Pharmacokinetic Interaction between Metformin and Verapamil in Rats: Inhibition of the OCT2-Mediated Renal Excretion of Metformin by Verapamil



Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
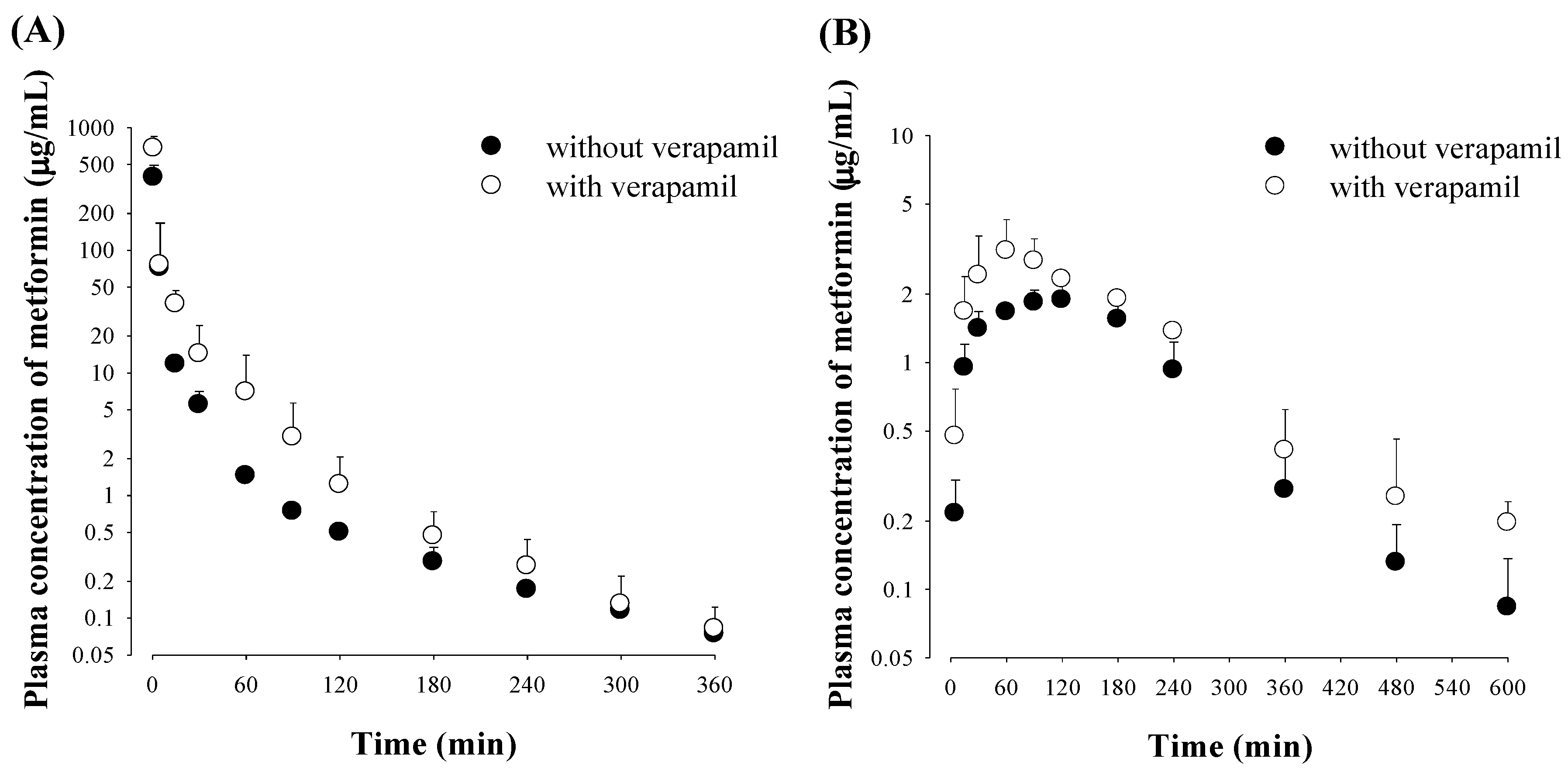
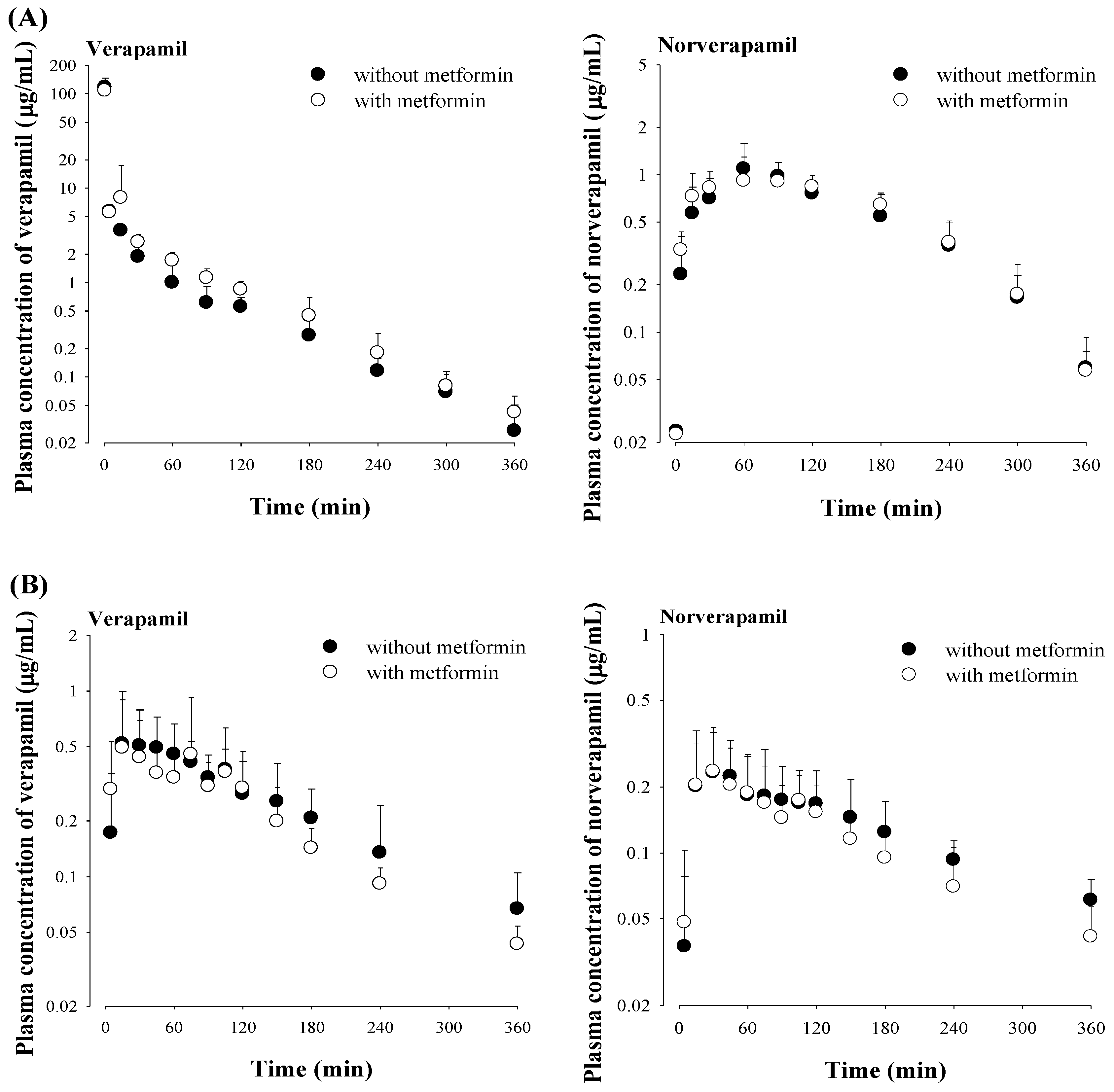
2.3. Pharmacokinetic Studies of Metformin, Verapamil and Both Drugs in Rats
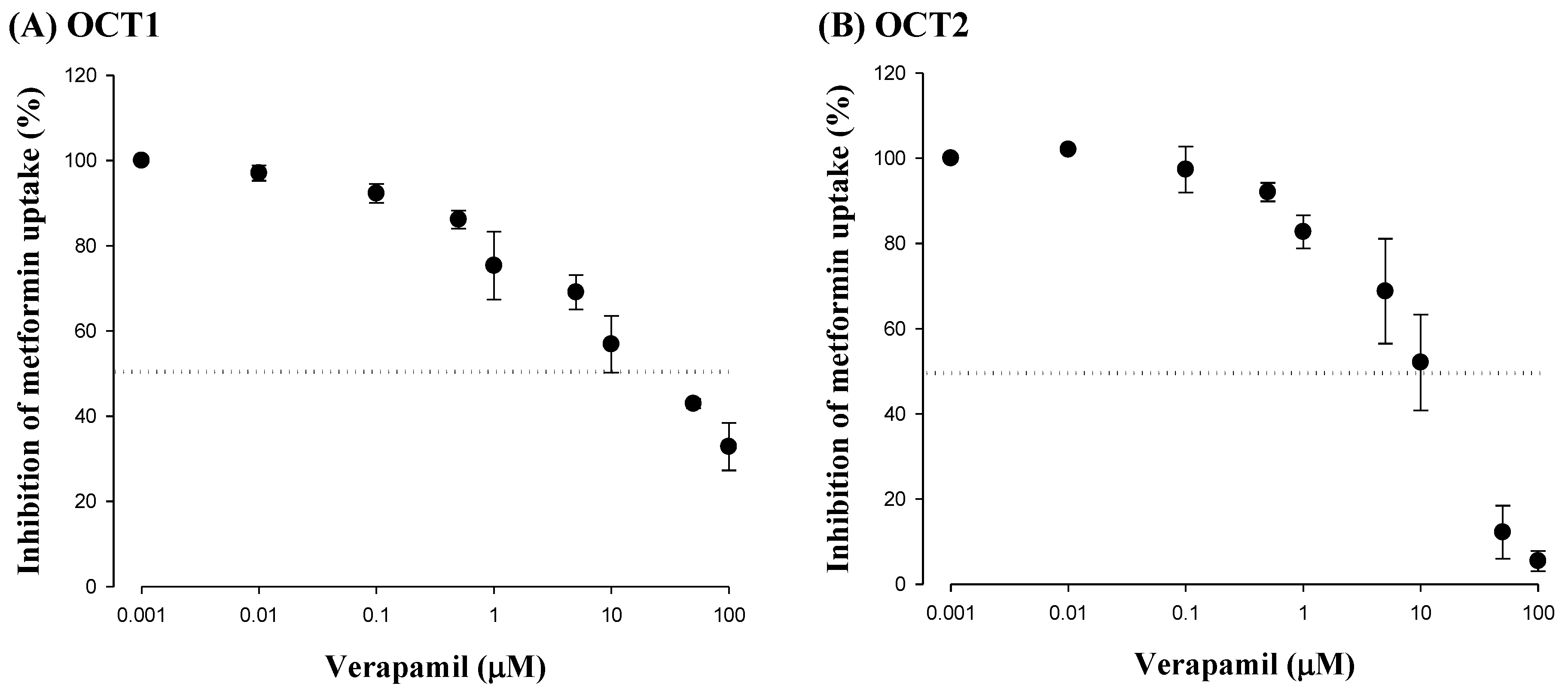
2.4. Effect of Verapamil on Metformin Uptake in HEK-293 Cells Overexpressing OCT1 or OCT2
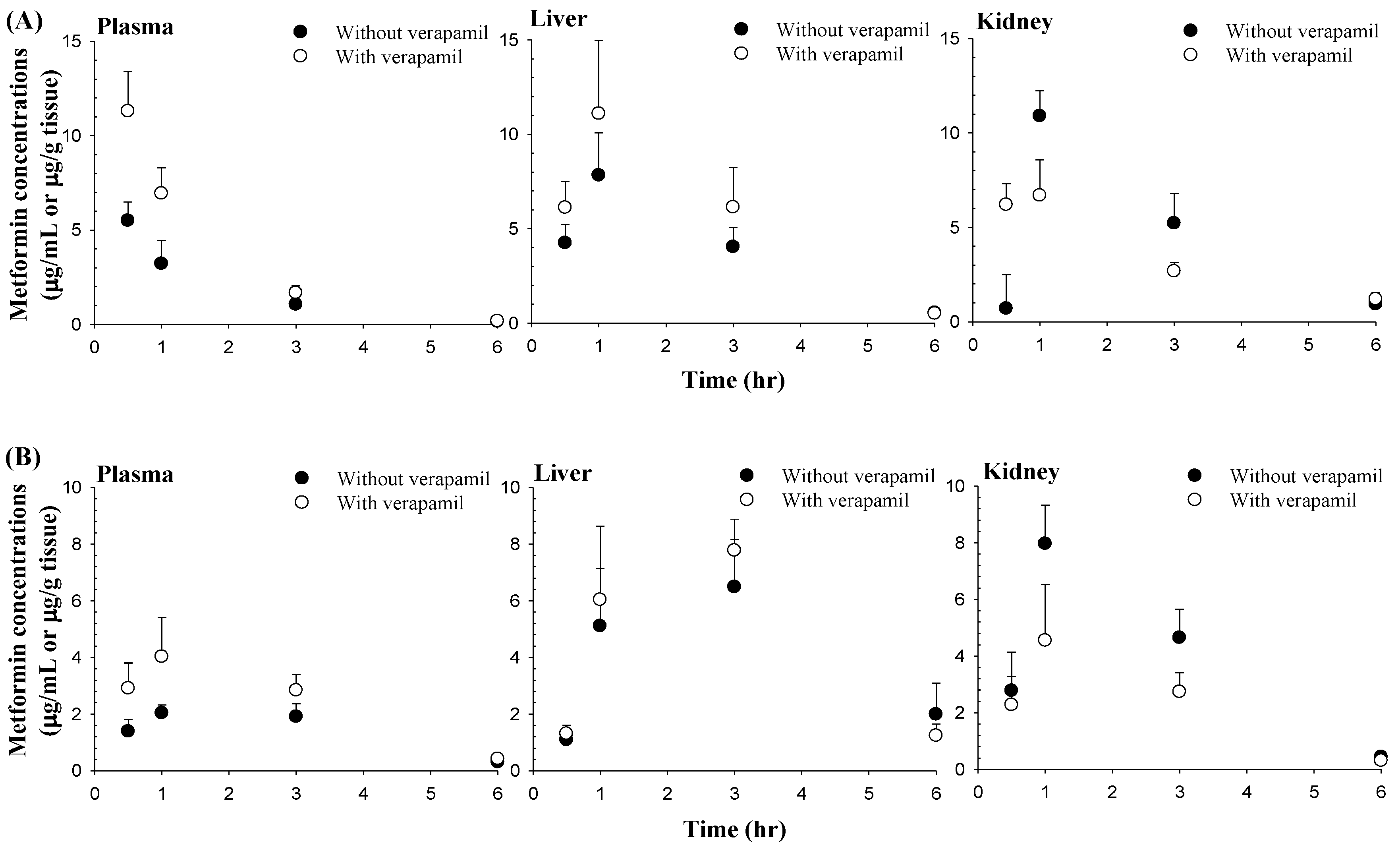
2.5. Effect of Verapamil on Metformin Concentration in the Liver and Kidneys
2.6. Effect of Metformin on Verapamil Metabolism in Rat Hepatic and Intestinal Microsomes
2.7. Rat Plasma Protein Binding of Metformin and Verapamil Using Equilibrium Dialysis
2.8. Analytical Methods for Metformin, Verapamil and Norverapamil
2.9. Pharmacokinetic Analysis
2.10. Statistical Analysis
3. Results
3.1. Effect of Verapamil on Metformin Pharmacokinetics
3.2. Effect of Verapamil on OCT1- or OCT2-Mediated Metformin Uptake in HEK-293 Cells Overexpressing OCT1 or OCT2
3.3. Effect of Verapamil on Metformin Concentrations in the Liver and Kidneys
3.4. Effect of Metformin on Verapamil Pharmacokinetics
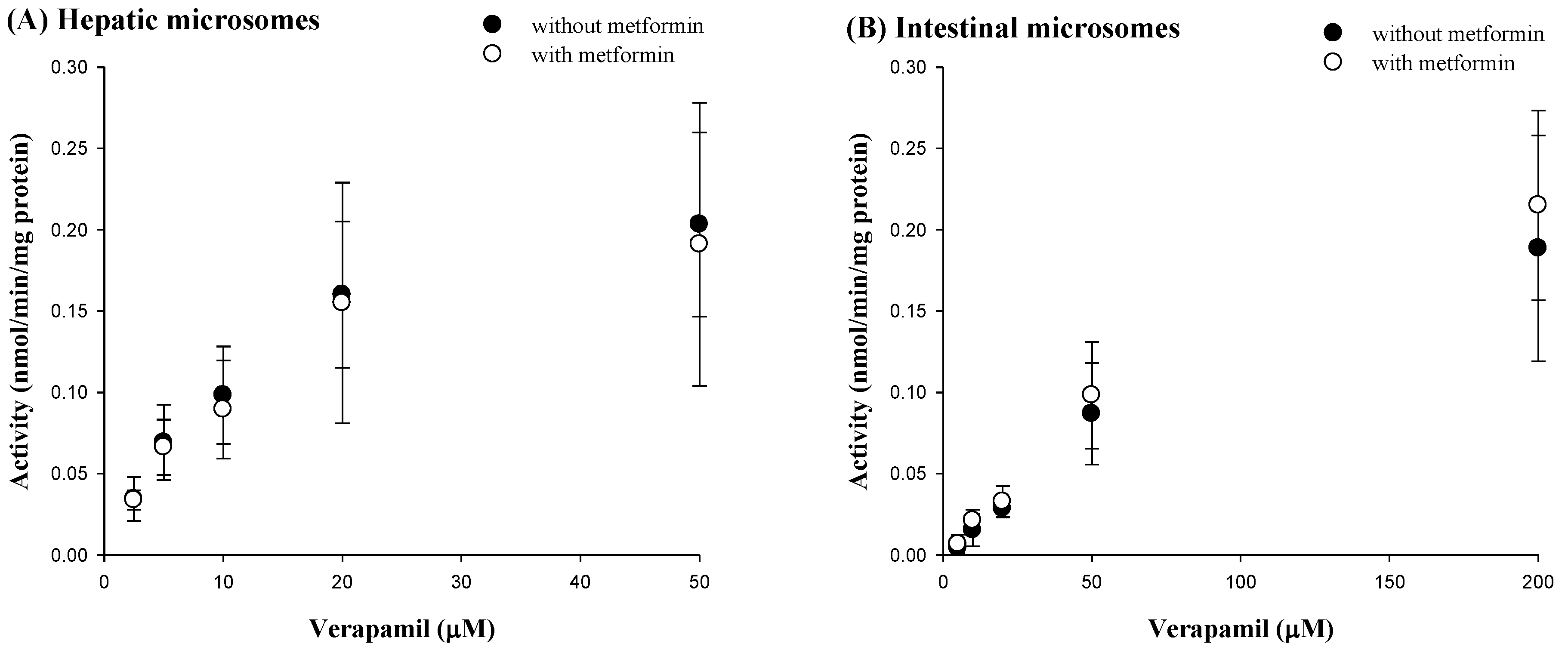
3.5. Effect of Metformin on Verapamil Metabolism in Rat Hepatic and Intestinal Microsomes
3.6. Rat Plasma Protein Binding of Verapamil and Metformin Using Equilibrium Dialysis
4. Discussion
0.0153 = ‘Funabs’ + (0.00831 × 0.309) with verapamil
0.00200 = ‘Funabs’ + (0.00117 × 0.182) with metformin
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Boer, I.H.; Bangalore, S.; Benetos, A.; Davis, A.M.; Michos, E.D.; Muntner, P.; Rossing, P.; Zoungas, S.; Bakris, G. Diabetes and hypertension: A position statement by the American Diabetes Association. Diabetes Care 2017, 40, 1273–1284. [Google Scholar] [CrossRef]
