Development of a Sustained Release Nano-In-Gel Delivery System for the Chemotactic and Angiogenic Growth Factor Stromal-Derived Factor 1α

 ,
,
Abstract
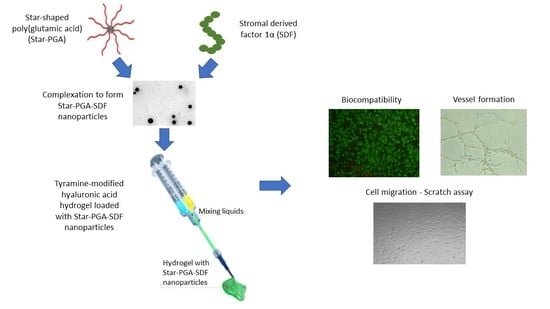
1. Introduction
2. Materials and Methods
2.1. Materials
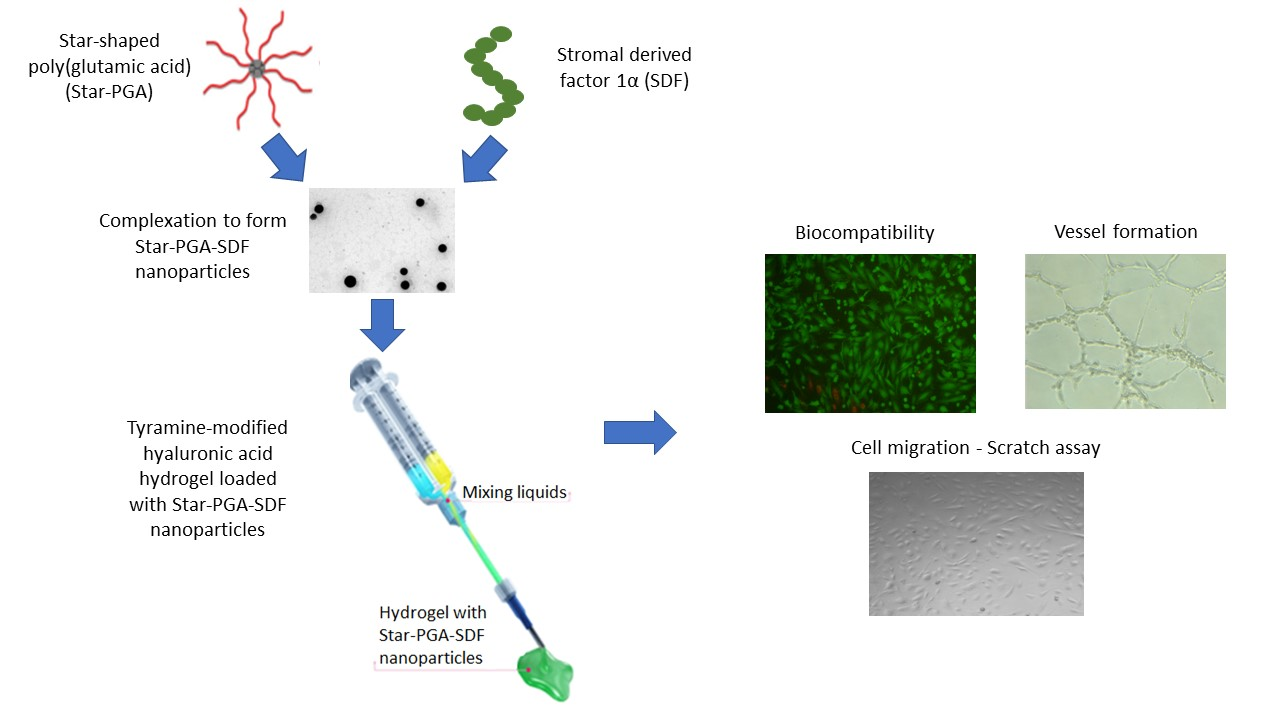
2.2. Nanoparticle Fabrication
2.3. Nanoparticle Size and Zeta Potential
2.4. Nanoparticle Tracking Analysis
2.5. Transmission Electron Microscopy
2.6. Nanoparticle Complexation Efficiency and Loading Content
2.7. In Vitro Biocomtapibility Testing of L-PGA–SDF and Star-PGA–SDF Nanoparticles
2.7.1. Culture of Human Umbilical Vein Endothelial Cells
2.7.2. Biocompatibility of L-PGA–SDF and Star-PGA–SDF
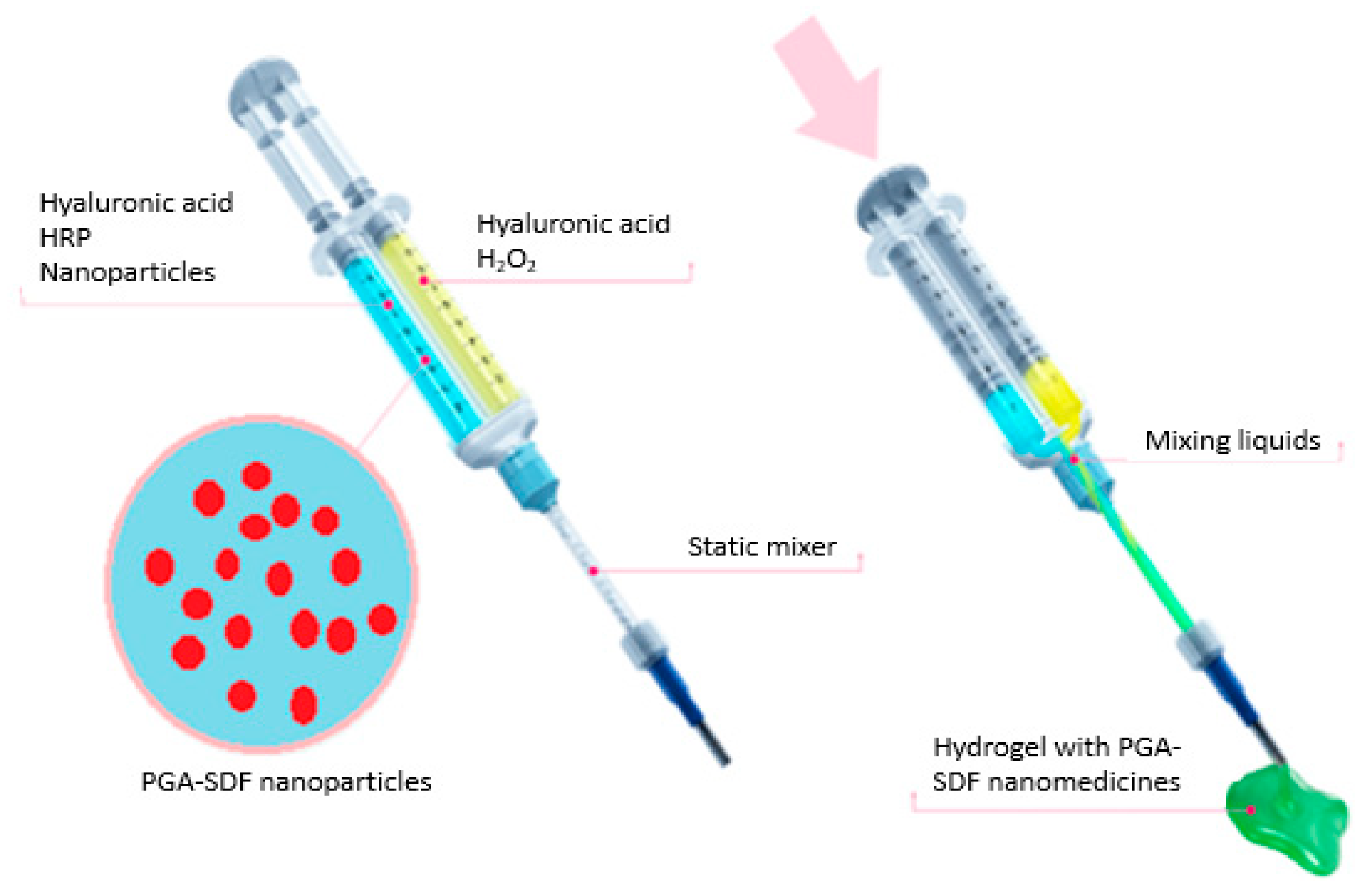
2.8. Preparation of Star-PGA–SDF Nanoparticle-Loaded Hyaluronic Acid Hydrogels
2.9. Release of SDF from a Star-PGA–SDF–HA–TA Nano-In-Gel Formulation
2.10. Biocompatibility of Star-PGA–SDF–HA–TA
2.11. Bioactivity of SDF Released From Star-PGA–SDF–HA–TA
2.11.1. Microvessel Formation—Matrigel® Assay
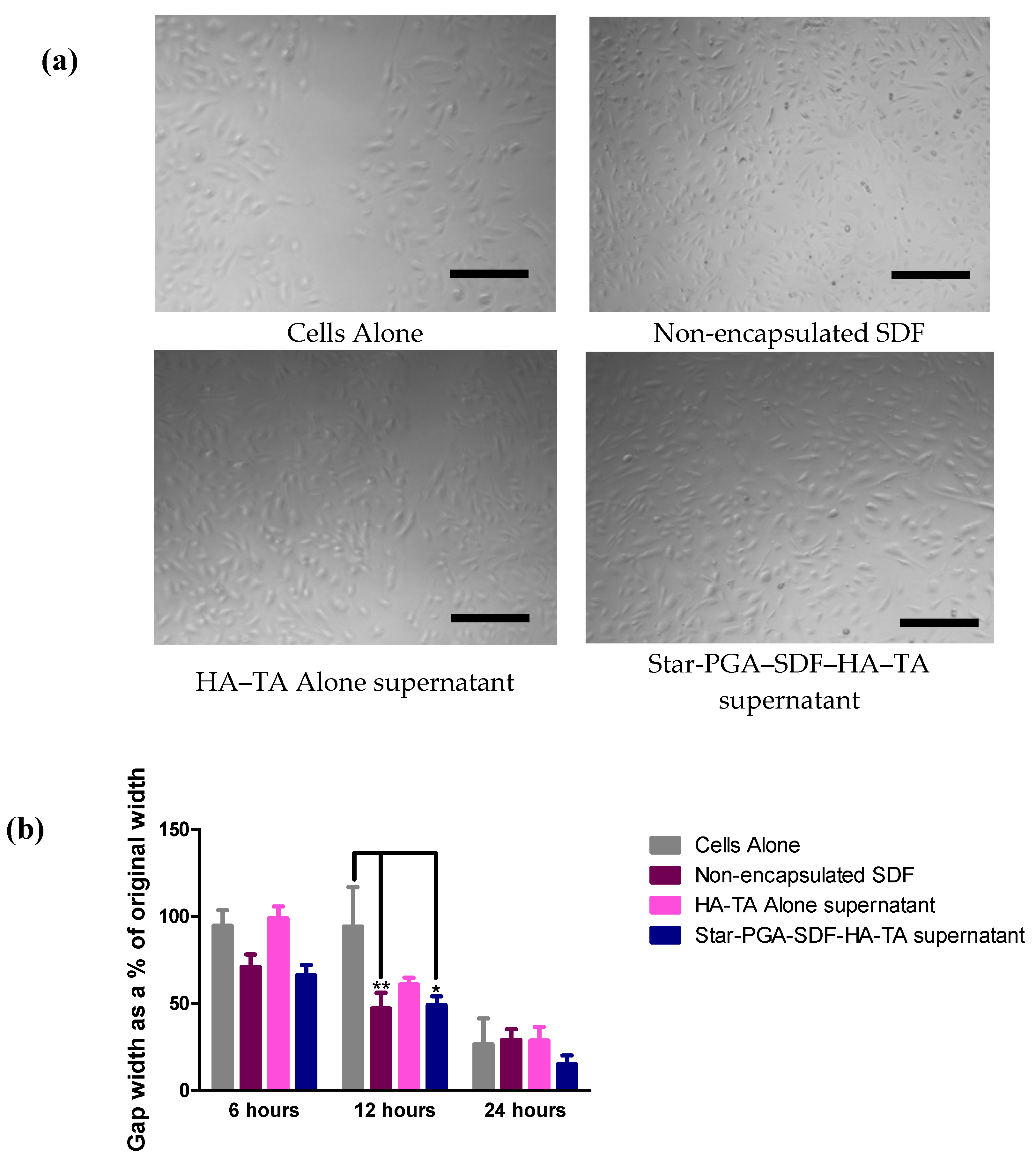
2.11.2. Cell Migration—Scratch Assay
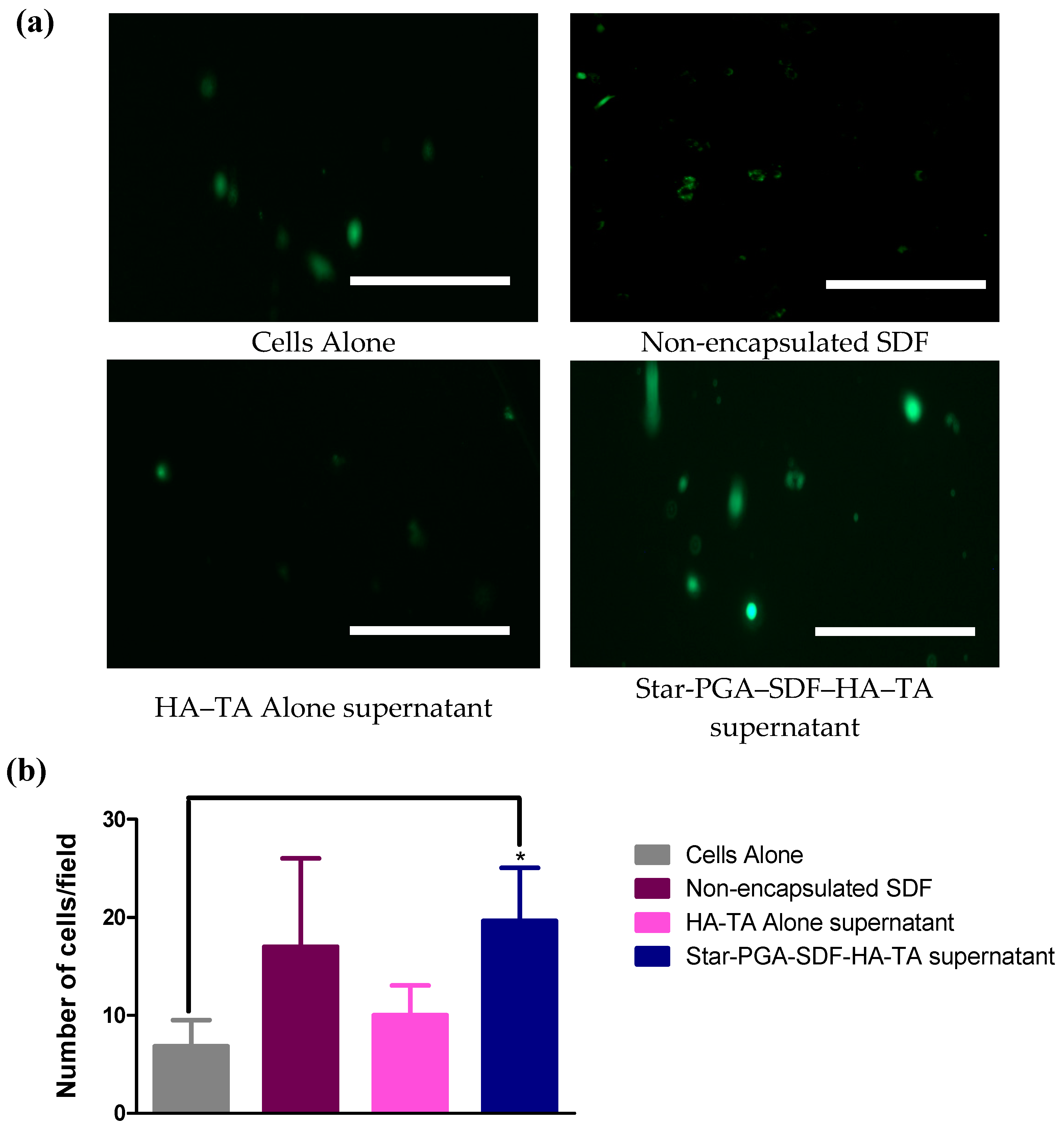
2.11.3. Cell Migration—Transwell® Migration Assay
2.12. Statistical Analysis
3. Results
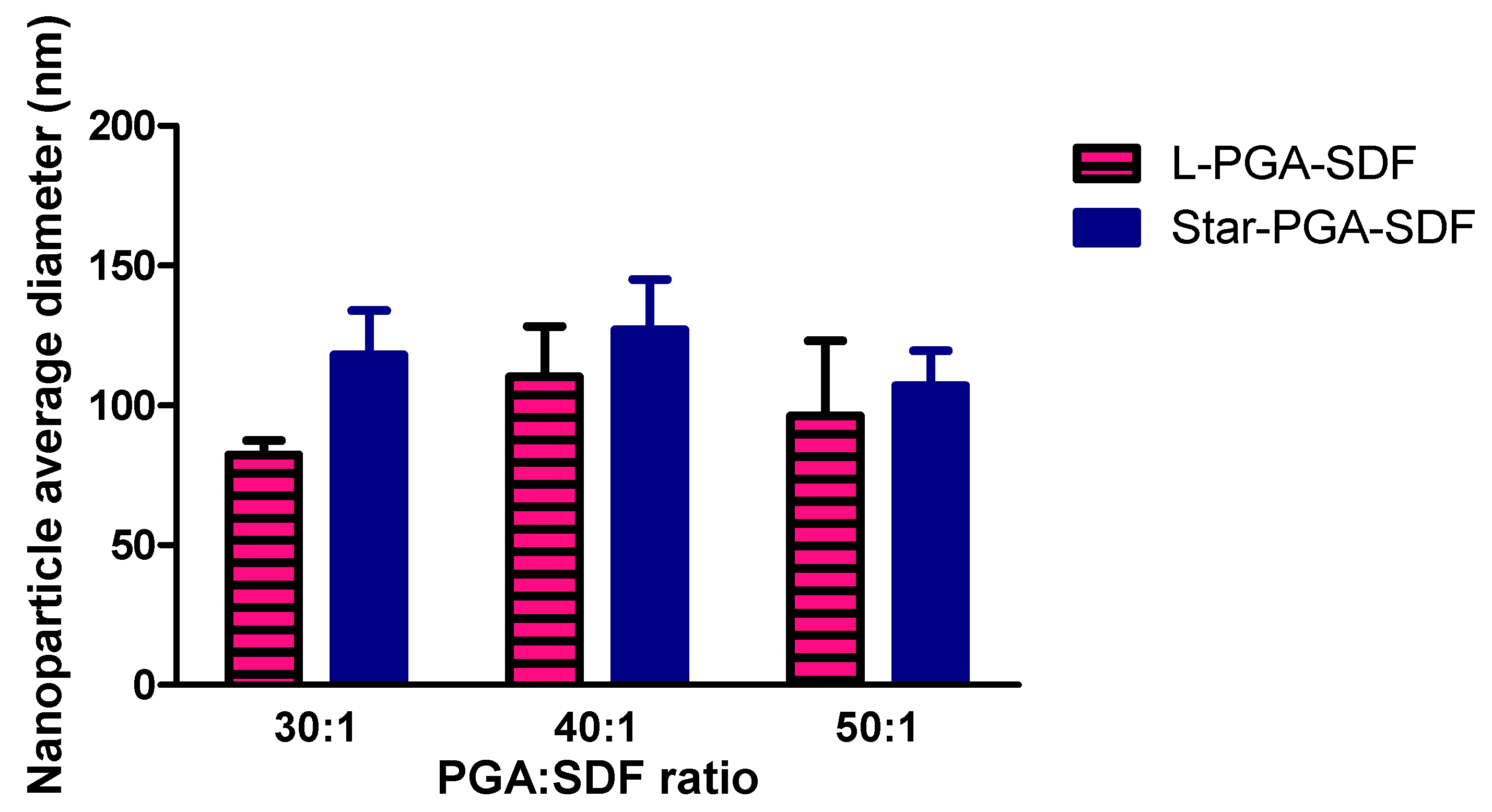
3.1. Nanoparticle Fabrication and Physicochemical Characterisation
3.2. Nanoparticle Tracking Analysis
3.3. Transmission Electron Microscopy
3.4. Nanoparticle Complexation Efficiency and Loading Content
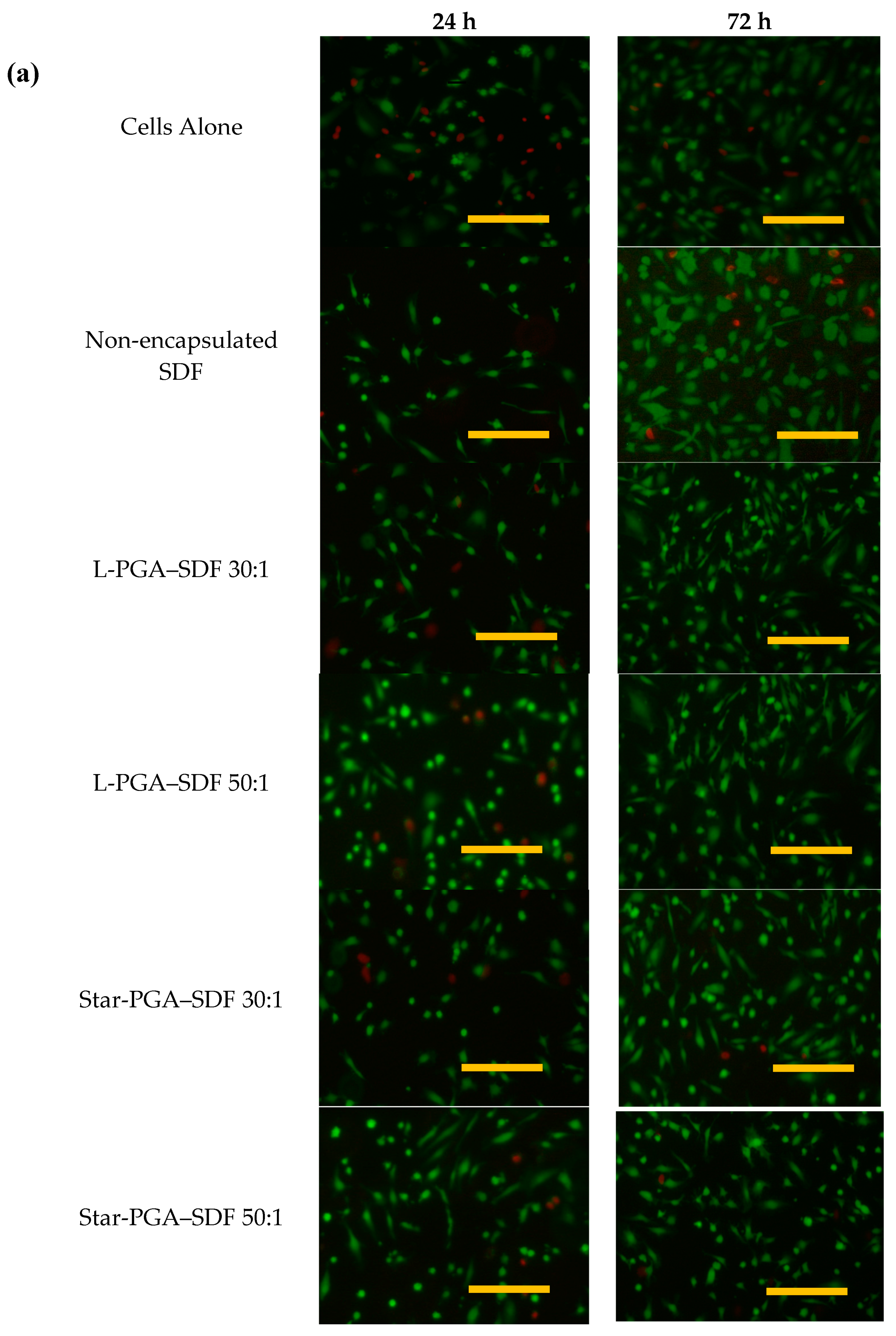
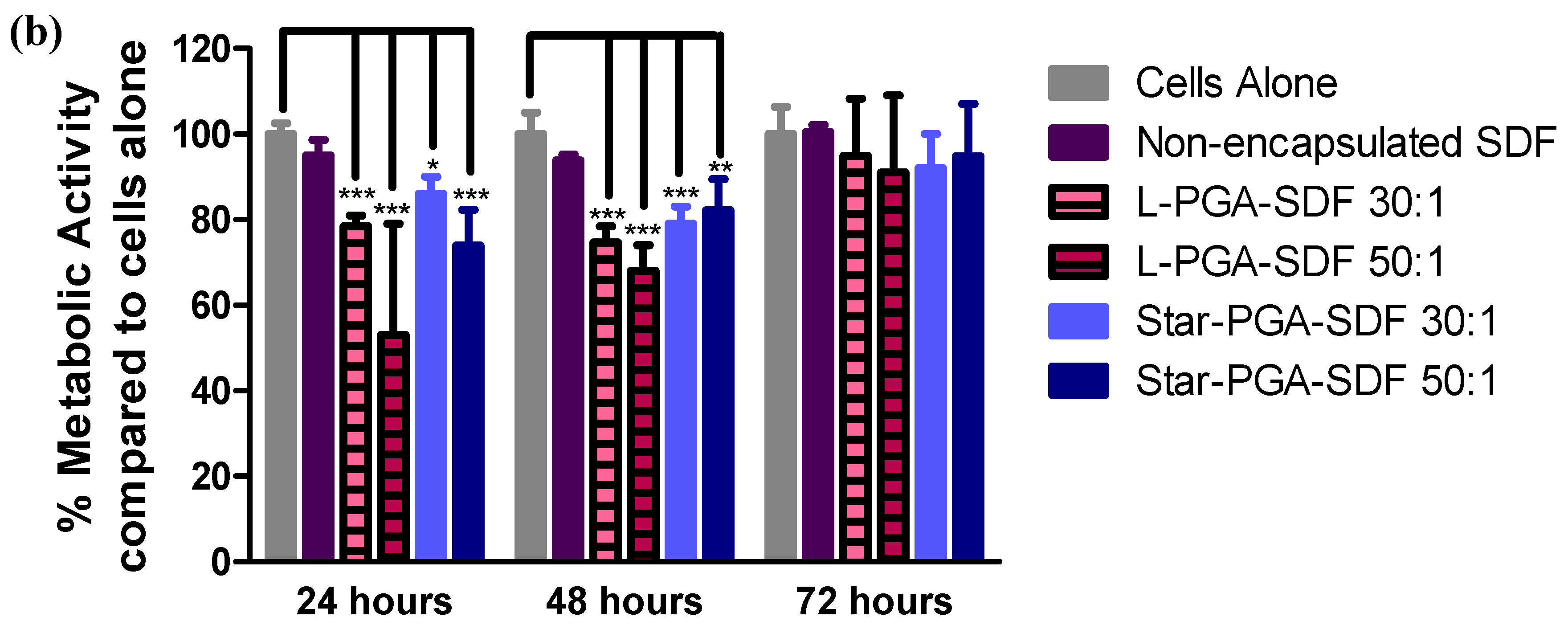
3.5. Biocompatibility of L-PGA–SDF and Star-PGA–SDF Nanoparticles
3.6. Formulation and Characterisation of Star-PGA–SDF–HA–TA
3.6.1. SDF Release from Star-PGA–SDF–HA–TA
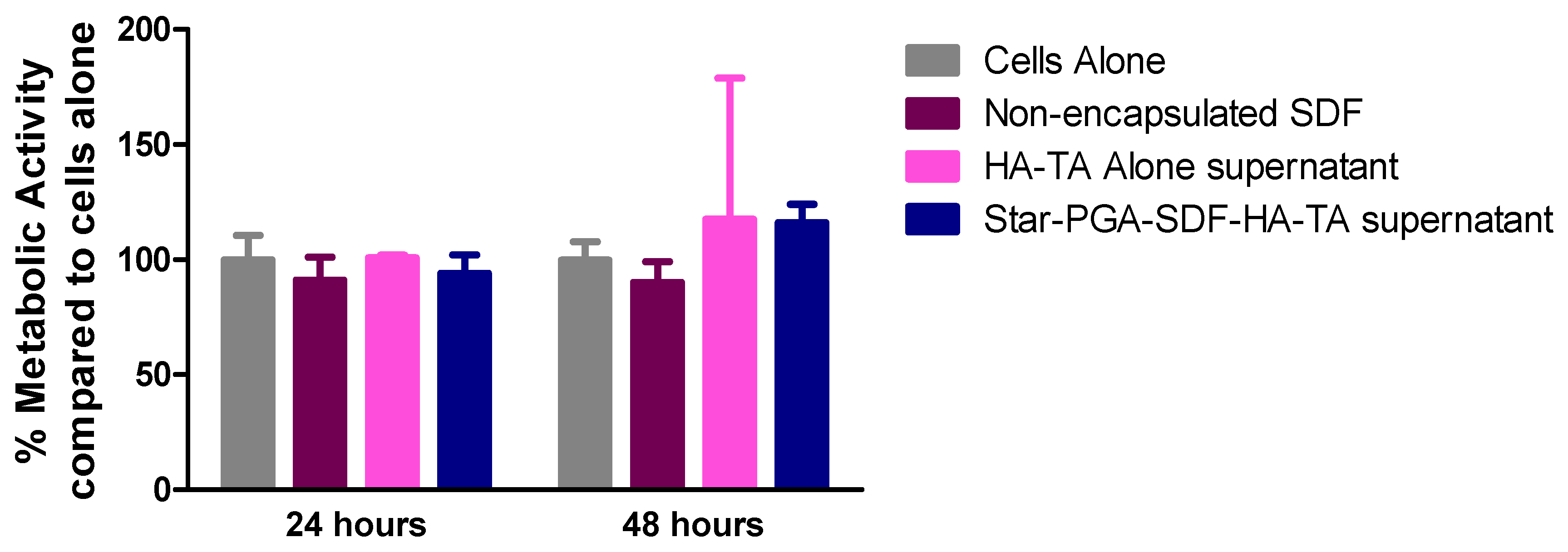
3.6.2. Biocompatibility of Star-PGA–SDF–HA–TA
3.7. Bioactivity of SDF Released from Star-PGA–SDF–HA–TA
3.7.1. Microvessel Formation—Matrigel® Assay
3.7.2. Cell Migration—Scratch Assay
3.7.3. Cell Migration–Transwell® Migration
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holmes, W.D.; Consler, T.G.; Dallas, W.S.; Rocque, W.J.; Willard, D.H. Solution Studies of Recombinant Human Stromal-Cell-Derived Factor-1. Protein Expr. Purif. 2001, 21, 367–377. [Google Scholar] [CrossRef]
