piggyBac-Based Non-Viral In Vivo Gene Delivery Useful for Production of Genetically Modified Animals and Organs



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
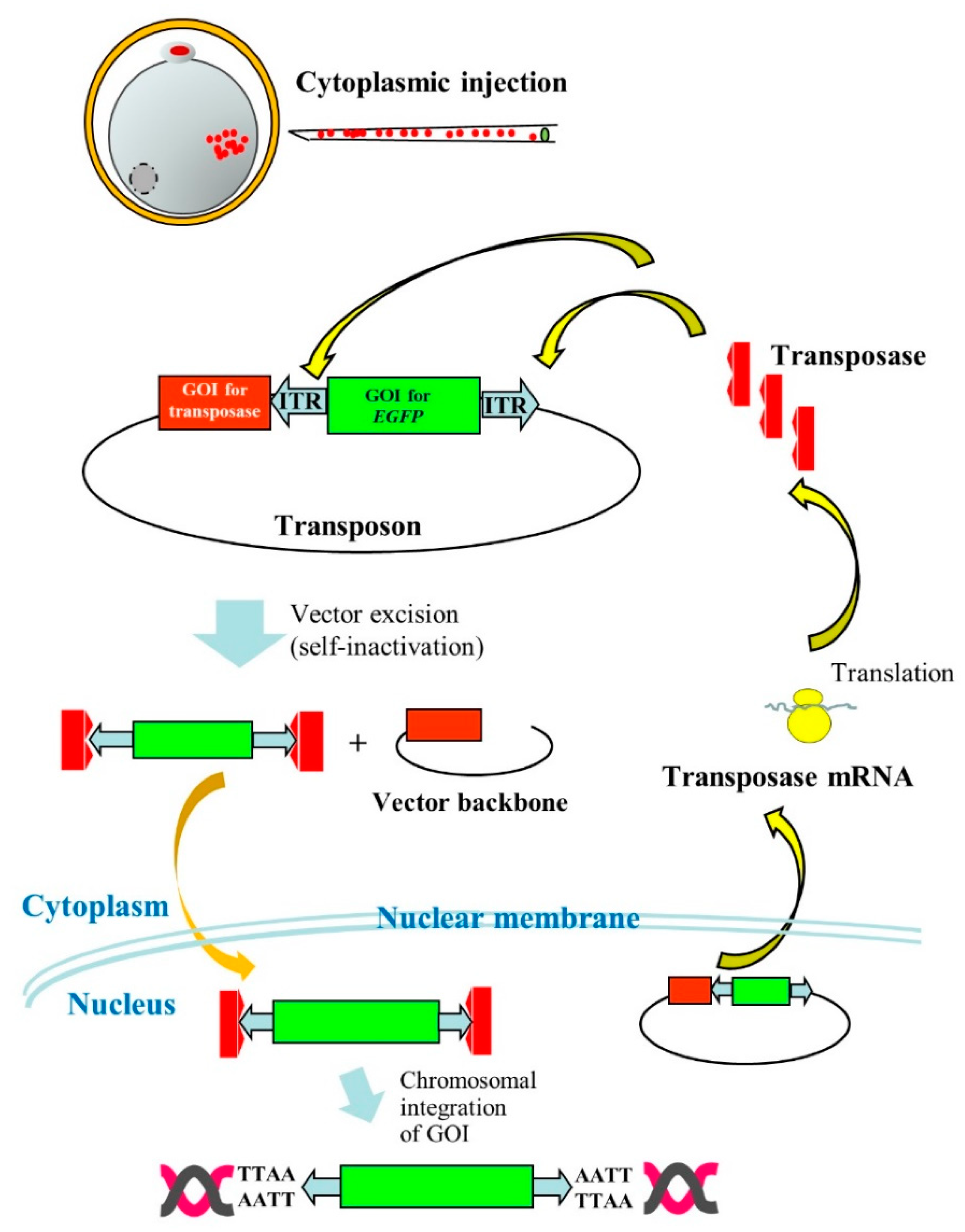
1. Introduction
2. Diverse Roles of piggyBac (PB)
2.1. Systemic Gene Delivery via Tail-Vein Injection of PB
2.2. Useful for Regulated Gene Expression In Vivo
2.3. Useful for Transgenic (Tg) Animal Production
2.4. Focal In Vivo PB Gene Delivery
2.4.1. Gene Delivery to Pancreas
2.4.2. Gene Delivery to Spleen
2.4.3. Gene Delivery to Oviducts
2.4.4. Gene Delivery to Muscle
2.4.5. Gene Delivery to Tail
2.4.6. Gene Delivery to Bladder
2.4.7. Gene Delivery to Brain
2.4.8. Gene Delivery to Kidney
2.4.9. Gene Delivery to Mammary Gland
2.4.10. Gene Delivery to Immune Cells
2.5. In Utero Gene Delivery
2.6. Application to Gene Therapy
3. Improvement of PB
3.1. Super PB Transposase
3.2. PB Transposase mRNA
3.3. Modification of Inverted Terminal Repeat (ITR)
3.4. Use of Insulators
3.5. Use of Epigenetic Regulatory Element
3.6. Hybrid Non-Viral/Viral Vector System
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated viral |
| ABCB4 | ATP binding cassette subfamily B member 4 |
| AV | Adenoviral |
| BAC | Bacterial artificial chromosome |
| CAG | Chicken β-actin-based promoter |
| CARs | Chimeric antigen receptors |
| CAT | Chloramphenicol acetyltransferase gene |
| CI | Cytoplasmic injection |
| CMV | Cytomegalovirus |
| DMD | Duchenne muscular dystrophy |
| DT-A | Diphtheria toxin-A chain |
| EGFP | Enhanced green fluorescent protein |
| EP | Electroporation |
| ES | Embryonic stem |
| FVIII | Factor VIII |
| GDNF | Glial cell line-derived neurotrophic factor |
| GFP | Green fluorescent protein |
| GLuc | Secretory Gaussia luciferase |
| GM | Genetically modified |
| gMAR | β-globin MAR |
| GOI | Gene of interest |
| HGD | Hydrodynamics gene delivery |
| IGF-1R | Insulin-like growth factor-1 receptor |
| iMAR | β-interferon MAR |
| iPS | Inducible pluripotent stem |
| ITR | Inverted terminal repeats |
| IUE | In utero EP |
| LacZ | Gene coding for β-galactosidase |
| LV | Lentiviral |
| MABs | Mesoangioblasts |
| MAR | Matrix attachment regions |
| MaSC | Mammary stem cell |
| MCDs | Malformations of cortical development |
| mCherry | Red fluorescent protein |
| NAs | Nucleic acids |
| NCre | Gene coding for Cre with nuclear localization signal |
| NOD-SCID | Non-obese diabetic-severe combined immunodeficiency |
| pA | Poly(A) sites |
| Pan | Pancreas |
| PB | piggyBac |
| PFIC3 | Progressive familial intrahepatic cholestasis type 3 |
| PI | Pronuclear microinjection |
| rAAV | Recombinant AAV |
| RFP | Red fluorescent protein |
| SB | Sleeping Beauty |
| SCNT | Somatic cell nuclear transfer |