- Ferrannini, E.; Cushman, W.C. Diabetes and hypertension: The bad companions. Lancet 2012, 380, 601–610. [Google Scholar] [CrossRef]
- Junior, V.C.; Fuchs, F.D.; Schaan, B.D.; Moreira, L.B.; Fuchs, S.C.; Gus, M. Effect of metformin on blood pressure in patients with hypertension: A randomized clinical trial. Endocrine 2019, 63, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Beckmman, J.A.; Creager, M.A.; Libby, P. Diabetes and antherosclerosis: Epidemiology, pathophysiology, and management. JAMA 2002, 287, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Hayward, R.A.; Reaven, P.D.; Wiitala, W.L.; Bahn, G.D.; Reda, D.J.; Ge, L.; McCarren, M.; Duckworth, W.C.; Emanuele, N.V.; VADT Investigators. Follow-up of glycemic control and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2015, 372, 2197–2206. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M.; Lithell, H.; Landsberg, L. Hypertension and associated metabolic abnormalities-the role of insulin resistance and the sympathoadrenale. N. Engl. J. Med. 1996, 334, 374–381. [Google Scholar] [CrossRef]
- Sun, D.; Zhou, T.; Heianza, Y.; Li, X.; Fan, M.; Fonseca, V.A.; Qi, L. Type 2 Diabetes and Hypertension: A Study on Bidirectional Causality. Circ. Res. 2019, 124, 930–937. [Google Scholar] [CrossRef]
- He, J.; Whelton, P.K. Epidemiology and prevention of hypertension. Med. Clin. N. Am. 1997, 81, 1077–1097. [Google Scholar] [CrossRef]
- Sharma, S.K.; Ruggeneti, P.; Remuzzi, G. Managing hypertension in diabetic patients-focus on trandolapril/verapamil combination. Vasc. Health Risk Manag. 2007, 3, 453–465. [Google Scholar]
- Dunn, D.J.; Peters, D.H. Metformin. A review of is pharmacological properties and therapeutic use in non-insulin-dependent diabetes mellitus. Drugs 1995, 49, 721–749. [Google Scholar] [CrossRef]