- Walentowicz-Sadlecka, M.; Sadlecki, P.; Bodnar, M.; Marszalek, A.; Walentowicz, P.; Sokup, A.; Wilińska-Jankowska, A.; Grabiec, M. Stromal Derived Factor-1 (SDF-1) and Its Receptors CXCR4 and CXCR7 in Endometrial Cancer Patients. PLoS ONE 2014, 9, e84629. [Google Scholar] [CrossRef]
- Ho, T.K.; Shiwen, X.; Abraham, D.; Tsui, J.; Baker, D. Stromal-Cell-Derived Factor-1 (SDF-1)/CXCL12 as Potential Target of Therapeutic Angiogenesis in Critical Leg Ischaemia. Cardiol. Res. Pract. 2012, 2012, 143209. Available online: http://www.ncbi.nlm.nih.gov/pubmed/22462026 (accessed on 9 June 2019). [CrossRef]
- Liu, X.; Duan, B.; Cheng, Z.; Jia, X.; Mao, L.; Fu, H.; Che, Y.; Ou, L.; Liu, L.; Kong, D. SDF-1/CXCR4 axis modulates bone marrow mesenchymal stem cell apoptosis, migration and cytokine secretion. Protein Cell 2011, 2, 845–854. [Google Scholar] [CrossRef]
- Song, M.; Jang, H.; Lee, J.; Kim, J.H.; Kim, S.H.; Sun, K.; Park, Y. Regeneration of chronic myocardial infarction by injectable hydrogels containing stem cell homing factor SDF-1 and angiogenic peptide Ac-SDKP. Biomaterials 2014, 35, 2436–2445. [Google Scholar] [CrossRef]
- Theiss, H.D.; Vallaster, M.; Rischpler, C.; Krieg, L.; Zaruba, M.M.; Brunner, S.; Vanchev, Y.; Fischer, R.; Gröbner, M.; Wollenweber, T.; et al. Dual stem cell therapy after myocardial infarction acts specifically by enhanced homing via the SDF-1/CXCR4 axis. Stem. Cell. Res. 2011, 7, 244–255. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1873506111000626 (accessed on 9 June 2019). [CrossRef]
- Zhang, M.; Mal, N.; Kiedrowski, M.; Chacko, M.; Askari, A.T.; Popović, Z.B.; Koc, O.N.; Penn, M.S. SDF-1 expression by mesenchymal stem cells results in trophic support of cardiac myocytes after myocardial infarction. FASEB J. 2007, 21, 3197–3207. [Google Scholar] [CrossRef]
- Jang, Y.-H.; Kim, J.-H.; Ban, C.; Ahn, K.; Cheong, J.-H.; Kim, H.-H.; Kim, J.-S.; Park, Y.-H.; Kim, J.; Chun, K.-J.; et al. Stromal Cell Derived Factor-1 (SDF-1) Targeting Reperfusion Reduces Myocardial Infarction in Isolated Rat Hearts. Cardiovasc. Ther. 2011, 30, 264–272. [Google Scholar] [CrossRef]
