Segmental-Dependent Solubility and Permeability as Key Factors Guiding Controlled Release Drug Product Development

 ,
, 
 and
and 
Abstract
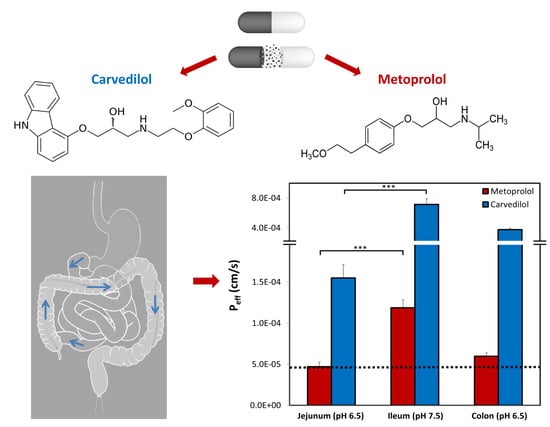
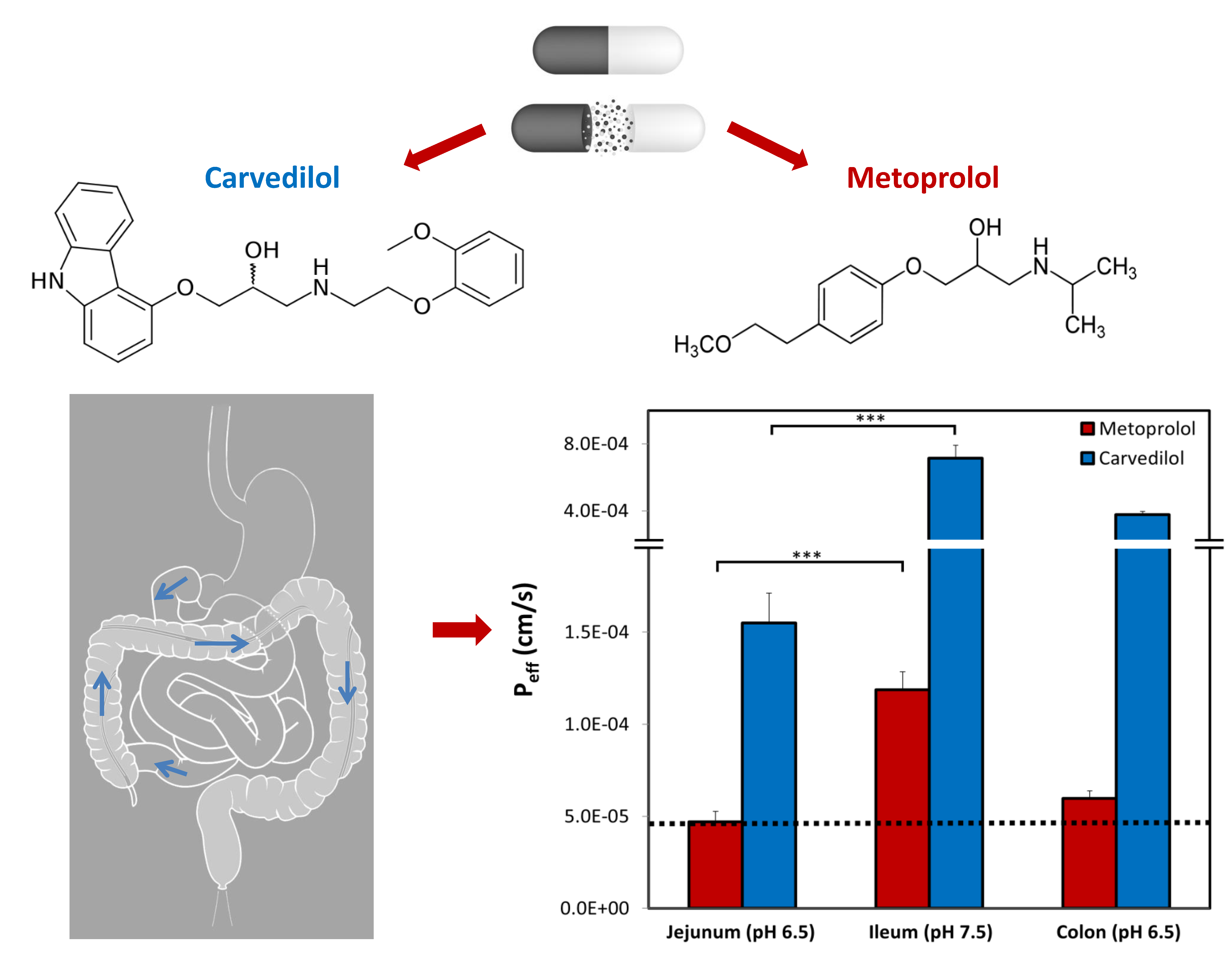
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Solubility
2.3. Octanol-Buffer Distribution Coefficients
2.4. Biorelevant pH-Dilution Dissolution Studies
2.5. Parallel Artificial Membrane Permeability Assay Studies
2.6. Rat Single-Pass Intestinal Perfusion (SPIP)
2.7. Physicochemical Analysis
2.8. Ultra-Performance Liquid Chromatography
2.9. Statistical Analysis
3. Results
3.1. Solubility
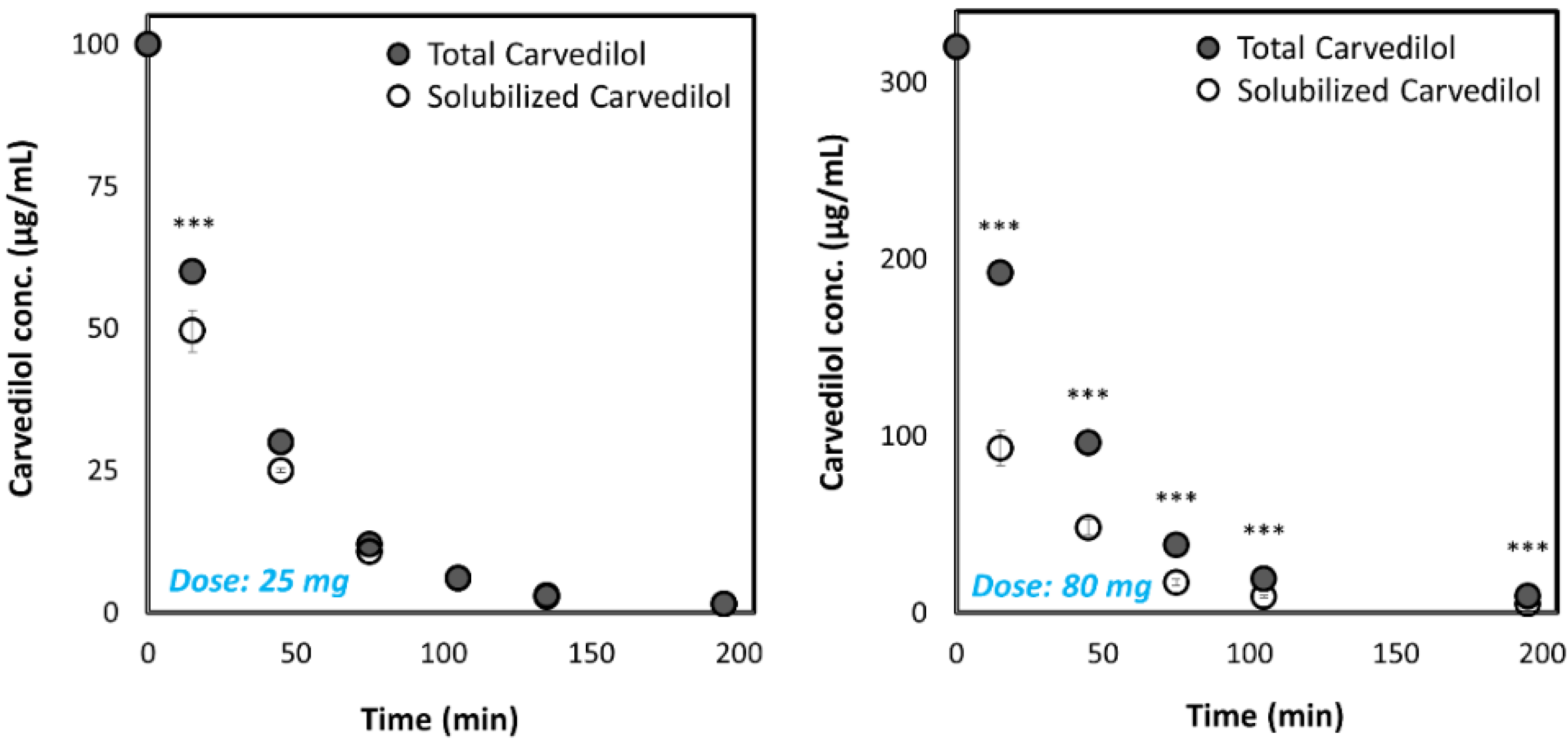
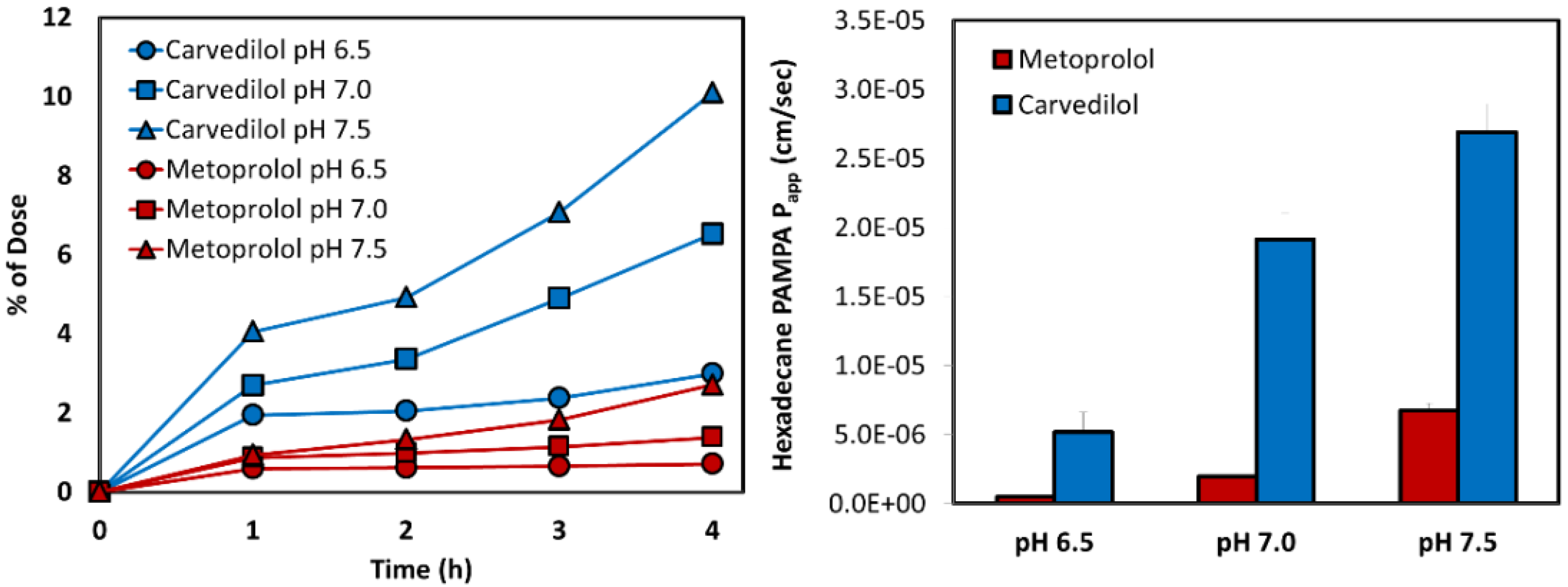
3.2. Biorelevant pH-Dilution Dissolution Studies
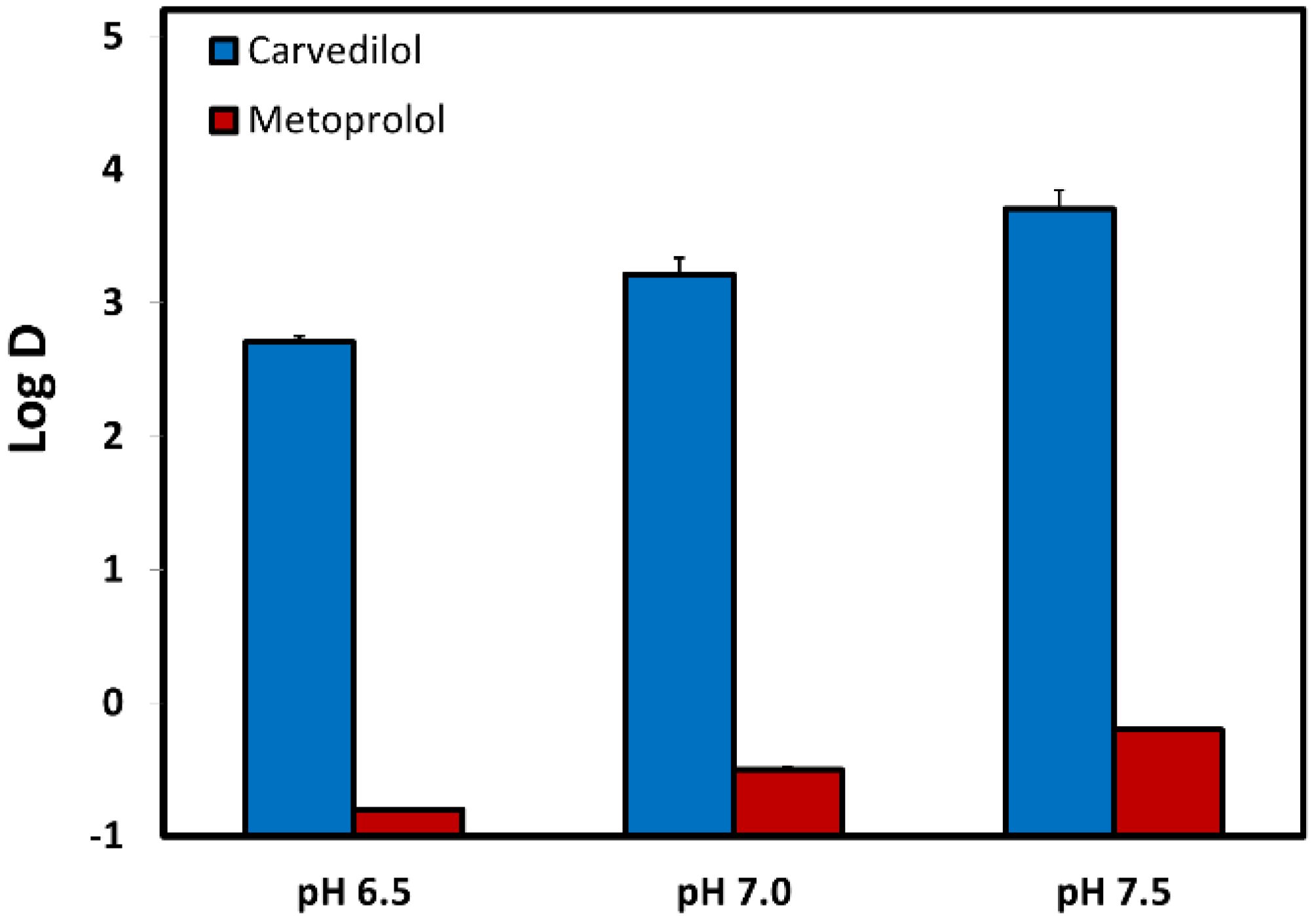
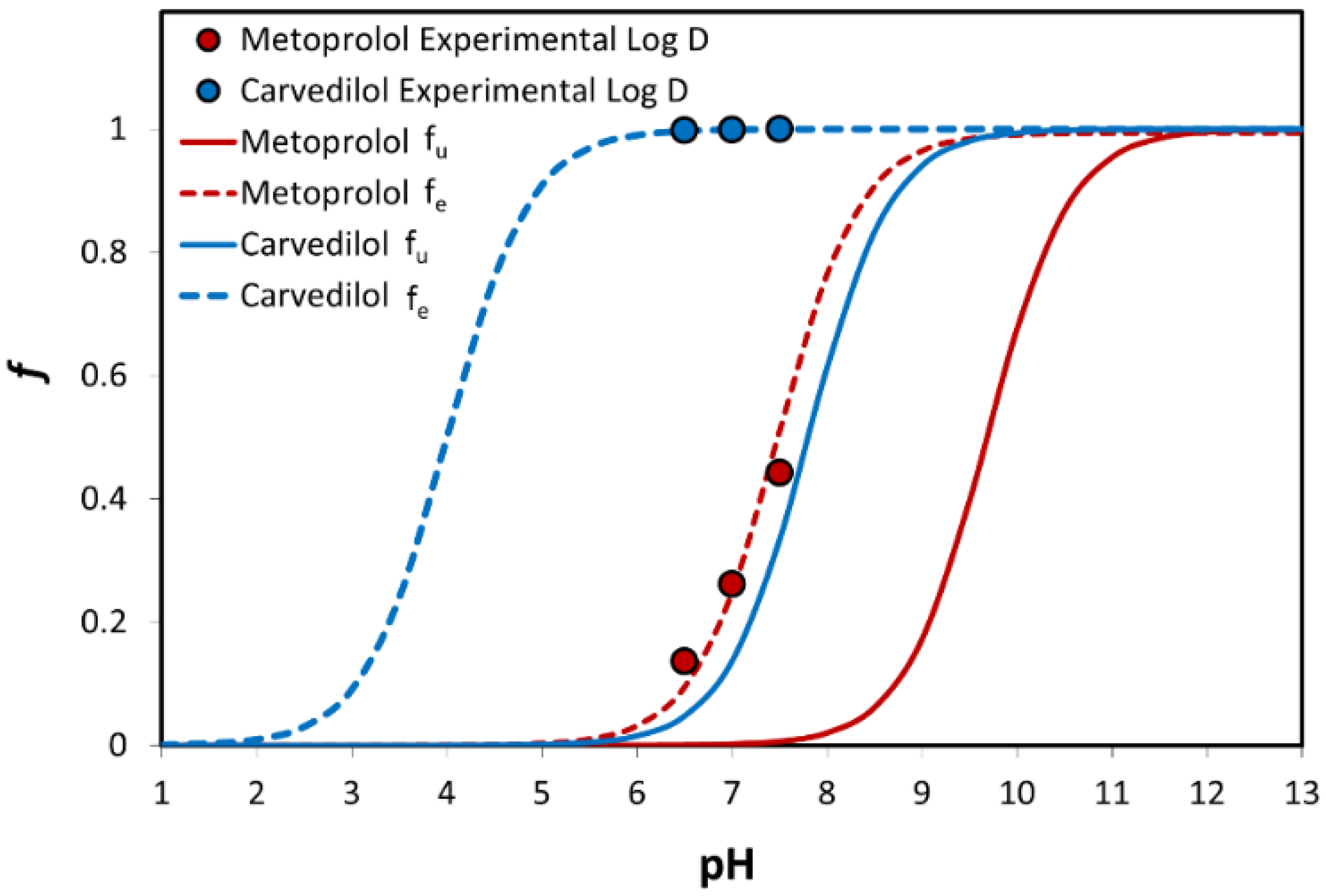
3.3. Log D
3.4. Physicochemical Analysis
3.5. PAMPA Assay
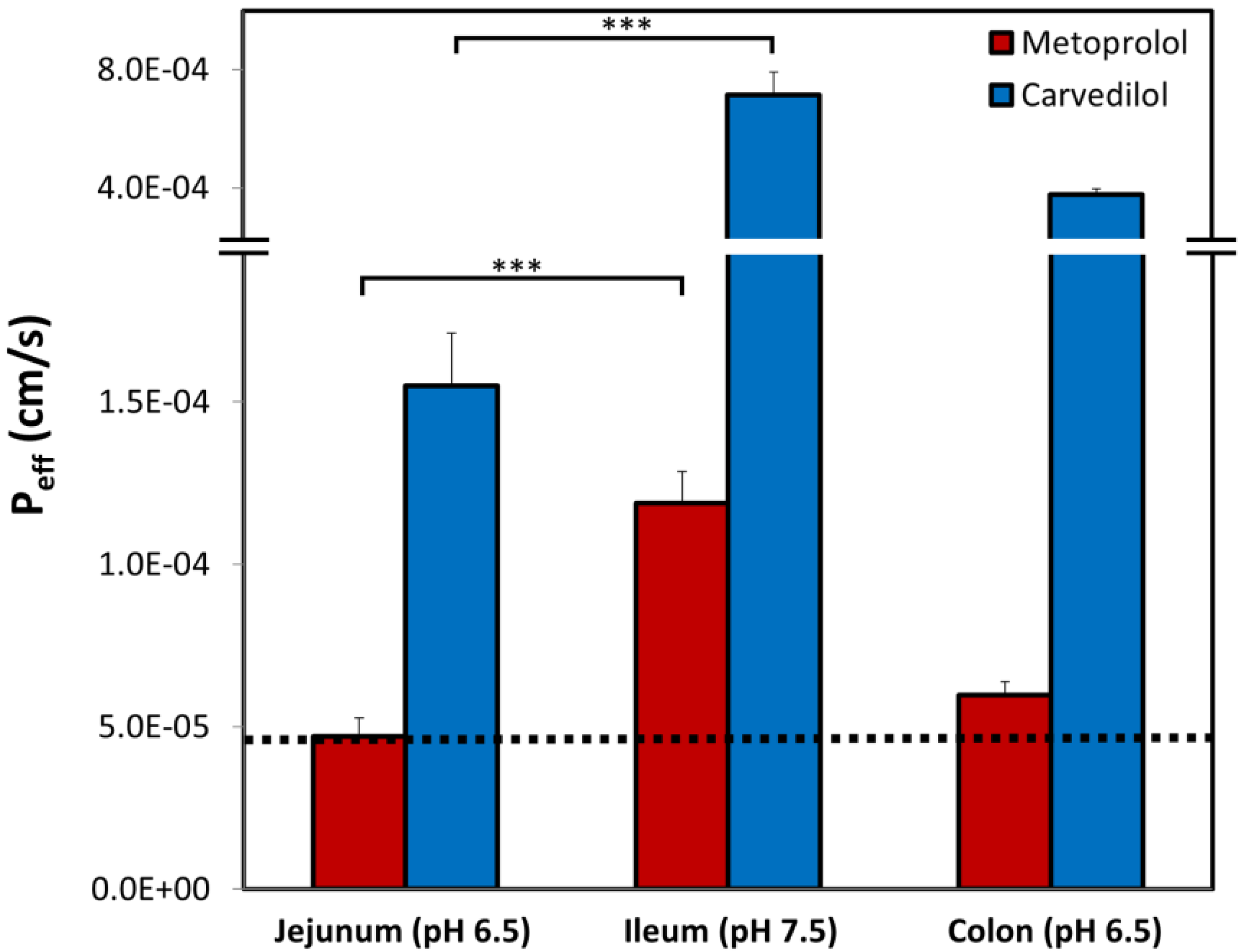
3.6. Rat Intestinal Perfusion Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dahan, A.; Miller, J.M.; Hilfinger, J.M.; Yamashita, S.; Yu, L.X.; Lennernas, H.; Amidon, G.L. High-permeability criterion for BCS classification: Segmental/pH dependent permeability considerations. Mol. Pharm. 2010, 7, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Lennernas, H. Intestinal Permeability and Drug Absorption: Predictive Experimental, Computational and In Vivo Approaches. Pharmaceutics 2019, 11, 411. [Google Scholar] [CrossRef] [PubMed]
- Fairstein, M.; Swissa, R.; Dahan, A. Regional-dependent intestinal permeability and BCS classification: Elucidation of pH-related complexity in rats using pseudoephedrine. AAPS J. 2013, 15, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernas, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Guideline on the Investigation of Bioequivalence; European Medicines Agency: Amsterdam, The Netherlands, 2010. [Google Scholar]
- U.S. Department of Health and Human Services, Food and Drug Administration; Center for Drug Evaluation and Research. Waiver of In-Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. Guidance for Industry; CDER: Silver Spring, MD, USA, 2017.
- Garcia-Arieta, A.; Gordon, J. Bioequivalence requirements in the European Union: Critical discussion. AAPS J. 2012, 14, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Wolk, O.; Zur, M.; Amidon, G.L.; Abrahamsson, B.; Cristofoletti, R.; Groot, D.W.; Kopp, S.; Langguth, P.; Polli, J.E.; et al. Biowaiver Monographs for Immediate-Release Solid Oral Dosage Forms: Codeine Phosphate. J. Pharm. Sci. 2014, 103, 1592–1600. [Google Scholar] [CrossRef]
- Ozawa, M.; Tsume, Y.; Zur, M.; Dahan, A.; Amidon, G.L. Intestinal permeability study of minoxidil: Assessment of minoxidil as a high permeability reference drug for biopharmaceutics classification. Mol. Pharm. 2015, 12, 204–211. [Google Scholar] [CrossRef]
- Dahan, A.; Lennernäs, H.; Amidon, G.L. The Fraction Dose Absorbed, in Humans, and High Jejunal Human Permeability Relationship. Mol. Pharm. 2012, 9, 1847–1851. [Google Scholar] [CrossRef]
- Lennernas, H. Regional intestinal drug permeation: Biopharmaceutics and drug development. Eur. J. Pharm. Sci. 2014, 57, 333–341. [Google Scholar] [CrossRef]
- Zur, M.; Gasparini, M.; Wolk, O.; Amidon, G.L.; Dahan, A. The Low/High BCS Permeability Class Boundary: Physicochemical Comparison of Metoprolol and Labetalol. Mol. Pharm. 2014, 11, 1707–1714. [Google Scholar] [CrossRef]
- Zur, M.; Hanson, A.S.; Dahan, A. The complexity of intestinal permeability: Assigning the correct BCS classification through careful data interpretation. Eur. J. Pharm. Sci 2014, 61, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Sabit, H.; Amidon, G.L. The H2 receptor antagonist nizatidine is a P-glycoprotein substrate: Characterization of its intestinal epithelial cell efflux transport. AAPS J. 2009, 11, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Lennernas, H. Human in vivo regional intestinal permeability: Importance for pharmaceutical drug development. Mol. Pharm. 2014, 11, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Ungell, A.L.; Nylander, S.; Bergstrand, S.; Sjoberg, A.; Lennernas, H. Membrane transport of drugs in different regions of the intestinal tract of the rat. J. Pharm. Sci. 1998, 87, 360–366. [Google Scholar] [CrossRef]
- Corrigan, O.I. The biopharmaceutic drug classification and drugs administered in extended release (ER) formulations. Adv. Exp. Med. Biol. 1997, 423, 111–128. [Google Scholar] [CrossRef]
- Tannergren, C.; Bergendal, A.; Lennernas, H.; Abrahamsson, B. Toward an increased understanding of the barriers to colonic drug absorption in humans: Implications for early controlled release candidate assessment. Mol. Pharm. 2009, 6, 60–73. [Google Scholar] [CrossRef]
- Xu, J.; Lin, Y.; Boulas, P.; Peterson, M.L. Low colonic absorption drugs: Risks and opportunities in the development of oral extended release products. Expert Opin. Drug Deliv. 2018, 15, 197–211. [Google Scholar] [CrossRef]
- Frishman, W.H. Carvedilol. N. Engl. J. Med. 1998, 339, 1759–1765. [Google Scholar] [CrossRef]
- Kukin, M.L. β-Blockers in Chronic Heart Failure: Considerations for Selecting an Agent. Mayo Clin. Proc. 2002, 77, 1199–1206. [Google Scholar] [CrossRef]
- Henderson, L.S.; Tenero, D.M.; Baidoo, C.A.; Campanile, A.M.; Harter, A.H.; Boyle, D.; Danoff, T.M. Pharmacokinetic and pharmacodynamic comparison of controlled-release carvedilol and immediate-release carvedilol at steady state in patients with hypertension. Am. J. Cardiol. 2006, 98, 17l–26l. [Google Scholar] [CrossRef]
- Packer, M.; Lukas, M.A.; Tenero, D.M.; Baidoo, C.A.; Greenberg, B.H. Pharmacokinetic profile of controlled-release carvedilol in patients with left ventricular dysfunction associated with chronic heart failure or after myocardial infarction. Am. J. Cardiol 2006, 98, 39l–45l. [Google Scholar] [CrossRef] [PubMed]
- Incecayir, T.; Tsume, Y.; Amidon, G.L. Comparison of the permeability of metoprolol and labetalol in rat, mouse, and Caco-2 cells: Use as a reference standard for BCS classification. Mol. Pharm. 2013, 10, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Brodde, O.E.; Kroemer, H.K. Drug-drug interactions of beta-adrenoceptor blockers. Arzneim. Forsch. 2003, 53, 814–822. [Google Scholar] [CrossRef]
- Baris, N.; Kalkan, S.; Guneri, S.; Bozdemir, V.; Guven, H. Influence of carvedilol on serum digoxin levels in heart failure: Is there any gender difference? Eur. J. Clin. Pharmacol. 2006, 62, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Giessmann, T.; Modess, C.; Hecker, U.; Zschiesche, M.; Dazert, P.; Kunert-Keil, C.; Warzok, R.; Engel, G.; Weitschies, W.; Cascorbi, I.; et al. CYP2D6 genotype and induction of intestinal drug transporters by rifampin predict presystemic clearance of carvedilol in healthy subjects. Clin. Pharmacol. Ther. 2004, 75, 213–222. [Google Scholar] [CrossRef]
- Hamed, R.; Awadallah, A.; Sunoqrot, S.; Tarawneh, O.; Nazzal, S.; AlBaraghthi, T.; Al Sayyad, J.; Abbas, A. pH-Dependent Solubility and Dissolution Behavior of Carvedilol-Case Example of a Weakly Basic BCS Class II Drug. AAPS PharmSciTech 2016, 17, 418–426. [Google Scholar] [CrossRef]
