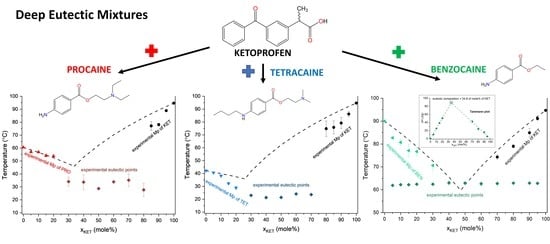
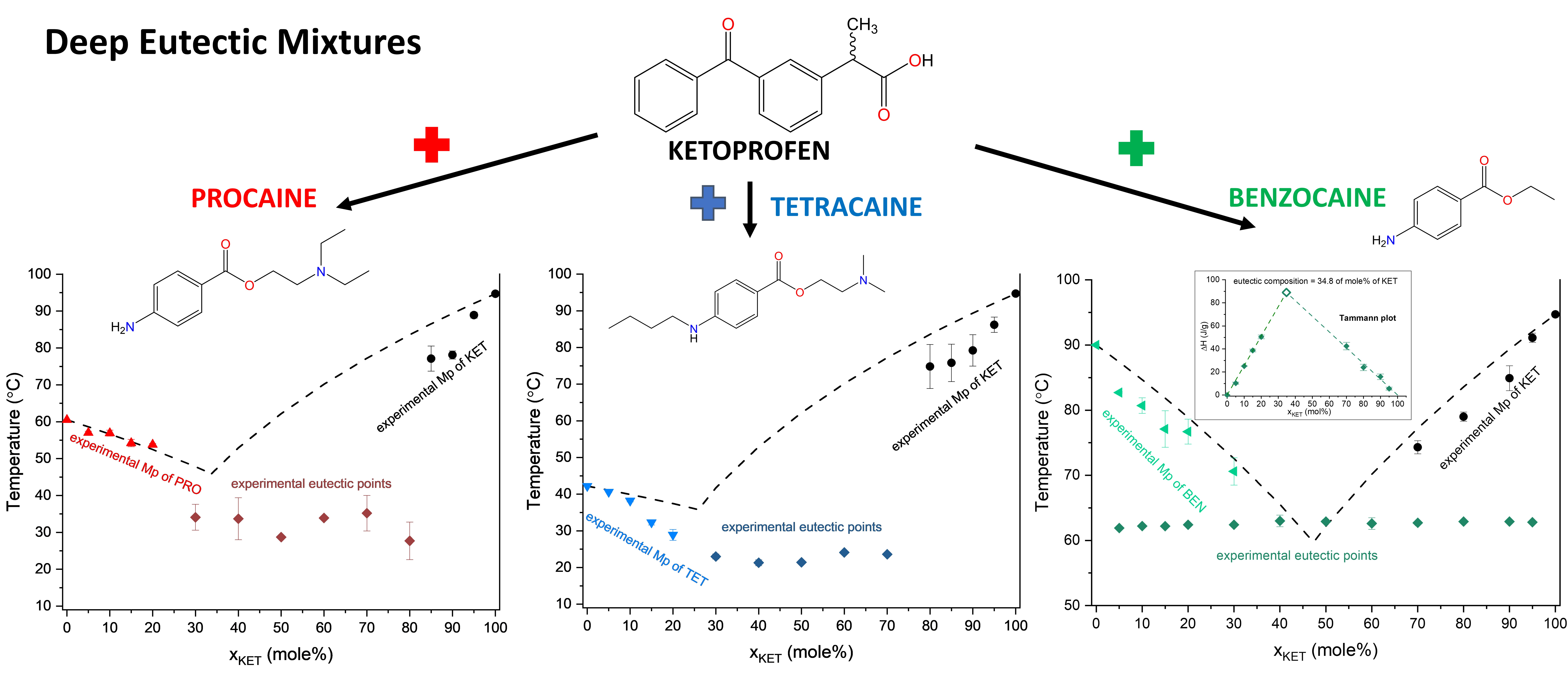
3.2. Ketoprofen-Procaine (KET-PRO) Systems
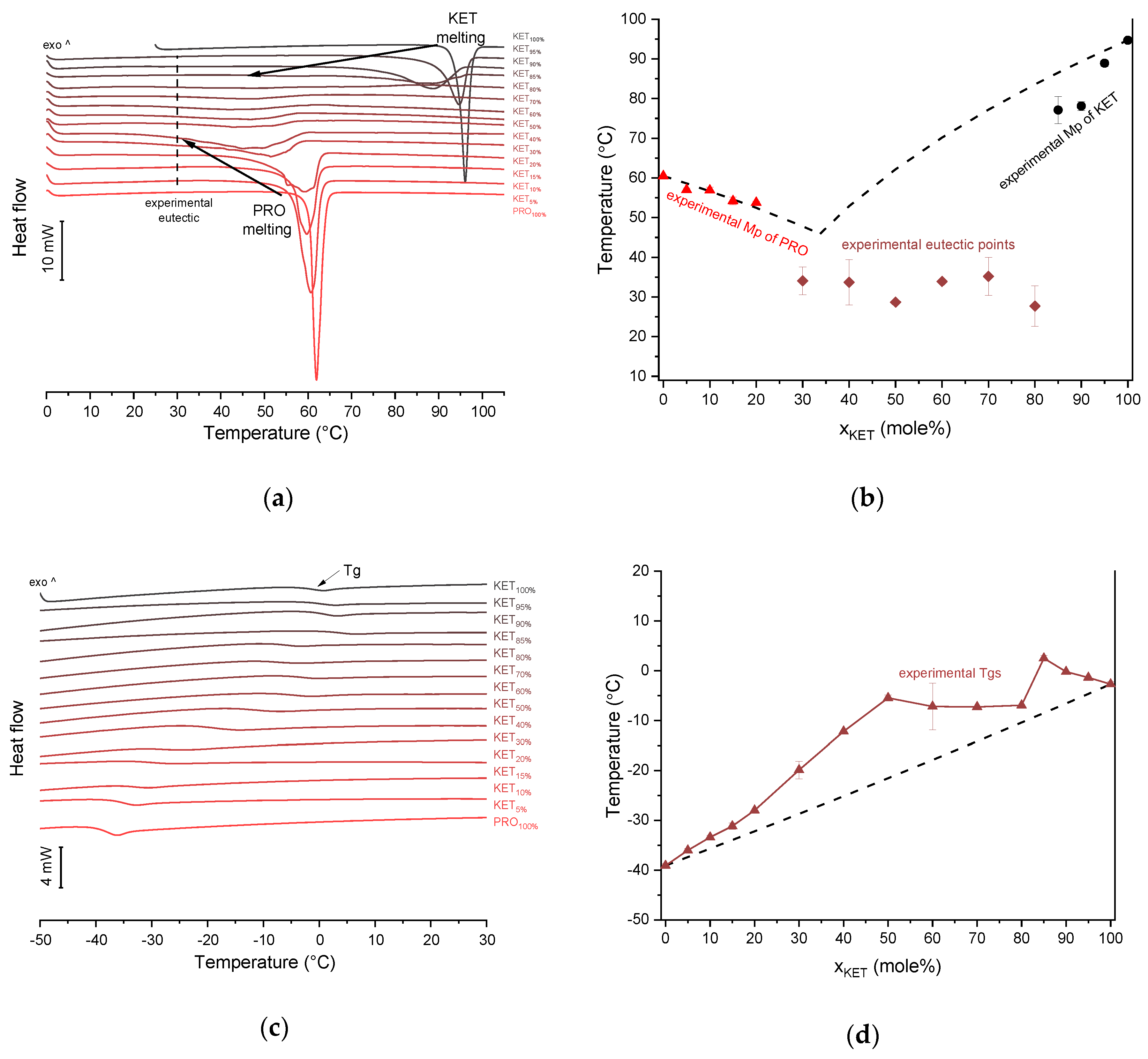
KET starting material in the first heating cycle showed a single melting endotherm with an onset at 94.7 ± 0.4 °C. PRO starting material in the first heating also exhibited a single melting endotherm (onset: 60.5 ± 0.1 °C). The heat treatment of the binary KET-PRO mixtures resulted in the formation of a eutectic phase (
Figure 2a) and a phase transition to viscous liquids. The Mps of KET and PRO decreased, and the melting endotherms became broader corresponding to their decreasing content in the mixtures. DSC traces of the KET-PRO samples containing 30–80 mol% of KET showed a very broad endotherm with an onset at around 30 °C, which had a low enthalpy of transition. This event can be considered as melting of the eutectic phase. The experimental Mps of PRO in samples containing 5–20 mol% of KET agreed with the predicted values calculated using the Schröder van Laar equation Equation (1), whereas the Mps of KET were lower (
Figure 2b). The experimental eutectic temperature was also lower than that determined from Equation (1), which was 46 °C. Therefore, the behavior of PRO and KET in KET-rich mixtures cannot be considered as that of an ideal system, where no strong intermolecular interactions take place. The calculated eutectic composition is 33 mol% of KET, but it was impossible to determine the experimental eutectic composition, due to an overlapping peak of PRO melting.