- Zhang, M.; Qiu, L.; Zhang, Y.; Xu, D.; Zheng, J.; Jiang, L. CXCL12 enhances angiogenesis through CXCR7 activation in human umbilical vein endothelial cells. Sci. Rep. 2017, 7, 8289. [Google Scholar] [CrossRef]
- Shah, A.; Mann, D.L. In search of new therapeutic targets and strategies for heart failure: Recent advances in basic science. Lancet 2011, 378, 704–712. [Google Scholar] [CrossRef]
- Cochain, C.; Channon, K.M.; Silvestre, J.-S. Angiogenesis in the Infarcted Myocardium. Antioxid. Redox Signal. 2013, 18, 1100–1113. [Google Scholar] [CrossRef]
- Wang, G.-Y.; Jiang, H.; Yu, Y.; He, F.; Liu, Y.-Y.; Wang, Z.-S.; Xiao, S.-C.; Tang, C.; Xia, Z.-F.; Ji, S.-Z.; et al. A new method of wound treatment: Targeted therapy of skin wounds with reactive oxygen species-responsive nanoparticles containing SDF-1α. Int. J. Nanomed. 2015, 10, 6571–6585. [Google Scholar] [CrossRef]
- Snell, R.J. Therapeutic Angiogenesis in the Management of Critical Limb Ischemia American College of Cardiology [Internet]. American College of Cardiology. 2016. Available online: https://www.acc.org/latest-in-cardiology/articles/2016/05/12/08/27/therapeutic-angiogenesis-in-the-management-of-critical-limb-ischemia (accessed on 9 June 2019).