| shRNA | Small hairpin RNA |
| Sp | Spleen |
| SV | Simian virus |
| Tg | Transgenic |
| TR | Transthyretin promoter |
| UUO | Unilateral ureteral obstruction |
| VE | Vascular endothelial |
References
- Chen, Y.H.; Keiser, M.S.; Davidson, B.L. Viral vectors for gene transfer. Curr. Protoc. Mouse Biol. 2018, 8, e58. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.; Gao, Y.-G.; Lin, X.; Li, Y.; Dang, K.; Tian, Y.; Zhang, W.-J.; Jiang, S.-F.; Qadir, A.; Qian, A.-R. The Development of functional non-viral vectors for gene delivery. Int. J. Mol. Sci. 2019, 20, 5491. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Song, Y.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999, 6, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Maehara, T.; Watanabe, S.; Ishihara, M.; Sato, M. Liver lobe and strain difference in gene expression after hydrodynamics-based gene delivery in mice. Anim. Biotechnol. 2015, 26, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Tipanee, J.; Chai, Y.C.; VandenDriessche, T.; Chuah, M.K. Preclinical and clinical advances in transposon-based gene therapy. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Di Matteo, M.; Mátrai, J.; Belay, E.; Firdissa, T.; Vandendriessche, T.; Chuah, M.K. PiggyBac toolbox. Methods Mol. Biol. 2012, 859, 241–254. [Google Scholar] [CrossRef]
- Nakamura, S.; Watanabe, S.; Ando, N.; Ishihara, M.; Sato, M. Transplacental gene delivery (TPGD) as a noninvasive tool for fetal gene manipulation in mice. Int. J. Mol. Sci. 2019, 20, 5926. [Google Scholar] [CrossRef]
- Ivics, Z.; Izsvák, Z. The expanding universe of transposon technologies for gene and cell engineering. Mob. DNA 2010, 1, 25. [Google Scholar] [CrossRef]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef]
- Narayanavari, S.A.; Chilkunda, S.S.; Ivics, Z.; Izsvák, Z. Sleeping Beauty transposition: From biology to applications. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 18–44. [Google Scholar] [CrossRef]
- Mirzaei, H.; Sahebkar, A.; Jaafari, M.R.; Hadjati, J.; Javanmard, S.H.; Mirzaei, H.R.; Salehi, R. PiggyBac as a novel vector in cancer gene therapy: Current perspective. Cancer Gene Ther. 2016, 23, 45–47. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.J.; Cary, L.; Boonvisudhi, K.; Wang, H.G. Assay for movement of lepidopteran transposon IFP2 in insect cells using a baculovirus genome as a target DNA. Virology 1995, 211, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.J.; Ciszczon, T.; Elick, T.; Bauser, C. Precise excision of TTAA-specific lepidopteran transposons piggyBac (IFP2) and tagalong (TFP3) from the baculovirus genome in cell lines from two species of Lepidoptera. Insect Mol. Biol. 1996, 5, 141–151. [Google Scholar] [CrossRef]
- Saridey, S.K.; Liu, L.; Doherty, J.E.; