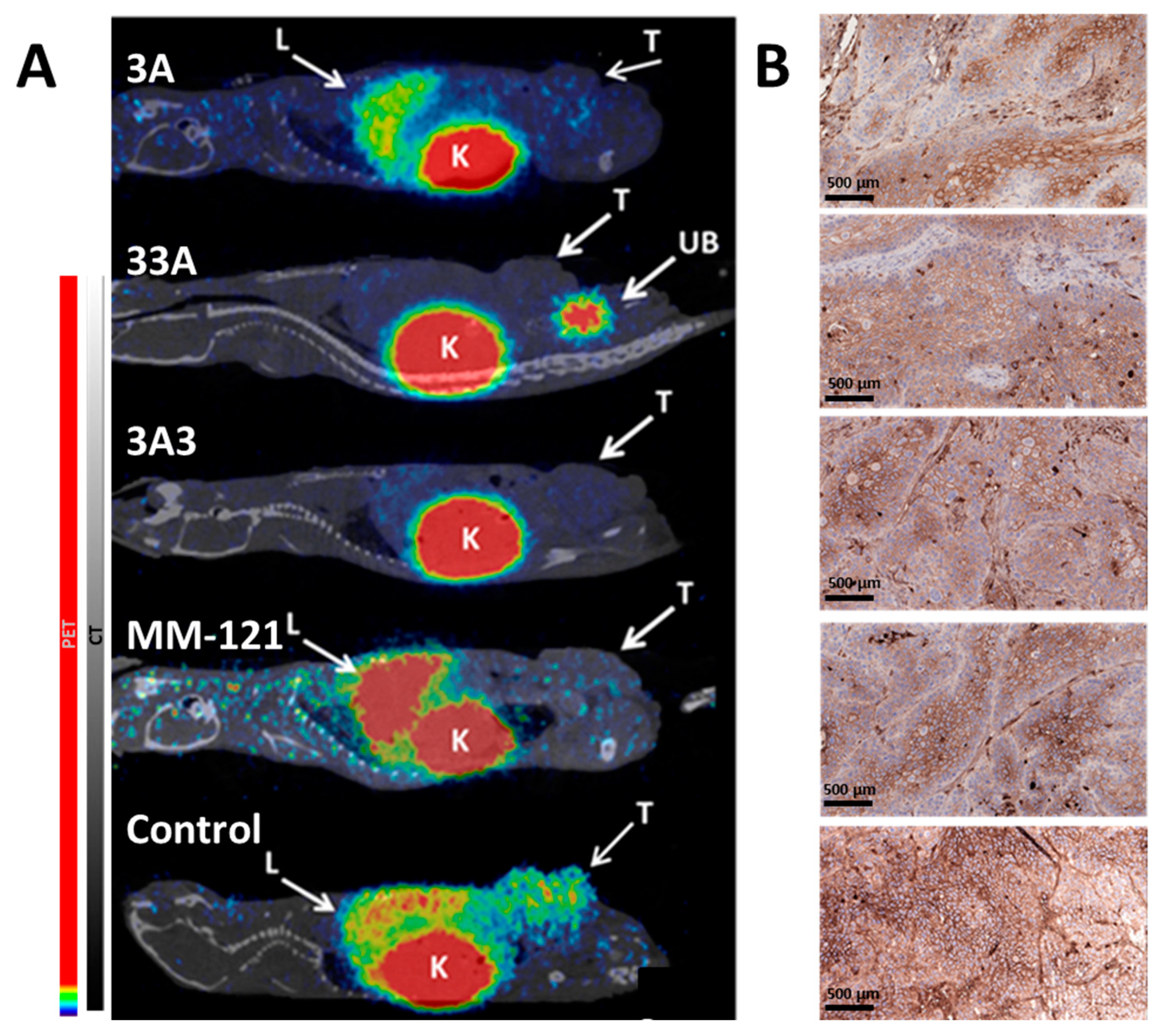
Female Balb/c nu/nu mice were implanted with a 100 µL suspension of 5 × 106 BxPC-3 cells in PBS, and one week later (day 0) mice were randomly sorted into five groups (n = 9–10 per group). Tumor volume was 45 ± 20 mm3 and mouse weight was 16 ± 1 g at the start of the experiments. Mice were i.p. injected with 150 μL conjugate solution in PBS containing 400 µg of 3A, 600 μg of 33A, 600 μg of 3A3, or 600 µg MM-121 three times per week. The control group was injected with PBS only. Tumor dimensions were measured using digital calipers and mice status was monitored twice per week. Mice were euthanized at a predetermined humane end point (tumor volume exceeding 1 cm3 or ulcerated, or when the animal’s weight was reduced by 10% within one week). The practical end point was 93 days after treatment started, with the last treatment being performed on day 90. HER3 occupancy was investigated using [68Ga]Ga-(HE)3-Z08698-NODAGA when tumors reached 700–800 mm3, as described below.