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin pathway: Pharmacokinetics and pharmacodynamics. Pharm. Genom. 2012, 22, 820–827. [Google Scholar] [CrossRef]
- Scheen, A.J. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 1996, 30, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Hermann, L.S. Clinical Pharmacology of Biguanides (Oral antidiabetes). In Handbook of Experimental Pharmacology; Kuhlmann, J., Puls, W., Eds.; Springer: New York, NY, USA, 1996; pp. 373–407. [Google Scholar]
- Snorgaard, O.; Kober, L.; Carlsen, J. The effect of metformin on blood pressure and metabolism in nondiabetic hypertensive patients. J. Intern. Med. 1997, 242, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Koepsell, H.; Winter, S.; Burk, O.; Klein, K.; Kerb, R.; Zanger, U.M.; Keppler, D.; Schwab, M.; Schaeffeler, E. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology 2009, 50, 1227–1240. [Google Scholar] [CrossRef]
- Nies, A.T.; Hofmann, U.; Resch, C.; Schaeffeler, E.; Ruis, M.; Schwab, M. Proton pump inhibitors inhibit metformin uptake by organic cation transporters (OCTs). PLoS ONE 2011, 6, e22163. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Kim, S.G.; Lee, M.G. Dose-independent pharmacokinetics of metformin in rats: Hepatic and gastrointestinal first-pass effects. J. Pharm. Sci. 2006, 95, 2543–2552. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Masuda, S.; Tanihara, Y.; Ueo, H.; Okuda, M.; Katsura, T.; Inui, K.I. Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab. Pharmacokinet. 2005, 20, 379–386. [Google Scholar] [CrossRef]
- Yang, S.; Dai, Y.; Liu, Z.; Wang, C.; Meng, Q.; Huo, X.; Sun, H.; Ma, X.; Peng, J.; Liu, K. Involvement of organic cation transporter 2 in the metformin-associated increased lactate levels caused by contrast-induced nephropathy. Biomed. Pharmacother. 2018, 106, 1760–1766. [Google Scholar] [CrossRef]