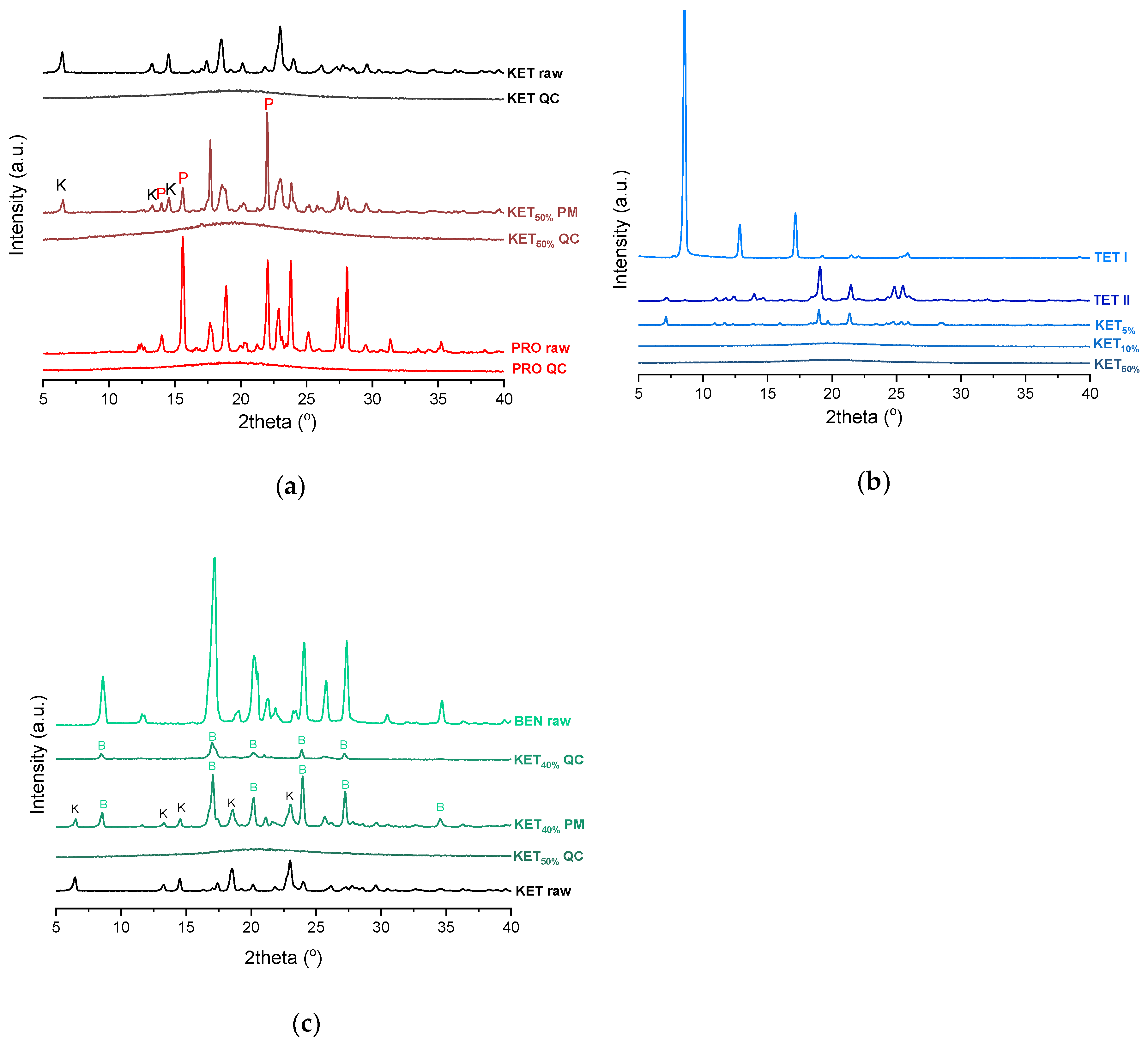
PXRD analysis of the KET-PRO physical mixtures prior to the heat treatment revealed the presence of crystalline substances with the peaks corresponding to both, PRO and KET starting materials (
Figure 3a). The phase behavior of an equimolar KET-PRO mixture was further investigated by HSDSC (
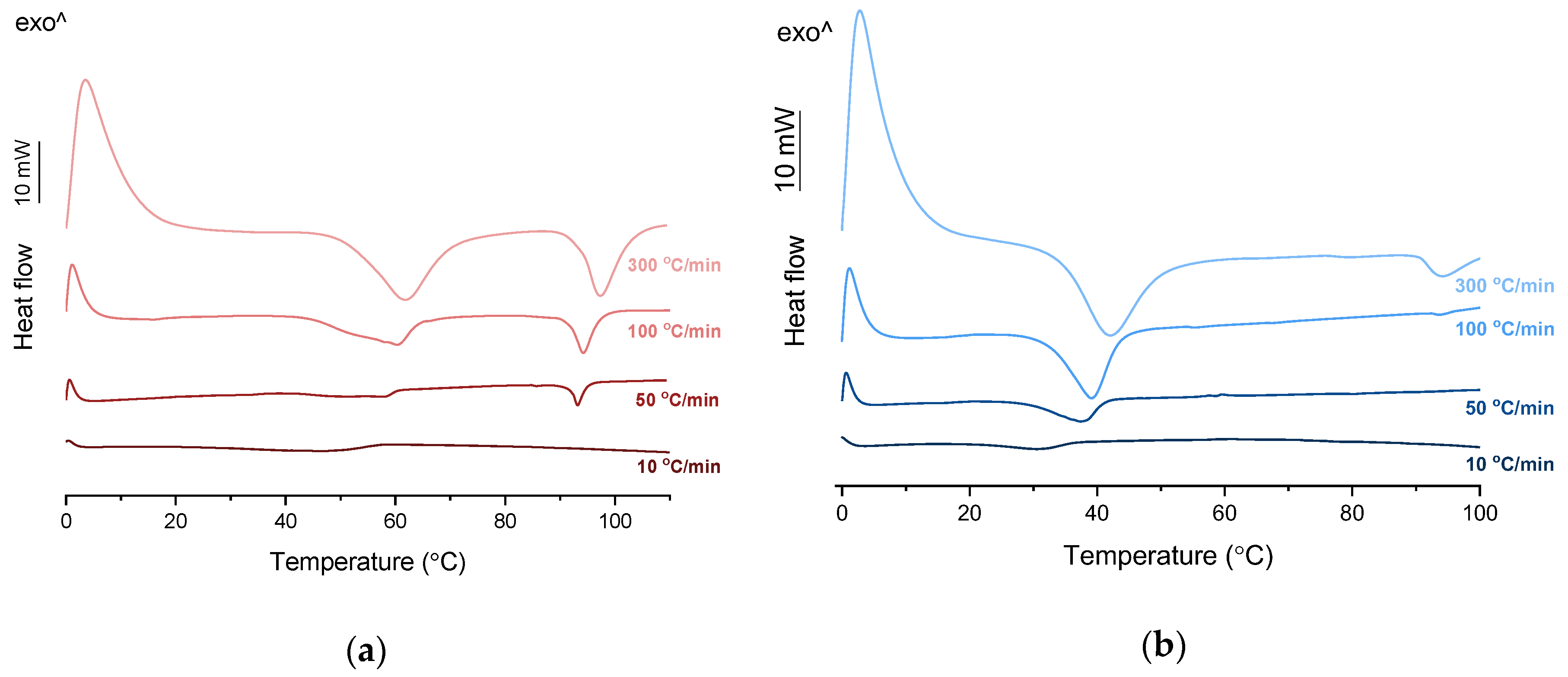
Figure 4a). When heated at a standard rate (10 °C/min), a very broad endotherm with an onset at 28.7 ± 0.4 °C was observed, which can be attributed to the eutectic melting followed by melting/dissolution of the excess of either KET or PRO. The onset of this eutectic endotherm moved from 40.8 ± 1.6 °C to 50.6 ± 1.5 °C when the heating rate increased from 50 °C/min to 300 °C/min and the peak became narrower. Moreover, in the equimolar mixture, at heating rates 50–300 °C/min, a second endotherm with an onset at approximately 92 °C appeared, which was attributed to KET. KET and PRO starting materials at heating rates 50–300 °C/min showed a single narrow melting endotherm with an onset at approximately 92 and 57 °C, respectively, independent on the heating rate (data not shown). Thus, the fast thermal treatment prevented the eutectic phase formation and a phase separation occurred, indicating that the eutectic was, indeed, composed of KET and PRO.