At the humane end point, samples from blood serum, kidney, liver, and tumor were collected for pathological examination. Blood serum was analyzed for the concentration of urea, creatinine, aspartate aminotransferase (AST), and alkaline phosphatase (ALP) at the Department of Pathology and Wildlife Diseases, National Veterinary Institute, Uppsala, Sweden. Histological examination was performed at the same department. Hemotoxylin, eosin (HE), and HER3 immunohistochemical (IHC) staining and slide scanning were performed at the Swedish SciLifeLab facilities, as previously described [
21].
2.11. Tumor Imaging
The labeling of (HE)
3-Z
08698-NODAGA with gallium-68 and micro positron emission tomography (microPET)/computed tomography (CT) imaging of HER3 expression in xenografted mice were done according to a published protocol [
27]. Briefly, whole body PET scans of the BxPC-3 xenografted mice were performed under general anesthesia in a nanoScan PET/MRI system (Mediso Medical Imaging Systems Ltd., Budapest, Hungary) 1 h post i.v. injection of 2 µg of the anti-HER3 affibody imaging probe [
68Ga]Ga-(HE)
3-Z
08698-NODAGA (1.6–7.3 MBq). CT acquisitions were performed using a nanoScan SPECT/CT system (Mediso Medical Imaging Systems Ltd., Budapest, Hungary) immediately after PET acquisition using the same bed position. PET scans were performed for 30 min. PET data were reconstructed into a static image using a Tera-Tomo™ 3D reconstruction engine. CT data were reconstructed using filtered back projection. PET and CT files were fused and analyzed using Nucline 2.03 Software. Imaging was performed one day after therapeutic injection.
4. Discussion
The overexpression of HER3 is a potential cause for therapy resistance and a driver for tumor progression in many cancers [
12]. HER3 has thus attracted attention as a molecular target for cancer therapy. While several HER3-targeting therapeutic agents are currently under preclinical and clinical development, no HER3-specific drugs or treatments have yet been approved [
12]. Efforts by our group and others have demonstrated in preclinical models that anti-HER3 affibody molecules could be an alternative for HER3-targeted therapy [
21,
27,
28]. For example, the affibody construct TAM-HER3 (an ABD flanked by two HER3-binding affibody molecules) delayed the growth of HER3 expressing tumors in a mouse model with equal potency as the HER3-targeting antibody MM-121 [
21], indicating the main mode of action is based on receptor blocking and is not Fc-mediated. It should also be noted that MM-121 (seribantumab) was in an IgG2 format for the clinical evaluations in order to minimize potential Fc-mediated adverse effects, indicating that the biological effect from MM-121 is largely driven by ligand blocking [
29]. In an effort to optimize the format of TAM-HER3, we hypothesized that the arrangement of the molecular building blocks could affect the therapeutic efficacy of the construct. The comparison of five different ABD-fused monovalent and bivalent anti-HER3 affibody constructs [
24] demonstrated that tumor uptake, clearance, and tumor retention were significantly influenced by valency and the position of the ABD.
In addition to the previously investigated TAM-HER3 (3A3), variants with an ABD fused to the C-terminus (3A, 33A) showed promising biodistribution profiles and inhibition of heregulin-induced phosphorylation in HER3-expressing BxPC-3 cells [
24]. Based on this, we decided to investigate the therapeutic efficacy of these HER3-targeting affibody constructs in vivo using a preclinical pancreatic cancer model, together with the therapeutic HER3-targeting monoclonal antibody MM-121 as a comparator.
Treatment with all affibody variants was well tolerated, as corroborated by animal weight curves, the analysis of blood serum, and liver and kidney pathology. Tumor growth data for MM-121 and the 3A3 construct were comparable to previously reported results [
21]. All three affibody variants demonstrated a therapeutic effect, e.g., significantly delayed tumor growth and prolonged survival compared to the control (PBS). This was in agreement with the results from in vitro experiments showing the dose-dependent inhibition of cell proliferation and HER3 phosphorylation. Furthermore, western blot analysis showed a downregulation of the downstream signaling molecules pAKT and pERK1/2, which was in good agreement with the downregulation of pHER3 observed from ELISA at the given concentrations.
While there was no significant difference between the affibody variants in vitro, the molecular design of the affibody molecules influenced the anti-proliferative efficacy and median survival in vivo. Among the tested variants, 33A was the least potent in terms of tumor growth inhibition and prolonging survival. Tumors in animals treated with 33A were significantly smaller (volume in mm
3) than in the control only 30 days after the start of treatment, compared to 14 days for 3A, 3A3, and MM-121. Furthermore, after 58 days, there was a significant separation in tumor volume curves between 33A- and 3A- or MM-121-treated groups. Animals treated with 33A also had a significantly shorter median survival than animals treated with 3A, 3A3, and MM-121. The lower in vivo efficacy of 33A could be explained by a more rapid blood clearance, and thus reduced bioavailability compared to 3A and 3A3, which was previously reported by our group [
24].
Comparing the tested affibody variants with the HER3-targeting affibody MM-121, we could conclude that treatment with the monovalent 3A variant was equally potent as the treatment with MM-121, both regarding absolute tumor volume and median survival. This could partially be attributed to the longer circulation of 3A in the blood [
24] and a higher monovalent affinity for HER3 compared to 3A3 and 33A. The slower clearance potentially provides a more constant supply to the tumor and the increased affinity could prolong its retention in tumors. A longer residence time in blood might also explain why MM-121 demonstrated a high in vivo efficacy, despite significantly lower IC50 values in the phospho-ELISA assay.
Multivalence did not appear to improve the therapeutic efficacy of our affibody molecules. This is particularly interesting, because our findings contradict data published by Schardt et al. [
27,
28], who investigated the therapeutic efficacy of multivalent anti-HER3 affibody constructs using the HER3-binding variant Z
05413. The authors hypothesized that dimerization would improve affinity through avidity and potentially increase the inhibition of HER3 phosphorylation, downstream signaling, and tumor progression and therefore improve the therapeutic efficacy [
27,
28]. Indeed, the authors demonstrated a significant reduction in tumor progression compared to controls for groups treated with bivalent affibody constructs but not with monovalent constructs [
27]. It should be noted that in the study, in vitro efficacy was dependent on cell lines being autocrine for heregulin [
28] and in vivo experiments were performed using an ovarian cancer model with OvCAR8-derived ADR-RES xenografts [
27]. The different in vivo model could be a possible explanation for the deviating results, as the density of HER3 receptors could affect the ability of bivalent variants to bind two HER3 receptors simultaneously. In the present study, the lower HER3 receptor density on BxPC-3 cells might prevent the bivalent binding of the 33A and 3A3 constructs, in which case the smaller size of 3A could be beneficial for efficient tumor penetration and extravasation.