- Abbasi, M.M.; Valizadeh, H.; Hamishehkar, H.; Zakeri-Milani, P. Inhibition of P-glycoprotein expression and function by anti-diabetic drugs gliclazide, metformin, and pioglitazone in vitro and in situ. Res. Pharm. Sci. 2016, 11, 177–186. [Google Scholar]
- Krausova, L.; Stejskalova, L.; Wang, H.; Vrzal, R.; Dvorak, Z.; Maniƒ, S.; Pavek, P. Metformin suppresses pregnane X receptor (PXR)-regulated transactivation of CYP3A4 gene. Biochem. Pharmacol. 2011, 82, 1771–1780. [Google Scholar] [CrossRef]
- Khodneva, Y.; Shalev, A.; Frank, S.J.; Carson, A.P.; Safford, M.M. Calcium channel blocker use is associated with lower fasting serum glucose among adults with diabetes from the REGARDS study. Diabetes Res. Clin. Pract. 2016, 115, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Kuo, S.C.; Chang, Y.Y.; Chen, T.T.; Wang, K.W.K. Verapamil use is associated with reduction of newly diagnosed diabetes mellitus. J. Clin. Endocrinol. Metab. 2017, 102, 2604–2610. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.E.H.; Rojdmark, S. Improvement of glucose tolerance by verapamil in patients with non-insulin dependent diabetes mellitus. Acta Med. Scandina. 2009, 210, 27–33. [Google Scholar] [CrossRef]
- Cooper-Dehoff, R.; Cohen, J.D.; Bakris, G.L.; Messerli, F.H.; Erdine, S.; Hewkin, A.C.; Kupfer, S.; Pepine, C.J. Predictors of development of diabetes mellitus in patients with coronary artery disease taking antihypertensive medications (findings from the International Verapamil SR-Trandolapril Study [INVEST]). Am. J. Cardiol. 2006, 98, 890–894. [Google Scholar] [CrossRef] [PubMed]
- Cooper-DeHoff, R.M.; Aranda, J.M., Jr.; Gaxiola, E.; Cangiano, J.L.; Garcia-Barreto, D.; Conti, C.R.; Hewkin, A.; Pepine, C.J. Blood pressure control and cardiovascular outcomes in high-risk Hispanic patients–findings from the International Verapamil SR/Trandolapril Study (INVEST). Am. Heart J. 2006, 151, 1072–1079. [Google Scholar] [CrossRef]