KET did not crystallize either during the fast cooling step or during the second heating (10 °C/min) as evidenced by the lack of the endothermic events and by a glass transition event (
Tg) in the second heating cycle (midpoint: −2.7 ± 0.5 °C) (
Figure 2c,d). Similarly, PRO did not crystallize either during cooling or during the second heating cycle and exhibited a
Tg at −39.1 ± 0.4 °C. Quench cooled (QC) PRO and KET were disordered by PXRD (
Figure 3a). In the phase diagram of the heat-treated KET-PRO samples (
Figure 2c) only one phase transition was observed, i.e., the
Tg, consistent with the disordered “halo” pattern recorded by PXRD (
Figure 3a). The presence of one
Tg in the binary mixtures implies mixing of the components at the molecular level. The
Tg values, which were dependent on mixture composition (
Figure 2d), indicate that heat-treated KET-PRO mixtures formed supercooled liquids. The experimental
Tg values of samples containing at least 30 mol% of KET were larger than the predicted values calculated by the Fox equation (Equation (2)), thereby implying strong interactions between PRO and KET in the glassy state, which could involve proton transfer or at least strong H-bonds. KET-PRO supercooled liquids showed no tendency to crystallize when heated at 10 °C/min supported by the lack of crystallization exotherms and melting endotherms during the second heating. Moreover, slower heating and/or cooling rates (2 °C/min) did not induce crystallization.
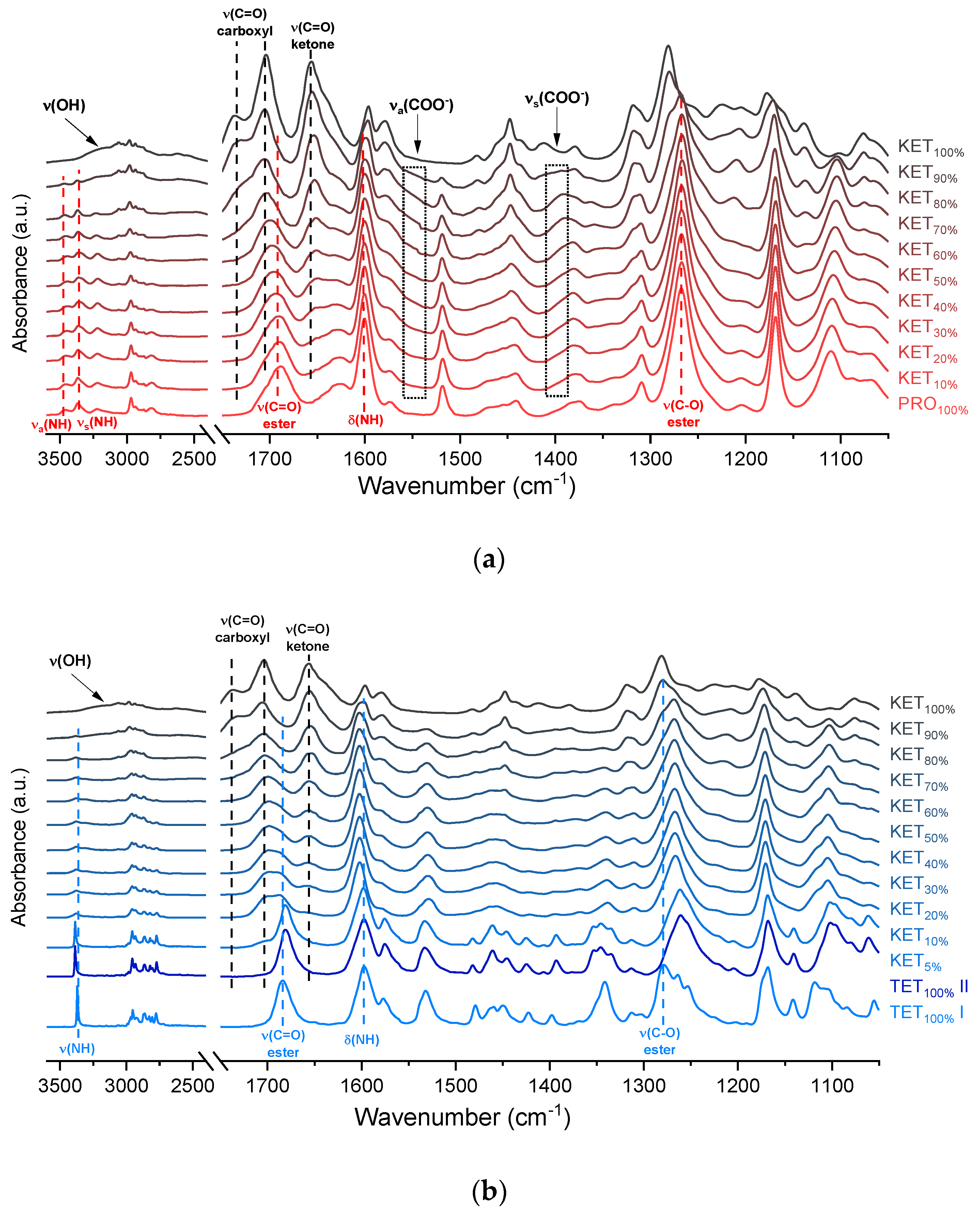
The infrared spectra of QC PRO, KET and KET-PRO mixtures are shown in
Figure 5a. The band at 1695 cm
−1 in the crystalline KET (starting material) can be ascribed to the C=O stretching of the carboxylic groups of KET molecules organized in dimers in the crystal lattice [
33]. In the QC KET, which contained carboxylic acid monomers due to release of KET molecules from crystal lattice, this band shifted towards higher wavenumbers, giving a peak with the center at 1704 cm
−1 and shoulder at 1737 cm
−1. The band at 1655 cm
−1, assigned to the stretching mode of the ketone group [
33], was observed at 1656 cm
−1 in QC KET. The broad peak in the range 2200–3400 cm
−1 observed in both, KET starting material and QC KET can be assigned to the O–H stretching [
33]. It overlaps with the alkane C–H stretching band at 2800–3000 cm
−1 [
34]. In the spectrum of crystalline PRO, two sharp peaks at 3463 and 3366 cm
−1 can be ascribed to the N–H stretching bands [
35]. The NH
2 groups in the crystal structure form intermolecular hydrogen bond with the ester C=O group [
36]. The intensity of these N–H stretching bands decreased drastically in the QC PRO. The band at 1600 cm
−1 can be attributed to the NH
2 scissoring mode [
35]. The band at 1665 cm
−1 in PRO starting material is probably the ester C=O stretching [
35]. In the spectrum of QC PRO this band shifted towards higher wavenumbers (1689 cm
−1). The band at 1272 cm
−1, the most intensive band in the spectrum of PRO starting material, can be attributed to the ester C–O stretching [
35]. The band at 1272 cm
−1, most likely of the C–O stretching, shifted towards lower wavenumbers (1267 cm
−1) in the spectrum of heated PRO. In the spectra of the QC KET-PRO mixtures, the doublet of N–H stretching bands shifted towards lower wavenumbers (3444 and 3361 cm
−1 for equimolar mixture), but in the samples containing 70–90 mol% of KET it shifted back towards higher wavenumbers. The broad O–H stretching band was present in the sample containing 90 mol% of KET, but it was difficult to observe it in the sample containing 80 mol% of KET. The position of the N–H scissoring band (1600 cm