- Varma, M.V.; Gardner, I.; Steyn, S.J.; Nkansah, P.; Rotter, C.J.; Whitney-Pickett, C.; Zhang, H.; Di, L.; Cram, M.; Fenner, K.S.; et al. pH-Dependent Solubility and Permeability Criteria for Provisional Biopharmaceutics Classification (BCS and BDDCS) in Early Drug Discovery. Mol. Pharm. 2012, 9, 1199–1212. [Google Scholar] [CrossRef]
- Zur, M.; Cohen, N.; Agbaria, R.; Dahan, A. The biopharmaceutics of successful controlled release drug product: Segmental-dependent permeability of glipizide vs. metoprolol throughout the intestinal tract. Int. J. Pharm. 2015, 489, 304–310. [Google Scholar] [CrossRef]
- Beig, A.; Miller, J.M.; Lindley, D.; Dahan, A. Striking the Optimal Solubility-Permeability Balance in Oral Formulation Development for Lipophilic Drugs: Maximizing Carbamazepine Blood Levels. Mol. Pharm. 2017, 14, 319–327. [Google Scholar] [CrossRef]
- Fine-Shamir, N.; Beig, A.; Zur, M.; Lindley, D.; Miller, J.M.; Dahan, A. Toward Successful Cyclodextrin Based Solubility-Enabling Formulations for Oral Delivery of Lipophilic Drugs: Solubility–Permeability Trade-Off, Biorelevant Dissolution, and the Unstirred Water Layer. Mol. Pharm. 2017, 14, 2138–2146. [Google Scholar] [CrossRef]
- Fine-Shamir, N.; Dahan, A. Methacrylate-Copolymer Eudragit EPO as a Solubility-Enabling Excipient for Anionic Drugs: Investigation of Drug Solubility, Intestinal Permeability, and Their Interplay. Mol. Pharm. 2019, 16, 2884–2891. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; West, B.T.; Amidon, G.L. Segmental-dependent membrane permeability along the intestine following oral drug administration: Evaluation of a triple single-pass intestinal perfusion (TSPIP) approach in the rat. Eur. J. Pharm. Sci. 2009, 36, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Lozoya-Agullo, I.; Zur, M.; Fine-Shamir, N.; Markovic, M.; Cohen, Y.; Porat, D.; Gonzalez-Alvarez, I.; Gonzalez-Alvarez, M.; Merino-Sanjuan, M.; Bermejo, M.; et al. Investigating drug absorption from the colon: Single-pass vs. Doluisio approaches to in-situ rat large-intestinal perfusion. Int. J. Pharm. 