- Hamann, S.R.; Blouin, R.A.; McAllister, R.G. Clinical pharmacokinetics of verapamil. Clin. Pharmacokinet. 1984, 9, 26–41. [Google Scholar] [CrossRef]
- Hanada, K.; Ikemi, Y.; Kkita, K.; Mihara, K.; Ogata, H. Stereoselective first-pass metabolism of verapamil in the small intestine and liver in rats. Drug Metab. Dispos. 2008, 36, 2037–2042. [Google Scholar] [CrossRef]
- Hong, S.P.; Chang, K.S.; Koh, Y.Y.; Choi, D.H.; Choi, J.S. Effects of lovastatin on the pharmacokinetics of verapamil and its active metabolite, norverapamil in rats: Possible role of p-glycoprotein inhibition by lovastatin. Arch. Pharm. Res. 2009, 32, 1447–1452. [Google Scholar] [CrossRef]
- Uchida, Y.; Ohtsuki, S.; Terasaki, T. Pharmacoproteomics-based reconstruction of in vivo P-glycoprotein function at blood-brain barrier and brain distribution of substrate verapamil in pentylenetetrazole-kindled epilepsy, spontaneous epilepsy, and phenytoin treatment models. Drug Metab. Dispos. 2014, 42, 1719–1726. [Google Scholar] [CrossRef]
- Zolk, O.; Solbach, T.F.; Konig, J.; Fromm, M.F. Functional characterization of the human organic cation transporter 2 Variant p.270Ala>Ser. Drug Metab. Dispos. 2009, 37, 1312–1318. [Google Scholar] [CrossRef]
- Cho, S.K.; Kim, C.O.; Park, E.S.; Chung, J.-Y. Verapamil decreases the glucose-lowering effect of metformin in healthy volunteers. Br. J. Clin. Pharmacol. 2014, 78, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Han, S.Y.; Chae, H.S.; You, B.H.; Chin, Y.W.; Kim, H.; Choi, H.S.; Choi, Y.H. Lonicera japonica extract increases metformin distribution in the liver without change of systemic exposed metformin in rats. J. Ethnopharmacol. 2019, 238, 111892. [Google Scholar] [CrossRef] [PubMed]