−1) did not change in QC KET-PRO mixtures, neither did the position of the ester C–O stretching band. The ketone C=O stretching band shifted slightly towards lower wavenumbers (1652 cm
−1 in the equimolar mixture), and in the sample containing 10 mol% of KET it was no longer observed. In the case of the ester C=O band of PRO (1689 cm
−1) and carboxyl C=O band of KET (1704 cm
−1), the gradual change of one band into another was observed, in the equimolar sample the position of the ‘hybrid’ band was 1697 cm
−1. The shoulder of the KET carboxyl C=O monomers shifted towards lower wavenumbers in samples containing 90–70 mol% of KET and it was no longer observed in the sample containing 60 mol% of KET. Hence, the carboxyl group of KET is strongly involved in interactions with PRO molecules. The carboxylate group bands should appear at 1650–1550 cm
−1 and 1400 cm
−1, arising from asymmetric and symmetric stretching, respectively, with the former that should be intensive and the latter weak. An increased absorbance in the region above 1550 cm
−1 was observed in the spectra of QC KET-PRO samples. It was the most intensive in samples containing 60–70 mol% of KET. Hence, it is possible that a fraction of KET molecules is ionized in the supercooled mixtures and the proton is transferred probably to tertiary amine of PRO.
3.3. Ketoprofen-Tetracaine (KET-TET) Systems
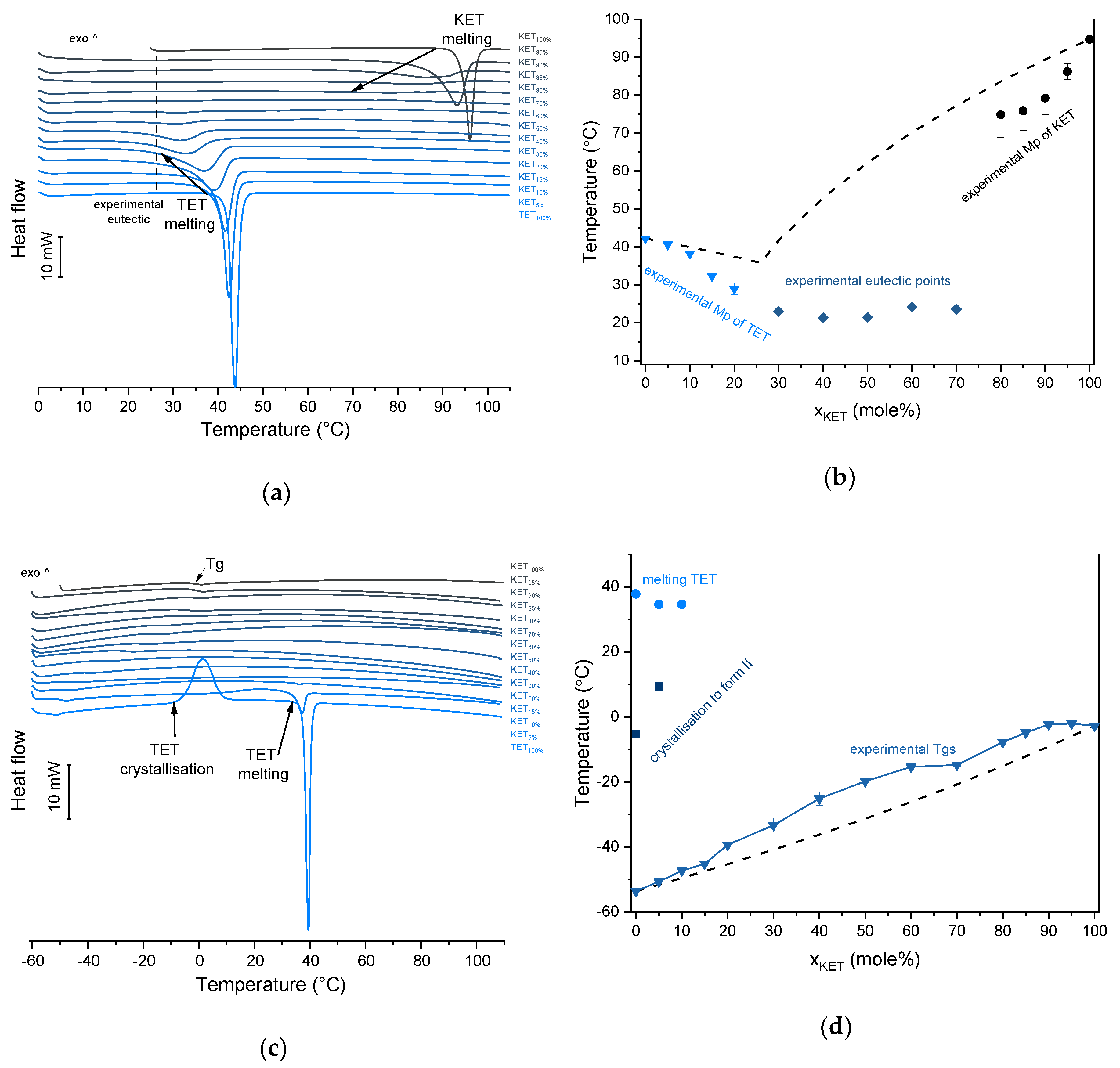
TET showed a single melting endotherm (onset: 42.2 ± 0.0 °C) in the first heating (
Figure 6a,b). The Mp of KET and TET decreased and the melting endotherms became broader corresponding to the decreasing quantity of the drugs in the sample. The KET-TET samples with 5–10 mol% of KET showed an Mp similar to that calculated using the Schröder van Laar equation, whereas all other experimental Mps were lower than those predicted for ideal systems. Similar to the KET-PRO mixtures, the formation of a eutectic phase was observed. A decrease in Mp in mixtures containing 15–20 mol% of KET may be explained by accelerated dissolution of TET crystals in the presence of the liquid phase. The eutectic point is probably close to that observed for 30–70 mol% of KET mixtures with an onset of approximately 21–23 °C, hence it is lower than the value calculated for an ideal system (35.8 °C). The theoretical eutectic composition determined from Equation (1) was 26 mol% of KET, but it was impossible to determine the experimental composition, due to overlapping peak of the TET melting endotherm (
Figure 6a). PXRD revealed that the equimolar sample (unheated) displayed a pattern corresponding to the mixture of crystalline TET and KET (
Figure 3b). To better understand the phase behavior of KET-TET systems, the equimolar physical mixture was heated at different rates. At 300 and 100 °C/min two melting endotherms were observed; the first one that appeared at approximately 33.7 ± 0.9 °C, and the second one had an onset above 90 °C (melting of KET) (
Figure 4b). When the heating rate was reduced to 50 °C/min, the first endotherm appeared at lower temperature (30.6 ± 1.3 °C) and was broader, and the melting endotherm of KET was no longer observed. Upon heating at 50–300 °C/min TET showed a sharp melting event at 38 °C (data not shown). Hence, the eutectic and either KET or TET were present in the sample, but at a standard heating rate of 10 °C/min dissolution of KET and/or TET crystals dispersed in the liquid phase can take place resulting in a broad and flat endotherm.