- Zamproni, L.N.; Mundim, M.; Porcionatto, M.; Rieux, A.D. Injection of SDF-1 loaded nanoparticles following traumatic brain injury stimulates neural stem cell recruitment. Int. J. Pharm. 2017, 519, 323–331. [Google Scholar] [CrossRef]
- Dutta, D.; Fauer, C.; Mulleneux, H.L.; Stabenfeldt, S.E. Tunable controlled release of bioactive SDF-1α via specific protein interactions within fibrin/nanoparticle composites. J. Mater. Chem. B 2015, 3, 7963–7973. [Google Scholar] [CrossRef]
- Mi, L.; Liu, H.; Gao, Y.; Miao, H.; Ruan, J. Injectable nanoparticles/hydrogels composite as sustained release system with stromal cell-derived factor-1α for calvarial bone regeneration. Int. J. Boil. Macromol. 2017, 101, 341–347. [Google Scholar] [CrossRef]
- O’Brien, F.J. Biomaterials & scaffolds for tissue engineering. Mater. Today Internet 2011, 14, 88–95. Available online: https://www.sciencedirect.com/science/article/pii/S136970211170058X (accessed on 9 June 2019).
- Andreas, K.; Sittinger, M.; Ringe, J. Toward in situ tissue engineering: Chemokine-guided stem cell recruitment. Trends Biotechnol. 2014, 32, 483–492. [Google Scholar] [CrossRef]
- Kim, J.J.; Hou, L.; Huang, N.F. Vascularization of three-dimensional engineered tissues for regenerative medicine applications. Acta Biomater. 2016, 41, 17–26. [Google Scholar] [CrossRef]
- Auger, F.A.; Gibot, L.; Lacroix, D. The Pivotal Role of Vascularization in Tissue Engineering. Annu. Rev. Biomed. Eng. 2013, 15, 177–200. [Google Scholar] [CrossRef]
- Laiva, A.L.; Raftery, R.M.; Keogh, M.; O’Brien, F.J. Pro-angiogenic impact of SDF-1α gene-activated collagen-based scaffolds in stem cell driven angiogenesis. Int. J. Pharm. 2018, 544, 372–379. [Google Scholar] [CrossRef]
- Zwingenberger, S.; Langanke, R.; Vater, C.; Lee, G.; Niederlohmann, E.; Sensenschmidt, M.; Jacobi, A.; Bernhardt, R.; Muders, M.; Rammelt, S.; et al. The effect of SDF-1α on low dose BMP-2 mediated bone regeneration by release from heparinized mineralized collagen type I matrix scaffolds in a murine critical size bone defect model. J. Biomed. Mater. Res. Part A 2016, 104, 2126–2134. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, S.; Jalili-Firoozinezhad, S.; Ashtiani, M.K.; Le Carrou, G.; Tajbakhsh, S.; Baharvand, H. Effect of chemical immobilization of SDF-1α into muscle-derived scaffolds on angiogenesis and muscle progenitor recruitment. J. Tissue Eng. Regen. Med. 2017, 12, e438–e450. [Google Scholar] [CrossRef] [PubMed]
- Schantz, J.-T.; Chim, H.; Whiteman, M. Cell Guidance in Tissue Engineering: SDF-1 Mediates Site-Directed Homing of Mesenchymal Stem Cells within Three-Dimensional Polycaprolactone Scaffolds. Tissue Eng. 2007, 13, 2615–2624. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Tao, J.; Zhu, S.; Cai, Y.; Mao, Q.; Yu, N.; Dai, J.; Ouyang, H. Radially oriented collagen scaffold with SDF-1 promotes osteochondral repair by facilitating cell homing. Biomaterials 2015, 39, 114–123. [Google Scholar] [CrossRef]
- Kirkpatrick, B.; Nguyen, L.; Kondrikova, G.; Herberg, S.; Hill, W.D. Stability of human stromal-derived factor-1alpha (CXCL12alpha) after blood sampling. Ann. Clin. Lab. Sci. 2010, 40, 257–260. [Google Scholar]
- Baumann, L.; Prokoph, S.; Gabriel, C.; Freudenberg, U.; Werner, C.; Beck-Sickinger, A.G. A novel, biased-like SDF-1 derivative acts synergistically with starPEG-based heparin hydrogels and improves eEPC migration in vitro. J. Control. Release 2012, 162, 68–75. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, Y.; Li, Q.; Chen, B.; Hou, X.; Xiao, Z.; Dai, J. Controlled Release of Collagen-Binding SDF-1α Improves Cardiac Function after Myocardial Infarction by Recruiting Endogenous Stem Cells. Sci. Rep. 2016, 6, 26683. [Google Scholar] [CrossRef]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef]
- Mansor, M.H.; Najberg, M.; Contini, A.; Alvarez-Lorenzo, C.; Garcion, E.; Jérôme, C.; Boury, F. Development of a non-toxic and non-denaturing formulation process for encapsulation of SDF-1α into PLGA/PEG-PLGA nanoparticles to achieve sustained release. Eur. J. Pharm. Biopharm. 2018, 125, 38–50. [Google Scholar] [CrossRef]
- Wang, B.; Tan, L.; Deng, D.; Lu, T.; Zhou, C.; Li, Z.; Tang, Z.; Wu, Z.; Tang, H. Novel stable cytokine delivery system & nbsp; in physiological pH solution: Chitosan oligosaccharide/heparin nanoparticles. Int. J. Nanomed. Internet 2015, 10, 3417. Available online: http://www.ncbi.nlm.nih.gov/pubmed/26056441 (accessed on 9 June 2019).