- You, B.H.; Chin, Y.W.; Kim, H.; Choi, H.S.; Choi, Y.H. Houttuynia cordata extract increased exposure and liver concentrations of metformin through OCTs and MATEs in rats. Phytother. Res. 2018, 32, 1004–1013. [Google Scholar] [CrossRef]
- Liang, X.; Giacomini, K.M. Transporters involved in metformin pharmacokinetics and treatment response. J. Pharm. Sci. 2017, 106, 2245–2250. [Google Scholar] [CrossRef]
- Tornio, A.; Filppula, A.M.; Niemi, M.; Backman, J.T. Clinical studies on drug-drug interactions involving metabolism and transport: Methodology, pitfalls, and interpretation. Clin. Pharmacol. Ther. 2019, 105, 1345–1361. [Google Scholar] [CrossRef]
- Iwersen, S.; Schmoldt, A. A specific hydroxysteroid UGT is responsible for the conjugation of aliphatic alcohols in rats: An estimation of the importance of glucuronidation versus oxidation. Alcohol 1998, 15, 185–192. [Google Scholar] [CrossRef]
- Peng, J.Z.; Remmel, R.P.; Sawchuk, R.J. Inhibition of murine cytochrome P4501A by tacrine: In vitro studies. Drug Metab. Dispos. 2004, 32, 805–812. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Duggleby, R.G. Analysis of enzyme progress curves by nonlinear regression. Methods Enzymol. 1995, 249, 61–90. [Google Scholar]
- Gibaldi, M.; Perrier, D. Pharmacokinetics, 2nd ed.; Marcel Dekker: New York, NY, USA, 1982. [Google Scholar]
- Manitpisitkul, P.; Chiou, W.L. Intravenous verapamil kinetics in rats: Marked arteriovenous concentration difference and comparison with humans. Biopharm. Drug Dispos. 1993, 14, 555–566. [Google Scholar] [CrossRef]
- US Food and Drug Administration, Center for Drug Evaluation and Research. Clinical Drug Interaction Studies-Study Design, Data Analysis, and Clinical Implications Guideline for Industry. 2017. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm292362.pdf (accessed on 31 October 2017).