After rapid cooling, TET formed a supercooled liquid with a glass transition at −53.6 ± 0.1 °C (
Figure 6c,d). Upon second heating, supercooled TET showed a crystallization exotherm (onset: −5.2 ± 0.3 °C, Δ
H = 102.8 ± 2.7 J/g) followed by a melting endotherm (onset: 37.8 ± 0.1 °C, Δ
H = 116.4 ± 0.7 J/g). Since the value of crystallization Δ
H is approximately 88% of melting Δ
H, the crystallization occurs mainly during re-heating, but it is possible that the sample partially crystallized during the cooling step. The melting of TET in the second cycle occurred at a lower temperature compared with that from the first heating cycle (onsets 37.8 ± 0.2 °C and 42.2 ± 0.1 °C, respectively). Moreover, the PXRD pattern of QC TET that was heated past the crystallization in the DSC was different than that of the starting material (
Figure 3b), indicating a different polymorphic form. Two polymorphs, II and I, have been reported for TET with melting points at 37 °C and 42 °C, respectively [
37]. Hence, the starting material is most likely polymorph I (TET I), whereas the product obtained by crystallization during the second heating (heating of supercooled TET) is form II (TET II). The sample containing 5 mol% of KET formed a supercooled liquid and upon heating only 15% of TET crystallized into TET II, the rest of TET remained in the supercooled state. The TET crystallization peak for this sample appeared at higher temperatures (onset: 9.3 ± 4.4 °C) and was broader compared with that of 100% TET. The enthalpy of crystallization was the same as the enthalpy of melting, hence the crystallization process took place during heating. The QC sample containing 10 mol% of KET was disordered by PXRD, but a small quantity of crystalline TET (less than 1%) was detected by DSC. All other QC KET-TET mixtures (15–95 mol% of KET) exhibited only one phase change, a
Tg, indicating mixing TET and KET at molecular level. The
Tg values were dependent on the composition and they increased corresponding to an increasing quantity of KET, but in the KET-rich samples (90 and 95 mol% of KET) the Tgs reached a maximum and this temperature was the same as that of 100% KET. The Tgs of the QC mixtures samples containing at least 20 mol% of KET were higher than those calculated from Fox equation (
Figure 6d), thereby implying important deviations from the behavior of an ideal system due to interactions between KET and TET.
The infrared spectrum of TET starting material (TET I) showed a single sharp band at 3371 cm
−1 ascribed to the N–H stretching of the secondary aromatic amino group (
Figure 5b), which shifted towards a higher wavenumber in TET II (3387 cm
−1). In the spectrum of TET I the stretching of C=O and C–O ester bonds produced bands at 1684 cm
−1 and 1279 cm
−1, respectively, [
35,
38] and they shifted to lower wavenumbers (1681 cm
−1 and 1261 cm
−1, respectively) in TET II. The presence of TET II was also observed in the spectrum of the mixture containing 5 mol% of KET. In the sample comprising 10 mol% of KET broadening and a dramatic decrease in the intensity of the N–H stretching band (3375 cm
−1) was observed due to TET molecules being able to interact with KET. The O–H stretching band was detected in the sample containing 90 mol% of KET, and it was difficult to observe it in the samples containing a smaller amount of KET. The position of the ester stretching C–O band was depended on the solid-state properties of the sample: it shifted from 1279 cm
−1 (TET I) to 1261 cm
−1 in TET II, and in the QC KET-TET samples it was present at 1267 cm
−1. A decreased intensity of the ester C=O stretching band was observed in the QC samples. In the sample containing 10 mol% of KET it was localized at 1688 cm
−1 and it shifted towards a higher wavenumber corresponding to an increasing KET concentration and overlapped with the carboxyl C=O stretching band. The latter band shifted towards a lower wavenumber and its intensity decreased corresponding to a decreasing KET concentration. The shoulder of the carboxyl C=O stretching band localized at 1737 cm
−1 (100% KET) also shifted towards lower wavenumbers and was no longer observed in the sample containing 60 mol% of KET, similar to the KET-PRO system. The IR results indicate that TET interact with KET by strong hydrogen bonding, and the carboxyl group of KET is involved in the interactions. Interactions between KET and TET are sufficiently strong to break the crystal lattice of TET even at KET concentrations as low as 10 mol%. However, in contrast to KET-PRO, in the KET-TET samples carboxylate anion was not detected.
3.4. Ketoprofen-Benzocaine (KET-BEN) Systems
BEN exhibits polymorphism and three polymorphic forms have been described to date. Form I, formerly known as form β, is a monoclinic P 2
1/c polymorph (Z = 4) [
39,
40]. Form II, formerly known as α, is orthorhombic with space group P2
12
12
1 (Z = 4) [
40,
41]. Form III is another monoclinic P2
1 polymorph (Z = 8) [
40,
42]. Polymorph II (referred to as Mod I
0 by Schmidt, [
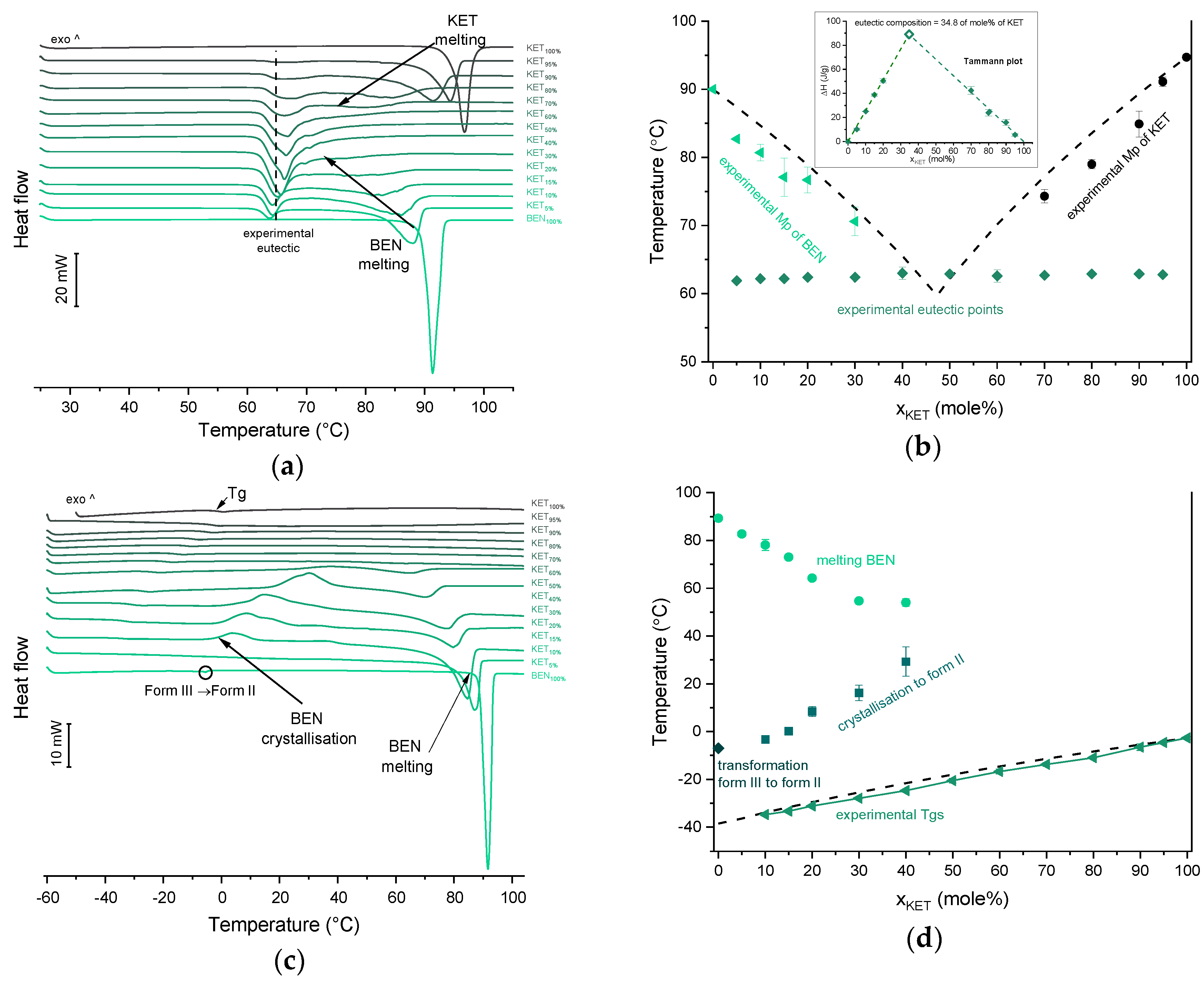
43]) is thermodynamically stable under ambient conditions, and is present in commercial products. The starting material used in our study was form II. In the first heating cycle BEN showed a single melting endotherm (onset: 90.0 ± 0.1 °C). The KET-BEN mixtures formed a eutectic phase (