- Bader, A.R.; Li, T.; Wang, W.; Kohane, D.S.; Loscalzo, J.; Zhang, Y.-Y. Preparation and Characterization of SDF-1α-Chitosan-Dextran Sulfate Nanoparticles. J. Vis. Exp. 2015, 95, e52323. Available online: http://www.jove.com/video/52323/preparation-characterization-sdf-1-chitosan-dextran-sulfate (accessed on 9 June 2019).
- Zhu, G.; Mallery, S.R.; Schwendeman, S.P. Stabilization of proteins encapsulated in injectable poly (lactide- co-glycolide). Nat. Biotechnol. 2000, 18, 52–57. [Google Scholar] [CrossRef]
- Yan, Y.; Li, J.; Zheng, J.; Pan, Y.; Wang, J.; He, X.; Zhang, L.; Liu, D. Poly(l-lysine)-based star-block copolymers as pH-responsive nanocarriers for anionic drugs. Colloids Surf. B Biointerfaces 2012, 95, 137–143. [Google Scholar] [CrossRef]
- Byrne, M.; Victory, D.; Hibbitts, A.; Lanigan, M.; Heise, A.; Cryan, S.-A. Molecular weight and architectural dependence of well-defined star-shaped poly(lysine) as a gene delivery vector. Biomater. Sci. 2013, 1, 1223. [Google Scholar] [CrossRef]
- Walsh, D.P.; Murphy, R.D.; Panarella, A.; Raftery, R.M.; Cavanagh, B.; Simpson, J.C.; O’Brien, F.J.; Heise, A.; Cryan, S.-A. Bioinspired Star-Shaped Poly(l-lysine) Polypeptides: Efficient Polymeric Nanocarriers for the Delivery of DNA to Mesenchymal Stem Cells. Mol. Pharm. 2018, 15, 1878–1891. [Google Scholar] [CrossRef]
- Yan, Y.; Wei, D.; Li, J.; Zheng, J.; Shi, G.; Luo, W.; Pan, Y.; Wang, J.; Zhang, L.; He, X.; et al. A poly(l-lysine)-based hydrophilic star block co-polymer as a protein nanocarrier with facile encapsulation and pH-responsive release. Acta Biomater. 2012, 8, 2113–2120. [Google Scholar] [CrossRef]
- Byrne, M.; Thornton, P.; Cryan, S.-A.; Heise, A. Star polypeptides by NCA polymerisation from dendritic initiators: Synthesis and enzyme controlled payload release. Polym. Chem. 2012, 3, 2825. [Google Scholar] [CrossRef]
- Dalonneau, F.; Liu, X.Q.; Sadir, R.; Almodóvar, J.; Mertani, H.C.; Brückert, F.; Albiges-Rizo, C.; Weidenhaupt, M.; Lortat-Jacob, H.; Picart, C. The effect of delivering the chemokine SDF-1α in a matrix-bound manner on myogenesis. Biomaterials 2014, 35, 4525–4535. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef]
- Vermonden, T.; Censi, R.; Hennink, W.E. Hydrogels for Protein Delivery. Chem. Rev. 2012, 112, 2853–2888. [Google Scholar] [CrossRef]
- Holland, T.A.; Tabata, Y.; Mikos, A.G. Dual growth factor delivery from degradable oligo(poly(ethylene glycol) fumarate) hydrogel scaffolds for cartilage tissue engineering. J. Control. Release 2005, 101, 111–125. [Google Scholar] [CrossRef]
- Lee, F.; Chung, J.E.; Kurisawa, M. An injectable hyaluronic acid–tyramine hydrogel system for protein delivery. J. Control. Release 2009, 134, 186–193. [Google Scholar] [CrossRef]
- Kim, K.; Park, S.; Yang, J.-A.; Jeon, J.-H.; Bhang, S.; Kim, B.-S.; Hahn, S.K. Injectable hyaluronic acid–tyramine hydrogels for the treatment of rheumatoid arthritis. Acta Biomater. 2011, 7, 666–674. [Google Scholar] [CrossRef]
- Lee, F.; Chung, J.E.; Kurisawa, M. An injectable enzymatically crosslinked hyaluronic acid–tyramine hydrogel system with independent tuning of mechanical strength and gelation rate. Soft Matter 2008, 4, 880. [Google Scholar] [CrossRef]
- Dolan, E.B.; Kovarova, L.; O’Neill, H.; Pravda, M.; Sulakova, R.; Scigalkova, I.; Velebny, V.; Daro, D.; Braun, N.; Cooney, G.M.; et al. Advanced Material Catheter (AMCath), a minimally invasive endocardial catheter for the delivery of fast-gelling covalently cross-linked hyaluronic acid hydrogels. J. Biomater. Appl. 2018, 33, 681–692. [Google Scholar] [CrossRef]
- O’Dwyer, J.; Murphy, R.; Dolan, E.B.; Kovarova, L.; Pravda, M.; Velebny, V.; Heise, A.; Duffy, G.P.; Cryan, S.-A. Development of a nanomedicine-loaded hydrogel for sustained delivery of an angiogenic growth factor to the ischaemic myocardium. Drug Deliv. Transl. Res. 2019, 10, 440–454. [Google Scholar] [CrossRef]
- Shen, S.; Wu, Y.; Liu, Y.; Wu, D. High drug-loading nanomedicines: Progress, current status, and prospects. Int. J. Nanomed. 2017, 12, 4085–4109. [Google Scholar] [CrossRef]
- Petit, I.; Jin, D.; Rafii, S. The SDF-1–CXCR4 signaling pathway: A molecular hub modulating neo-angiogenesis. Trends Immunol. 2007, 28, 299–307. [Google Scholar] [CrossRef]
- Huang, C.; Gu, H.; Zhang, W.; Manukyan, M.C.; Shou, W.; Wang, M. SDF-1/CXCR4 mediates acute protection of cardiac function through myocardial STAT3 signaling following global ischemia/reperfusion injury. Am. J. Physiol. Heart. Circ. Physiol. 2011, 301, H1496–H1505. Available online: http://www.ncbi.nlm.nih.gov/pubmed/21821779 (accessed on 24 August 2019). [CrossRef]
- European Medicines Agency. Guideline on Process Validation for Finished Products Information and Data to be Provided in Regulatory Submissions [Internet]. 2016. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-process-validation-finished-products-information-data-be-provided-regulatory-submissions_en.pdf (accessed on 16 May 2020).