2017, 527, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Wolk, O.; Markovic, M.; Porat, D.; Fine-Shamir, N.; Zur, M.; Beig, A.; Dahan, A. Segmental-Dependent Intestinal Drug Permeability: Development and Model Validation of In Silico Predictions Guided by In Vivo Permeability Values. J. Pharm. Sci. 2019, 108, 316–325. [Google Scholar] [CrossRef]
- Lozoya-Agullo, I.; Zur, M.; Beig, A.; Fine, N.; Cohen, Y.; Gonzalez-Alvarez, M.; Merino-Sanjuan, M.; Gonzalez-Alvarez, I.; Bermejo, M.; Dahan, A. Segmental-dependent permeability throughout the small intestine following oral drug administration: Single-pass vs. Doluisio approach to in-situ rat perfusion. Int. J. Pharm. 2016, 515, 201–208. [Google Scholar] [CrossRef]
- Lozoya-Agullo, I.; Zur, M.; Wolk, O.; Beig, A.; Gonzalez-Alvarez, I.; Gonzalez-Alvarez, M.; Merino-Sanjuan, M.; Bermejo, M.; Dahan, A. In-situ intestinal rat perfusions for human Fabs prediction and BCS permeability class determination: Investigation of the single-pass vs. the Doluisio experimental approaches. Int. J. Pharm. 2015, 480, 1–7. [Google Scholar] [CrossRef]
- Tugcu-Demiroz, F.; Gonzalez-Alvarez, I.; Gonzalez-Alvarez, M.; Bermejo, M. Validation of phenol red versus gravimetric method for water reabsorption correction and study of gender differences in Doluisio’s absorption technique. Eur. J. Pharm. Sci. 2014, 62, 105–110. [Google Scholar] [CrossRef]
- Wagner, J.G.; Sedman, A.J. Quantitaton of rate of gastrointestinal and buccal absorption of acidic and basic drugs based on extraction theory. J. Pharmacokinet. Biopharm. 1973, 1, 23–50. [Google Scholar] [CrossRef][Green Version]