Figure 7a). Both, KET and BEN melted at lower temperatures and had broader melting endotherms corresponding to their decreasing content in the mixture. Experimental Mps of KET and BEN showed an agreement with values calculated using the Schröder van Laar equation. All binary KET-BEN mixtures showed eutectic peaks with an onset at approximately 62–63 °C. The shape of the eutectic peak depended on the composition: the peaks were broader in the KET-rich samples than in the BEN-rich samples. It was difficult to discern individual peaks in the mixtures containing 30–70 mol% of KET. Thus, to construct the Tammann plot (Δ
H of the eutectic peak as a function composition), only the mixtures containing 5–20 and 80–95 mol% of KET were taken. The eutectic composition determined using the Tammann plot was 34.8 of mol% of KET (
Figure 7b). This composition contains less KET that the theoretical eutectic composition determined by Equation (1), which is 47 mol% KET. Assuming that this eutectic phase was a simple mechanical mix, the enthalpy of mixing (Δ
Hmix) was calculated using Equation (4) [
23]:
where (Δ
Hfus)
exp is the heat of fusion of the eutectic peak at the eutectic composition obtained from the Tammann plot,
x is the mole fraction of the component (KET or BEN) and Δ
Hfus is the heat of fusion of the pure constituent (KET or BEN). The value of Δ
Hmix for the KET-BEN system was negative, –2.33 kJ/mol, indicating the presence of weak intermolecular forces leading to cluster formation [
44]. The KET-BEN physical mixtures showed peaks characteristic of starting materials in X-ray diffractograms (
Figure 3c).
In the second heating cycle BEN melting endotherm was observed at the same temperature as in the first heating, but it was preceded by a small endotherm at −7.0 ± 0.1 °C (Δ
H = 1.0 ± 0.2 J/g) (
Figure 7c,d). Interestingly, Gana et al., [
45] observed a similar endothermic event, which was attributed to a phase transition of form III into form II. Forms II and III are enantiotropic under ambient conditions. Form III is stable at low temperature and form II is stable at higher temperature and melts eventually. Form III becomes more stable with increasing pressure at ambient temperature [
45]. Form II was detected in the QC BEN by PXRD (
Figure 3c). After a rapid cooling step of the KET-BEN melt, a supercooled liquid was formed, except for the mixture containing 5 mol% of KET. A
Tg was detectable down to the KET content of 10 mol%. A very strong linear relationship was observed between the composition (mol% of either BEN or KET) and the
Tg (
R2 = 0.999). It was not achievable to obtain BEN 100% in the supercooled state, because it crystallized during cooling despite the very fast cooling rate employed. Hence, to determine the calculated Tgs using the Fox equation, the
Tg of BEN 100% (−38.5 °C) was determined by extrapolation. The experimental Tgs showed a good agreement with the calculated Tgs. This implies the behavior of an ideal mixture, in which the tendency of two kinds of molecules (BEN and KET) to transfer from the glassy state to the supercooled liquid state is unchanged. The presence of only one
Tg in the binary mixtures containing at least 10 mol% of KET indicates that KET and BEN are miscible. The amount of crystalline of BEN decreased from 86.0 ± 0.7% (5 mol% of KET) to 15.2 ± 7.8% (40 mol% of KET), and crystalline BEN was no longer observed in samples containing at least 50 mol% of KET. QC mixtures that contained at least 50 mol% of KET showed a halo-pattern characteristic of disordered materials (
Figure 3c). Hence, the increasing concentration of KET increased the amount of BEN entrapped in the supercooled state. In the sample containing 5 mol% of KET, crystallization of BEN took place during cooling. The crystallization of BEN was viewed during the second heating for mixtures containing 40–90 mol% of KET. The onset temperature of the crystallization peak increased corresponding to a decreasing BEN concentration in the sample. The percentage of BEN that crystallized during heating increased from 23% (10 mol% of KET) to approximately 100% in mixtures containing 20–40 mol% of KET. X-ray diffractograms confirmed that it is BEN that crystallizes, because only BEN peaks were observed in the X-ray diffractograms of heated, previously QC KET-BEN systems (KET ≤ 40 mol%) (
Figure 3c). Some samples, such as that containing 10 mol% of KET, showed a multistep crystallization process, which could be the interfacial crystallization (first part of the peak) that was followed by bulk crystallization (second part of the peak). PXRD patterns of this QC sample that was heated in the second heating cycle to either 20 °C or 50 °C were the same and showed peaks characteristic for BEN (data not shown). The BEN crystallization peak was followed by the melting peak with an onset temperature decreasing corresponding to a decreasing BEN content, from 78.1 ± 2.3 °C to 54.0 ± 1.6 °C for KET 10 and 40 mol%, respectively. Δ
H values of the BEN melting peak in the second heating cycle were markedly higher than those observed in the first heating cycle for the mixtures containing 5–30 mol% of KET.