The observation of more prevalent cystic structures in tumors in the control group and more apoptotic cells and inflammations in the treated groups suggest that the evaluated constructs have a therapeutic effect on cell growth and survival, which is in agreement with the in vitro data demonstrating the inhibition of HER3 phosphorylation and of downstream signaling molecules. In the histopathological examination of tumor material, it was found that treatment with the affibody constructs and MM-121 cause the downregulation of HER3, together with inflammatory cell reactions in the periphery of treated tumors (but not in the center of tumor cell islands), indicating a limited penetration depth of the molecules. The downregulation of HER3 expression in response to HER3-targeted therapy has previously been observed [
21,
27,
30]. In contrast to our findings however, Schardt et al. [
27,
28] reported HER3 downregulation mainly by the multimeric HER3-targeting affibody constructs, but no or little effect was observed for the monomeric affibody. It was hypothesized that multivalence is required to trigger the sequestration of HER3 receptors, but from our data it could be speculated that the multivalence of the therapeutic agent is not essential for the downregulation of HER3 and for achieving a therapeutic effect. The larger size of the multimeric affibody constructs (3A3, 33A) could possibly negate the positive effects of potential avidity by reducing tissue penetration and extravasation compared to the monovalent 3A. Smaller size is one of the potential advantages of using smaller scaffold proteins rather than antibodies.
In addition to the evaluation of the therapeutic efficacy of the affibody constructs, we also demonstrated the feasibility of the radiolabeled anti-HER3 conjugate
68Ga-(HE)
3-Z
HER3-NODAGA as a companion diagnostic agent of HER3 expression and of the monitoring of receptor occupancy.
68Ga-(HE)
3-Z
HER3-NODAGA was developed and preclinically evaluated for the imaging of HER3 expression [
31]. As expected, the control animal showed activity uptake in the non-treated HER3 expressing xenografts, as well as the liver and kidneys. The renal pathway is the main elimination pathway for affibody molecules and the liver naturally expresses mErbB3 (murine ortholog of HER3), resulting in tracer accumulation in these organs. No activity uptake of
68Ga-(HE)
3-Z
HER3-NODAGA was observed in the tumors of animals in the treated groups, demonstrating the occupation of HER3 receptors by the therapeutic agents. PET imaging showed no differences between the bivalent affibody constructs, but revealed hepatic uptake of
68Ga-(HE)
3-Z
HER3-NODAGA in animals treated with MM-121 and 3A. MM-121 is not cross-reactive to mErbB3, which explains the activity accumulation in the liver. The hepatic uptake was lower in animals treated with 3A than in control animals or animals treated with MM-121. This low, but not negligible, hepatic uptake could be explained by differences in cellular processing in tumor and normal cells after the binding of anti-HER3 affibody molecules. Despite similar values of binding affinities both to mErbB3 and MSA for all tested affibody constructs (
Table 1), their blood clearance rate and hepatic uptake differed dramatically [
24]. The monovalent variant 3A had significantly higher blood concentration and significantly lower hepatic uptake 24 h p.i. than the bivalent variants 33A and 3A3 [
24]. It has previously been shown that a trimeric anti-HER3 affibody (without an ABD) had a 10-fold higher hepatic uptake than a monomeric affibody, supposedly due to the slower dissociation of the multivalent affibody and HER3 sequestration [
32]. It could be speculated that the bivalent therapeutic variants have a similar effect, and as a result, the hepatic uptake of
68Ga-(HE)
3-Z
HER3-NODAGA was observed in the 3A treated group, but not the 3A3 and 33A groups. Additionally, the images confirmed that the affibody constructs and MM-121 bind to the same epitope of the HER3 receptor, as was shown previously in vitro [
33]. This demonstrates that anti-HER3 affibody-based imaging agents could be used as highly sensitive and specific diagnostics for both patient stratification and therapy monitoring in therapy studies involving MM-121.
These results extend the work conducted by Schardt et al. evaluating the efficacy of mono- and bivalent HER3-targeting affibody constructs in an ovarian cancer xenograft model as monotherapies and in combination with carboplatin [
27]. This study demonstrates the plausibility of affibody molecules as HER3-targeting therapeutic agents for the inhibition of HER3-mediated tumor growth and corroborates previously obtained in vivo efficacy data pertaining to the structurally similar TAM-HER3 [
21].



 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}