- Winne, D. Shift of pH-absorption curves. J. Pharmacokinet. Biopharm. 1977, 5, 53–94. [Google Scholar] [CrossRef]
- Teksin, Z.S.; Hom, K.; Balakrishnan, A.; Polli, J.E. Ion pair-mediated transport of metoprolol across a three lipid-component PAMPA system. J. Control. Release 2006, 116, 50–57. [Google Scholar] [CrossRef]
- Tsume, Y.; Mudie, D.M.; Langguth, P.; Amidon, G.E.; Amidon, G.L. The Biopharmaceutics Classification System: Subclasses for in vivo predictive dissolution (IPD) methodology and IVIVC. Eur. J. Pharm. Sci. 2014, 57, 152–163. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Database. Carvedilol, CID=2585. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Carvedilol (accessed on 5 December 2019).
- Henchoz, Y.; Guillarme, D.; Martel, S.; Rudaz, S.; Veuthey, J.-L.; Carrupt, P.-A. Fast log P determination by ultra-high-pressure liquid chromatography coupled with UV and mass spectrometry detections. Anal. Bioanal. Chem. 2009, 394, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Database. Metoprolol, CID=4171. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Metoprolol (accessed on 5 December 2019).
- Lennernas, H. Human intestinal permeability. J. Pharm. Sci. 1998, 87, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Lennernas, H. Animal data: The contributions of the Ussing Chamber and perfusion systems to predicting human oral drug delivery in vivo. Adv. Drug Deliv. Rev. 2007, 59, 1103–1120. [Google Scholar] [CrossRef] [PubMed]
- COREG CR™ (Carvedilol Phosphate) Extended-Release Capsules Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/022012s003lbl.pdf (accessed on 13 January 2020).
- Metoprolol SuccinateTM Extended-Release Tablets, Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2006/019962s032lbl.pdf (accessed on 13 January 2020).