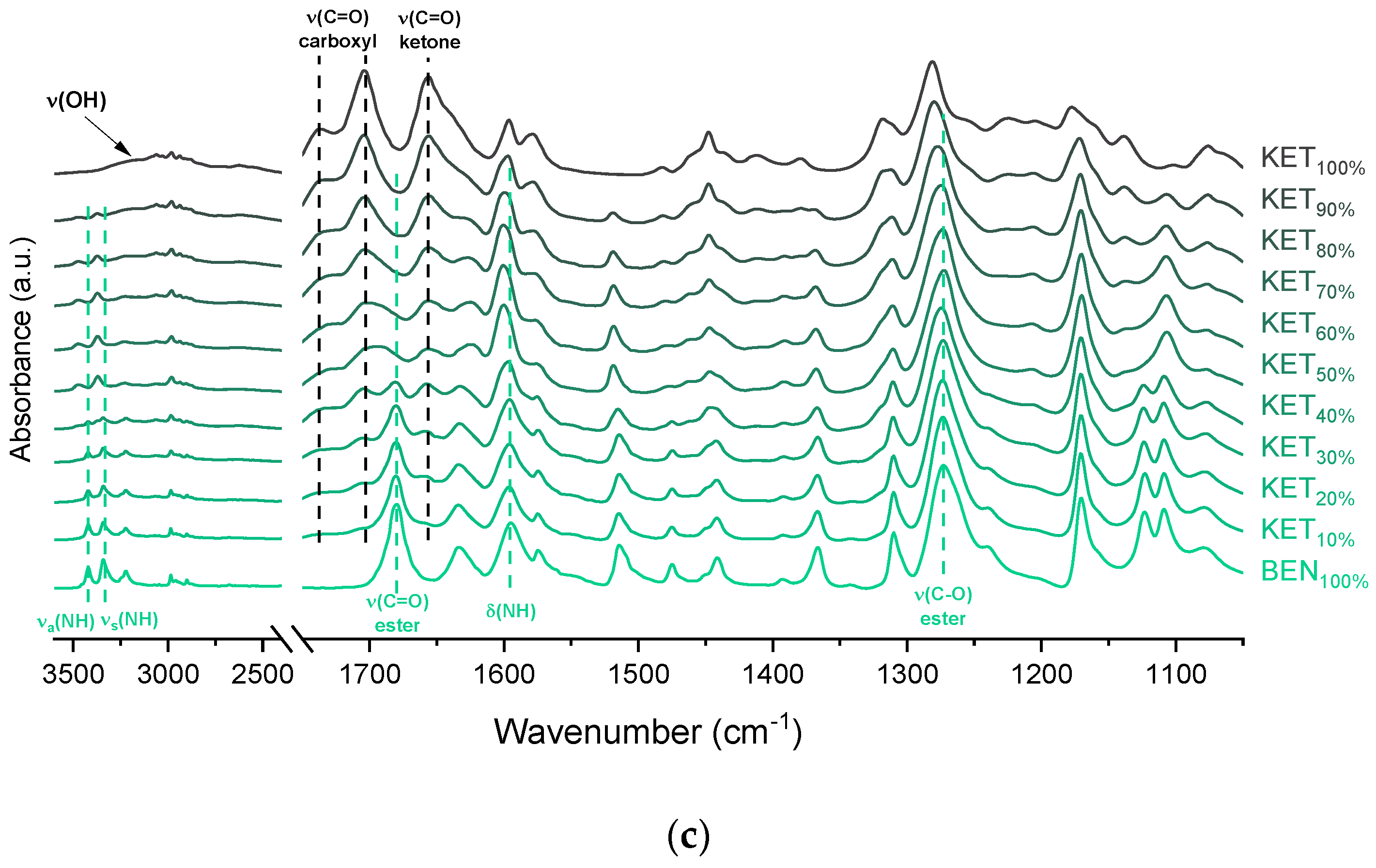
The interactions between BEN and KET were further examined by ATR-FTIR (
Figure 5c). The doublet consisting of two bands at 3421 and 3340 cm
−1 can be ascribed to asymmetric and symmetric N–H stretching bands, respectively [
35,
40]. The scissoring band of NH
2 is present at 1595 cm
−1. The C=O and C–O bonds of aromatic esters produce bands at 1680 and 1273 cm
−1, respectively [
35]. The latter may overlap with the C–N stretching band characteristic of aromatic amines in the range 1240–1366 cm
−1 [
35]. In the spectra of QC KET-BEN mixtures the positions of bands at 1273 cm
−1 of the ester C-O stretching and at 1656 cm
−1 of the stretching of ketone C=O group did not change markedly. In mixtures containing at least 50 mol% of KET the bands at 3421 and 3340 cm
−1 of the asymmetric and symmetric N–H stretching bands disappeared, and new bands at 3472 and 3371 cm
−1 were observed. The latter bands became noticeable in samples containing 20–30 mol% of KET. Interestingly, in the sample comprising KET 40 mol% both, the two former (3421 and 3340 cm
−1) and the two latter (3472 and 3371 cm
−1) bands were easily observed. The presence of those bands is in a good agreement with the crystallinity of BEN determined by PXRD and DSC. The packing of BEN molecules in crystals is stabilized by N–H···O hydrogen bonds [
42]. Hence, the bands at 3421 and 3340 cm
−1 correspond to N–H stretches of the NH
2 group in the crystal lattice that interact via H-bonding with the C=O moiety of the ester groups, and the increase in wavenumber to 3472 and 3371 cm
−1 is probably due to the liberation of the NH
2 groups from the crystal lattice due to interactions with KET. This is consistent with the fact that the band of the ester C=O stretching of BEN (1680 cm
−1) was no longer detectable in the equimolar KET-BEN mixture. The peaks characteristic of the NH
2 group in the crystal lattice disappear in the samples that do not contain a detectable amount of crystalline BEN (at least 50 mol% of KET). The intensity of the bands characteristic of ‘supercooled’ NH
2 groups also agree well with the content of noncrystalline BEN in the sample. The band of the NH scissoring at 1595 cm
−1 shifted towards higher wavenumbers (1600 cm
−1) in the QC samples containing 50–80 mol% of KET, the band shifted to 1597 cm
−1 for the sample containing 90 mol% of KET because it was dominated by the C–C stretching band of KET at 1596 cm
−1 [
34]. The intensity of the broad O–H stretching band decreased corresponding to a decreasing KET concentration. Interestingly, this band was observed in the sample containing 60 mol% of KET, whereas in the PRO and TET mixtures it was only observed at KET 90 mol%. Also, both shoulders of the C=O stretching group band of the carboxyl acid monomer of KET (1704 and 1737 cm
−1) moved to lower wavenumber corresponding to a decreasing KET concentration (1693 and 1727 cm
−1 for the equimolar KET-BEN mixture). It can be concluded that KET interacts BEN via H-bonding, as suggested by the negative value of Δ
Hmix, thereby decreasing the interactions between BEN molecules in the crystal lattice and leading to entrapment of BEN in the supercooled state.
3.5. DFT Studies
Molecular modelling studies were carried out to determine the likelihood and mode of intermolecular interactions in the binary systems. The global reactivity parameters are listed in
Table 2. For LAs, the higher HOMO energy was calculated for TET, therefore this molecule should be the best electron donor, while the lowest LUMO energy was determined for BEN and PRO, implying the best electron acceptor properties of these two substances [
46]. The values of I and A also suggested the same characteristics. Electronegativity values indicate if a molecule is a Lewis acid (large
χ) or a base (low
χ), therefore it can be said that KET will act as a Lewis acid (also consistent with the high value of
ω) [
47]. The molecule that is deemed as most reactive, based on the chemical hardness is TET followed closely by PRO. The global reactivity parameters, however, do not show a similar trend as that observed from the experimental studies, where PRO interacted with KET most strongly, followed by TET and BEN. It is because the calculated values do not include any effects of ionization (proton transfer), that possibly occur between KET and PRO.