- Davies, B.; Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Burt, H.J.; Neuhoff, S.; Almond, L.; Gaohua, L.; Harwood, M.D.; Jamei, M.; Rostami-Hodjegan, A.; Tucker, G.T.; Rowland-Yeo, K. Metformin and cimetidine: Physiologically based pharmacokinetic modelling to investigate transporter mediated drug-drug interactions. Eur. J. Pharm. Sci. 2016, 88, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.W.; Bedwell, D.W.; Zamek-Gliszczynski, M.J. Ablation of both organic cation transporter (Oct)1 and Oct2 alters metformin pharmacokinetics but has no effect on tissue drug exposure and pharmacodynamics. Drug Metab. Dispos. 2012, 40, 1170–1177. [Google Scholar] [CrossRef]
- Zamek-Gliszczynski, M.J.; Bao, J.Q.; Day, J.S.; Higgins, J.W. Metformin sinusoidal efflux from the liver is consistent with negligible biliary excretion and absence of enterohepatic cycling. Drug Metab. Dispos. 2013, 41, 1967–1971. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Chiou, W.L. Evaluation of potential causes for the incomplete bioavailability of furosemide: Gastric first-pass metabolism. J. Pharmacokinet. Biopharm. 1983, 11, 623–640. [Google Scholar] [CrossRef] [PubMed]
- Eichelbaum, M.; Mikus, G.; Vog Dunn, D.J.; Peters Elgesang, B. Pharmacokinetics of (+)-, (–)- and (+/–)-verapamil after intravenous administration. Br. J. Clin. Pharmacol. 1984, 17, 453–458. [Google Scholar] [CrossRef]
- Wilkinson, G.R.; Shand, D.G. A physiological approach to hepatic drug clearance. Clin. Pharmacol. Ther. 1975, 18, 377–390. [Google Scholar] [CrossRef]
- Scarpello, J.H.; Howlett, H.C. Metformin therapy and clinical uses. Diabetes Vasc. Dis. Res. 2008, 5, 157–167. [Google Scholar] [CrossRef]
- US Food and Drug Administration, Center for Drug Evaluation and Reserach. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Center Drug Eval. Res. 2005, 7, 42346. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Intravenous (n = 6 for Each) | Parameter | Oral (n = 5 for Each) | ||
|---|---|---|---|---|---|
| Without Verapamil | With Verapamil | Without Verapamil | With Verapamil | ||
| Body weight (g) | 203 ± 8.80 | 209 ± 6.65 | Body weight (g) | 228 ± 19.2 | 217 ± 13.0 |
| AUC (μg min/mL) | 1590 ± 613 | 2650 ± 449 a | AUC (μg min/mL) | 472 ± 40.2 | 819 ± 82.3 b |
| Terminal half-life (min) | 92.4 ± 26.4 | 80.5 ± 34.6 | Terminal half-life (min) | 137 ± 44.4 | 101 ± 48.0 |
| MRT (min) | 21.4 ± 5.28 | 18.4 ± 7.69 | Cmax (μg/mL) | 2.00 ± 0.328 | 3.21 ± 1.04 c |
| VSS (mL/kg) | 349 ± 128 | 286 ± 92.4 | Tmax (min) | 60 (60–120) | 60 (60–120) |
| CL (mL/min/kg) | 24.2 ± 3.02 | 11.9 ± 2.00 b | CLR (mL/min/kg) | 45.6 ± 5.66 | 14.4 ± 5.45 b |
| CLNR (mL/min/kg) | 8.12 ± 3.03 | 7.13 ± 0.932 | Ae0–24 h (% of dose) | 71.3 ± 5.86 | 38.5 ±9.96 b |
| CLR (mL/min/kg) | 16.1 ± 3.18 | 4.72 ± 2.52 b | GI24 h (% of dose) | 3.57 ± 2.84 | 1.53 ± 1.57 |
| Ae0–24 h (% of dose) | 66.5 ± 11.3 | 37.9 ± 14.9 a | F (%) | 29.7 | 30.9 |
| GI24 h (% of dose) | 0.471 ± 0.782 | 0.831 ± 0.787 | |||
| Intravenous (n = 4 for Each) | Oral (n = 4 for Each) | |||
|---|---|---|---|---|
| Parameter | Without Verapamil | With Verapamil | Without Verapamil | With Verapamil |
| Plasma | ||||
| 0.5 | 5.50 ± 0.988 | 11.3 ± 2.09 a | 1.39 ± 0.415 | 2.91 ± 0.894 b |
| 1 | 3.22 ± 1.22 | 6.94 ± 1.34 a | 2.04 ± 0.281 | 4.03 ± 1.38 b |
| 3 | 1.06 ± 0.214 | 1.68 ± 0.377 b | 1.91 ± 0.455 | 2.84 ± 0.564 b |
| 6 | 0.158 ± 0.0530 | 0.184 ± 0.0789 | 0.306 ± 0.0724 | 0.421 ± 0.0570 b |
| Liver | ||||
| 0.5 | 4.25 ± 0.978 | 6.12 ± 1.39 | 1.09 ± 0.207 | 1.31 ± 0.300 |
| 1 | 7.84 ± 2.25 | 11.1 ± 3.84 | 5.11 ± 2.03 | 6.04 ± 2.60 |
| 3 | 4.04 ± 1.04 | 6.14 ± 2.10 | 6.49 ± 1.68 | 7.78 ± 1.11 |
| 6 | 0.553 ± 0.108 | 0.510 ± 0.222 | 1.99 ± 1.10 | 1.24 ± 0.407 |
| Kidney | ||||
| 0.5 | 0.706 ± 1.80 | 6.20 ± 1.12 | 2.78 ± 1.36 | 2.28 ± 1.01 |