- Fagerberg, J.H.; Tsinman, O.; Sun, N.; Tsinman, K.; Avdeef, A.; Bergström, C.A.S. Dissolution rate and apparent solubility of poorly soluble drugs in biorelevant dissolution media. Mol. Pharm. 2010, 7, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Stappaerts, J.; Wuyts, B.; Tack, J.; Annaert, P.; Augustijns, P. Human and simulated intestinal fluids as solvent systems to explore food effects on intestinal solubility and permeability. Eur. J. Pharm. Sci. 2014, 63, 178–186. [Google Scholar] [CrossRef]
- Loftsson, T.; Vogensen, S.B.; Desbos, C.; Jansook, P. Carvedilol: Solubilization and cyclodextrin complexation: A technical note. AAPS PharmSciTech 2008, 9, 425–430. [Google Scholar] [CrossRef]
- Sutton, S.C. Role of physiological intestinal water in oral absorption. AAPS J. 2009, 11, 277–285. [Google Scholar] [CrossRef]
- Nolte, K.; Backfisch, G.; Neidlein, R. In vitro absorption studies with carvedilol using a new model with porcine intestine called BM-RIMO (Boehringer-Mannheim ring model). Arzneim. Forsch. 1999, 49, 745–749. [Google Scholar] [CrossRef]
- Dahan, A.; Miller, J.M.; Amidon, G.L. Prediction of solubility and permeability class membership: Provisional BCS classification of the world’s top oral drugs. AAPS J. 2009, 11, 740–746. [Google Scholar] [CrossRef]
- Kim, J.S.; Mitchell, S.; Kijek, P.; Tsume, Y.; Hilfinger, J.; Amidon, G.L. The suitability of an in situ perfusion model for permeability determinations: Utility for BCS class I biowaiver requests. Mol. Pharm. 2006, 3, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Jobin, G.; Cortot, A.; Godbillon, J.; Duval, M.; Schoeller, J.P.; Hirtz, J.; Bernier, J.J. Investigation of drug absorption from the gastrointestinal tract of man. I. Metoprolol in the stomach, duodenum and jejunum. Br. J. Clin. Pharmacol. 1985, 2, 97s–105s. [Google Scholar] [CrossRef] [PubMed]
- Masaoka, Y.; Tanaka, Y.; Kataoka, M.; Sakuma, S.; Yamashita, S. Site of drug absorption after oral administration: Assessment of membrane permeability and luminal concentration of drugs in each segment of gastrointestinal tract. Eur. J. Pharm. Sci. 2006, 29, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.S.; Hardy, J.G.; Fara, J.W. Transit of pharmaceutical dosage forms through the small intestine. Gut 1986, 27, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Bart, J.; Dijkers, E.C.F.; Wegman, T.D.; de Vries, E.G.E.; van der Graaf, W.T.A.; Groen, H.J.M.; Vaalburg, W.; Willemsen, A.T.M.; Hendrikse, N.H. New positron emission tomography tracer [11C] carvedilol reveals P-glycoprotein modulation kinetics. Br. J. Clin. Pharmacol. 2005, 145, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Wessler, J.D.; Grip, L.T.; Mendell, J.; Giugliano, R.P. The P-glycoprotein transport system and cardiovascular drugs. J. Am. Coll. Cardiol. 2013, 61, 2495–2502. [Google Scholar] [CrossRef] [PubMed]
- Dahan, A.; Sabit, H.; Amidon, G.L. Multiple Efflux Pumps Are Involved in the Transepithelial Transport of Colchicine: Combined Effect of P-Glycoprotein and Multidrug Resistance-Associated Protein 2 Leads to Decreased Intestinal Absorption Throughout the Entire Small Intestine. Drug Metab. Dispos. 2009, 37, 2028–2036. [Google Scholar] [CrossRef]
- MacLean, C.; Moenning, U.; Reichel, A.; Fricker, G. Closing the Gaps: A Full Scan of the Intestinal Expression of P-Glycoprotein, Breast Cancer Resistance Protein, and Multidrug Resistance-Associated Protein 2 in Male and Female Rats. Drug Metab. Dispos. 2008, 36, 1249–1254. [Google Scholar] [CrossRef]
- Dahan, A.; Amidon, G.L. Segmental Dependent Transport of Low Permeability Compounds along the Small Intestine Due to P-Glycoprotein: The Role of Efflux Transport in the Oral Absorption of BCS Class III Drugs. Mol. Pharm. 2008, 6, 19–28. [Google Scholar] [CrossRef]
- International Transporter Consortium; Giacomini, K.M.; Huang, S.-M.; Tweedie, D.J.; Benet, L.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar]
- Tubic-Grozdanis, M.; Hilfinger, J.M.; Amidon, G.L.; Kim, J.S.; Kijek, P.; Staubach, P.; Langguth, P. Pharmacokinetics of the CYP 3A Substrate Simvastatin following Administration of Delayed Versus Immediate Release Oral Dosage Forms. Pharm. Res. 2008, 25, 1591–1600. [Google Scholar] [CrossRef] [PubMed]






| pH | Solubility (mg/mL) | Corresponding D0 |
|---|---|---|
| 1.0 | 0.021 (±0.004) | 4.720 |
| 4.0 | 2.320 (±1.090) | 0.043 |
| 7.5 | 0.035 (±0.002) | 2.880 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markovic, M.; Zur, M.; Fine-Shamir, N.; Haimov, E.; González-Álvarez, I.; Dahan, A. Segmental-Dependent Solubility and Permeability as Key Factors Guiding Controlled Release Drug Product Development. Pharmaceutics 2020, 12, 295. https://doi.org/10.3390/pharmaceutics12030295
Markovic M, Zur M, Fine-Shamir N, Haimov E, González-Álvarez I, Dahan A. Segmental-Dependent Solubility and Permeability as Key Factors Guiding Controlled Release Drug Product Development. Pharmaceutics. 2020; 12(3):295. https://doi.org/10.3390/pharmaceutics12030295
Chicago/Turabian StyleMarkovic, Milica, Moran Zur, Noa Fine-Shamir, Ester Haimov, Isabel González-Álvarez, and Arik Dahan. 2020. "Segmental-Dependent Solubility and Permeability as Key Factors Guiding Controlled Release Drug Product Development" Pharmaceutics 12, no. 3: 295. https://doi.org/10.3390/pharmaceutics12030295
APA StyleMarkovic, M., Zur, M., Fine-Shamir, N., Haimov, E., González-Álvarez, I., & Dahan, A. (2020). Segmental-Dependent Solubility and Permeability as Key Factors Guiding Controlled Release Drug Product Development. Pharmaceutics, 12(3), 295. https://doi.org/10.3390/pharmaceutics12030295