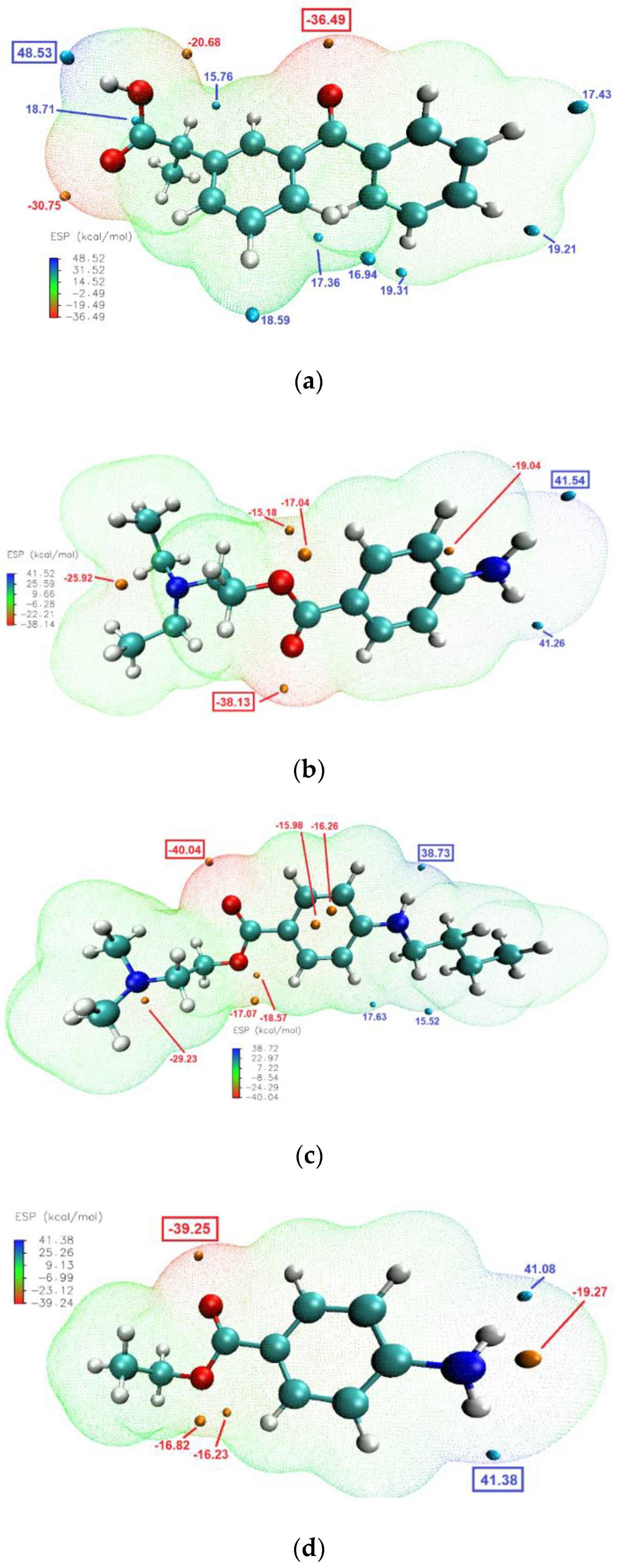
The molecular electrostatic potential (ESP) calculations can be vital in predicting intermolecular interactions [
48]. Mapping the ESP on molecular vdW surface of KET and the studied LAs (
Figure 8), clearly showed that the most likely intermolecular interactions between KET and each of the LAs will be between the –OH moiety of the carboxylic group of KET (ESP maximum of 48.5 kcal/mol) and the carbonyl of the ester moiety of the LA (ESP minimum of −38.13, −40.04 and −39.25 kcal/mol for PRO, TET and BEN, respectively). This is consistent with the results of the infrared analysis presented above, evidencing H-bond formation in the binary systems.
3.6. General Discussion
Multicomponent mixtures were successfully produced by the mechanosynthetic process, which involved mixing of an acid (KET) with a base (LA) in a range of molar ratios followed by heating. This process has important advantages. It does not necessitate the use of solvent, hence there is no need for product drying and determining the residual solvent content. Another advantage is the high product yield and purity because the process does not require product isolation and/or purification. Thus, the mechanosynthesis eliminates the drawbacks of commonly used metathesis reactions. The mechanosynthetic process described in this paper is applicable for NSAID-LA combinations and other DEMs/ILs. If the chosen combination contains substances that decompose upon melting (such as acetylsalicylic acid or diclofenac), solvent assisted grinding such as that described previously [
20] for ethambutol ILs/salts or aspirin cocrystals [
49] could be an alternative.
The heat treatment of a binary KET-LA physical mixtures resulted in the formation of eutectic phase. The thermograms of the binary KET-TET and KET-PRO mixtures contained broad endotherms with low intensity, except of mixtures containing very large excess of one of the components. It may be due to the fact that a part of dispersed crystals is surrounded by the liquid phase generated by the melting of the eutectic. This can induce the accelerated melting and/or dissolution of the component in excess (either KET or LA). Kataoka et al., [
14] observed a similar phenomenon for ibuprofen-lidocaine binary mixture. The formation of eutectic mixtures has been observed for another NSAID–LA pair: indomethacin-lidocaine [
18]. The eutectic peak in the KET-LA systems studied very often overlapped with the subsequent melting events. KET-PRO and KET-TET, except of LA-rich mixtures, showed important deviations from the behavior of an ideal system due to interactions between the KET and LA molecules, hence it was impossible to determine the eutectic composition. The behavior of the KET-BEN system, on the other hand, agreed with the theoretical predictions. The eutectic peaks were better separated from the succeeding thermal events compared with KET-TET and KET-PRO, and it was possible to determine the KET-BEN eutectic composition using the Tammann plot.
In the phase diagrams from the second heating, all KET-PRO samples were characterized by a single-phase transition from glass to the supercooled state. Similarly, only one
Tg was observed in the samples containing 5–85 mol% of TET and 5–50 mol% of BEN. The presence of only one
Tg confirms that KET and LAs are mixed at molecular level. KET-BEN and LA-rich KET-TET and KET-PRO mixtures showed a good agreement with theoretical predictions of
Tg by Fox equation, however in the other KET-TET and KET-PRO combinations a considerable increase in
Tg compared with predicted value was observed. This may imply the existence of strong KET-TET and KET-PRO interactions in the glassy state. During the second heating in the mixture containing 95 mol% of BEN only one melting was observed, whereas in the samples containing 60–90% of BEN and 95 mol% of TET three phase transitions were observed: a
Tg, followed by LA crystallization and LA melting. The crystallization was always to the pure LA (either TET or BEN). Active pharmaceutical ingredients can be categorized into three different classes based on their crystallization tendency [
16]. Despite the similarity of their structures, the tested LAs show a wide variation in crystallization tendency. BEN has been categorized as class I molecule, because it crystallizes during cooling from the undercooled melt state prior to the
Tg event [
16]. Hence, it has high crystallization tendency and low glass-forming ability. On the other hand, PRO and KET are class III molecules, with low crystallization tendency and high glass-forming ability for which no crystallization occurs upon either cooling to below
Tg or upon subsequent reheating up to the melting point [
16]. Baird et al. [
16] did not investigate TET, but based on our results it can be categorized as a class II molecule, for which no crystallization is observed upon cooling from the undercooled melt state to below
Tg, however, crystallization is observed during reheating above
Tg. Although BEN has a very low glass-forming ability, after mixing with KET it was possible to capture some BEN molecules in the supercooled state. Interactions between KET and TET are sufficiently strong to break the crystal lattice of TET even at KET concentrations as low as 10 mol%.
The preparation of protic ILs typically demands a sufficient pK
a difference between the acid and the base that could lead to an effective proton transfer and formation of an ion pair [
6]. The recommended difference of ΔpK
a of 10 is not possible to achieve for most APIs [
6], including NSAIDs and LAs. The pK
a is determined in a diluted aqueous solution, hence its application for tested multicomponent systems in the pure form that are not dissolved/dispersed in water, is limited. The ionization depends not only on the pK
a, but also on structural features of acid and base [
6,
8,
17]. The DSC and IR results show that interactions between KET and BEN are markedly weaker than those between KET and either TET or PRO. This is consistent with the fact that BEN is weaker base than either of TET or PRO. The interactions between PRO and KET seem to be stronger than those between TET and KET, because there are indications of the presence of a small level of carboxylate anions in the former mixture. Moreover, the positive deviations from the theoretical Tgs that are larger in KET-PRO system (a difference of 16 °C for the equimolar mixture) than in KET-TET system (a difference of 11 °C for mixtures containing 40–60 mol% of KET) imply stronger electrostatic interactions in the former. In systems composed of ciprofloxacin and Eudragit L100, particularly large positive
Tg deviations (45–48 °C) from theoretical predictions were observed due to ionic interactions [
21]. The low degree of proton transfer in the KET-LA systems tested is consistent with the previous observations for similar systems such as ibuprofen-lidocaine [
2]. When a tertiary amine is used as the base (PRO and TET in our study) the proton transfer may be severely restricted because of the lack of a satisfactory hydrogen bonding solvation environment for the anionic species formed [
2]. In conclusion, all KET-LA systems studies in this work are DEMs, i.e., mixtures of hydrogen bond donors and acceptors with intermolecular interactions via hydrogen bonding.
Although the salt preparation of the APIs with poor solubility in water is one of the most effective and developable approach to improve their solubility and dissolution rate, new salts of the API may be recognized as new chemical entities by the FDA and other healthcare authorities. Therefore, the absence of ion pair formation in the KET-LA systems could be beneficial from the regulatory point of view, as they should not be considered as new chemical entities.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}