- Chung, E.S.; Miller, L.; Patel, A.N.; Anderson, R.D.; Mendelsohn, F.O.; Traverse, J.; Silver, K.H.; Shin, J.; Ewald, G.; Farr, M.J.; et al. Changes in ventricular remodelling and clinical status during the year following a single administration of stromal cell-derived factor-1 non-viral gene therapy in chronic ischaemic heart failure patients: The STOP-HF randomized Phase II trial. Eur. Hear. J. 2015, 36, 2228–2238. [Google Scholar] [CrossRef]
- Duro-Castano, A.; England, R.M.; Razola, D.; Sanz, E.R.; Oteo, M.; Morcillo, M.A.; Vicent, M.J. Well-Defined Star-Shaped Polyglutamates with Improved Pharmacokinetic Profiles As Excellent Candidates for Biomedical Applications. Mol. Pharm. 2015, 12, 3639–3649. [Google Scholar] [CrossRef]
- Anderson, W.; Kozak, D.; Coleman, V.; Jämting, Å.K.; Trau, M. A comparative study of submicron particle sizing platforms: Accuracy, precision and resolution analysis of polydisperse particle size distributions. J. Colloid Interface Sci. 2013, 405, 322–330. [Google Scholar] [CrossRef]
- Filipe, V.; Hawe, A.; Jiskoot, W. Critical Evaluation of Nanoparticle Tracking Analysis (NTA) by NanoSight for the Measurement of Nanoparticles and Protein Aggregates. Pharm. Res. 2010, 27, 796–810. [Google Scholar] [CrossRef]
- Sukhanova, A.; Bozrova, S.; Sokolov, P.; Berestovoy, M.; Karaulov, A.V.; Nabiev, I. Dependence of Nanoparticle Toxicity on Their Physical and Chemical Properties. Nanoscale Res. Lett. 2018, 13, 44. [Google Scholar] [CrossRef]
- Yin, T.; Bader, A.R.; Hou, T.K.; Maron, B.A.; Kao, D.D.; Qian, R.; Kohane, D.S.; Handy, D.; Loscalzo, J.; Zhang, Y.-Y. SDF-1α in Glycan Nanoparticles Exhibits Full Activity and Reduces Pulmonary Hypertension in Rats. Biomacromolecules 2013, 14, 4009–4020. [Google Scholar] [CrossRef]
- Cross, D.P.; Wang, C. Stromal-derived factor-1 alpha-loaded PLGA microspheres for stem cell recruitment. Pharm. Res. 2011, 28, 2477–2489. [Google Scholar] [CrossRef]
- He, X.; Ma, J.; Jabbari, E. Migration of marrow stromal cells in response to sustained release of stromal-derived factor-1alpha from poly(lactide ethylene oxide fumarate) hydrogels. Int. J. Pharm. 2010, 390, 107–116. [Google Scholar] [CrossRef]
- Rabbany, S.Y.; Pastore, J.; Yamamoto, M.; Miller, T.; Rafii, S.; Aras, R.; Penn, M. Continuous Delivery of Stromal Cell-Derived Factor-1 from Alginate Scaffolds Accelerates Wound Healing. Cell Transplant. 2010, 19, 399–408. [Google Scholar] [CrossRef]
- Zhu, Y.; Hoshi, R.; Chen, S.; Yi, J.; Duan, C.; Galiano, R.D.; Zhang, H.F.; Ameer, G.A. Sustained release of stromal cell derived factor-1 from an antioxidant thermoresponsive hydrogel enhances dermal wound healing in diabetes. J. Control. Release 2016, 238, 114–122. [Google Scholar] [CrossRef]
- Fu, K.; Klibanov, A.; Langer, R. Protein stability in controlled-release systems. Nat. Biotechnol. 2000, 18, 24–25. [Google Scholar] [CrossRef]
- Unoki, N.; Murakami, T.; Nishijima, K.; Ogino, K.; van Rooijen, N.; Yoshimura, N. SDF-1/CXCR4 Contributes to the Activation of Tip Cells and Microglia in Retinal Angiogenesis. Investig. Opthalmology Vis. Sci. 2010, 51, 3362. Available online: http://www.ncbi.nlm.nih.gov/pubmed/20181837 (accessed on 9 June 2019). [CrossRef] [PubMed]
- Latifi-Pupovci, H.; Kuçi, Z.; Wehner, S.; Bönig, H.; Lieberz, R.; Klingebiel, T.; Bader, P.; Kuçi, S. In vitro migration and proliferation (“wound healing”) potential of mesenchymal stromal cells generated from human CD271(+) bone marrow mononuclear cells. J. Transl. Med. 2015, 13, 315. Available online: http://www.ncbi.nlm.nih.gov/pubmed/26407865 (accessed on 27 July 2019). [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | L-PGA:SDF Ratio | Z-Average Size (nm) | PDI | Zeta Potential (mV) |
| 30:1 | 275.0 | 0.6 | −6.8 | |
| 40:1 | 305.7 | 0.5 | −3.8 | |
| 50:1 | 255.2 | 0.6 | −3.5 | |
| (b) | Star-PGA:SDF Ratio | Z-Average Size (nm) | PDI | Zeta Potential (mV) |
| 30:1 | 279.4 | 0.6 | −3.1 | |
| 40:1 | 261.8 | 0.6 | −4.3 | |
| 50:1 | 275.8 | 0.3 | −4.0 |
| PGA:SDF Ratio | Complexation Efficiency | Protein Loading | ||
|---|---|---|---|---|
| L-PGA–SDF | Star-PGA–SDF | L-PGA–SDF | Star-PGA–SDF | |
| 30:1 | 99.97 | 100 | 1 | 0.63 |
| 40:1 | 99.96 | 99.98 | 0.76 | 0.47 |
| 50:1 | 99.96 | 99.98 | 0.61 | 0.38 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Dwyer, J.; Cullen, M.; Fattah, S.; Murphy, R.; Stefanovic, S.; Kovarova, L.; Pravda, M.; Velebny, V.; Heise, A.; Duffy, G.P.; et al. Development of a Sustained Release Nano-In-Gel Delivery System for the Chemotactic and Angiogenic Growth Factor Stromal-Derived Factor 1α. Pharmaceutics 2020, 12, 513. https://doi.org/10.3390/pharmaceutics12060513
O’Dwyer J, Cullen M, Fattah S, Murphy R, Stefanovic S, Kovarova L, Pravda M, Velebny V, Heise A, Duffy GP, et al. Development of a Sustained Release Nano-In-Gel Delivery System for the Chemotactic and Angiogenic Growth Factor Stromal-Derived Factor 1α. Pharmaceutics. 2020; 12(6):513. https://doi.org/10.3390/pharmaceutics12060513
Chicago/Turabian StyleO’Dwyer, Joanne, Megan Cullen, Sarinj Fattah, Robert Murphy, Smiljana Stefanovic, Lenka Kovarova, Martin Pravda, Vladimir Velebny, Andreas Heise, Garry P. Duffy, and et al. 2020. "Development of a Sustained Release Nano-In-Gel Delivery System for the Chemotactic and Angiogenic Growth Factor Stromal-Derived Factor 1α" Pharmaceutics 12, no. 6: 513. https://doi.org/10.3390/pharmaceutics12060513
APA StyleO’Dwyer, J., Cullen, M., Fattah, S., Murphy, R., Stefanovic, S., Kovarova, L., Pravda, M., Velebny, V., Heise, A., Duffy, G. P., & Cryan, S. A. (2020). Development of a Sustained Release Nano-In-Gel Delivery System for the Chemotactic and Angiogenic Growth Factor Stromal-Derived Factor 1α. Pharmaceutics, 12(6), 513. https://doi.org/10.3390/pharmaceutics12060513