| 1 | 10.9 ± 1.34 | 6.69 ± 1.87 b | 7.97 ± 1.36 | 4.55 ± 1.98 b |
| 3 | 5.22 ± 1.57 | 2.68 ± 0.471 b | 4.65 ± 1.01 | 2.74 ± 0.675 b |
| 6 | 0.935 ± 0.284 | 1.20 ± 0.342 | 0.443 ± 0.0640 | 0.309 ± 0.0767 b |
| Parameter | Intravenous (n = 6 for Each) | Parameter | Oral (n = 5 for Each) | ||
|---|---|---|---|---|---|
| Without Metformin | With Metformin | Without Metformin | With Metformin | ||
| Verapamil | Verapamil | ||||
| Body weight (g) | 205 ± 8.37 | 209 ± 6.65 | Body weight (g) | 214 ± 9.62 | 217 ± 13.0 |
| AUC (μg min/mL) | 441 ± 69.5 | 527 ± 134 | AUC (μg min/mL) | 103 ± 21.6 | 96.0 ± 30.6 |
| Terminal half-life (min) | 71.1 ± 19.0 | 79.1 ± 24.7 | Terminal half-life (min) | 128 ± 59.1 | 121 ± 57.5 |
| MRT (min) | 40.9 ± 7.75 | 42.4 ± 20.4 | Cmax (μg/mL) | 0.678 ± 0.386 | 0.714 ± 0.452 |
| VSS (mL/kg) | 1600 ± 861 | 1860 ± 535 | Tmax (min) | 45 (15–105) | 30 (15–75) |
| CL (mL/min/kg) | 46.3 ± 7.14 | 40.0 ± 9.45 | CLR (mL/min/kg) | 0.671 ± 0.748 | 0.489 ± 0.256 |
| CLNR (mL/min/kg) | 46.1 ± 7.08 | 39.8 ± 9.40 | Ae0–24 h (% of dose) | 0.308 ± 0.215 | 0.231 ± 0.102 |
| CLR (mL/min/kg) | 0.147 ± 0.121 | 0.108 ± 0.0726 | GI24 h (% of dose) | 0.291 ± 0.339 | 0.200 ± 0.135 |
| Ae0–24 h (% of dose) | 0.306 ± 0.219 | 0.258 ± 0.125 | F (%) | 23.4 | 18.2 |
| GI24 h (% of dose) | 0.148 ± 0.201 | 0.117 ± 0.0759 | |||
| Norverapamil | Norverapamil | ||||
| AUC (μg min/mL) | 188 ± 34.7 | 199 ± 38.7 | AUC (μg min/mL) | 68.3 ± 38.6 | 53.9 ± 24.8 |
| Terminal half-life (min) | 50.8 ± 10.2 | 63.3 ± 15.2 | Terminal half-life (min) | 168 ± 73.2 | 117 ± 44.5 |
| Cmax (μg/mL) | 1.24 ± 0.386 | 1.09 ± 0.308 | Cmax (μg/mL) | 0.267 ± 0.141 | 0.273 ± 0.123 |
| Tmax (min) | 60 (30−120) | 60 (30−90) | Tmax (min) | 45 (15–120) | 45 (15–105) |
| CLR (mL/min/kg) | 0.109 ± 0.102 | 0.0626 ± 0.0535 | CLR (mL/min/kg) | 0.336 ± 0.107 | 0.562 ± 0.445 |
| Ae0–24 h (% of dose) | 0.0872 ± 0.0691 | 0.0585 ± 0.0453 | Ae0–24 h (% of dose) | 0.104 ± 0.0567 | 0.113 ± 0.0522 |
| GI24 h (% of dose) | 0.146 ± 0.0841 | 0.126 ± 0.0927 | GI24 h (% of dose) | 0.0870 ± 0.0665 | 0.0782 ± 0.0514 |
| AUCnorverapamil/AUCverapamil | 43.0 ± 7.68 | 38.5 ± 11.6 | AUCnorverapamil/AUCverapamil | 87.3 ± 26.7 | 89.7 ± 27.0 |
| Parameter | Without Metformin | With Metformin |
|---|---|---|
| Hepatic | n = 5 for Each | |
| Km (μM) | 11.0 ± 3.11 | 13.8 ± 1.26 |
| Vmax (nmol/min/mg protein) | 0.225 ± 0.0951 | 0.289 ± 0.151 |
| CLint (μL/min/mg protein) | 0.0199 ± 0.00419 | 0.0205 ± 0.00911 |
| Intestinal | n = 4 for Each | |
| Km (μM) | 148 ± 53.5 | 171 ± 11.4 |
| Vmax (nmol/min/mg protein) | 0.332 ± 0.125 | 0.412 ± 0.0908 |
| CLint (μL/min/mg protein) | 0.00181 ± 0.00150 | 0.00240 ± 0.000409 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, S.Y.; Choi, Y.H. Pharmacokinetic Interaction between Metformin and Verapamil in Rats: Inhibition of the OCT2-Mediated Renal Excretion of Metformin by Verapamil. Pharmaceutics 2020, 12, 468. https://doi.org/10.3390/pharmaceutics12050468
Han SY, Choi YH. Pharmacokinetic Interaction between Metformin and Verapamil in Rats: Inhibition of the OCT2-Mediated Renal Excretion of Metformin by Verapamil. Pharmaceutics. 2020; 12(5):468. https://doi.org/10.3390/pharmaceutics12050468
Chicago/Turabian StyleHan, Seung Yon, and Young Hee Choi. 2020. "Pharmacokinetic Interaction between Metformin and Verapamil in Rats: Inhibition of the OCT2-Mediated Renal Excretion of Metformin by Verapamil" Pharmaceutics 12, no. 5: 468. https://doi.org/10.3390/pharmaceutics12050468
APA StyleHan, S. Y., & Choi, Y. H. (2020). Pharmacokinetic Interaction between Metformin and Verapamil in Rats: Inhibition of the OCT2-Mediated Renal Excretion of Metformin by Verapamil. Pharmaceutics, 12(5), 468. https://doi.org/10.3390/pharmaceutics12050468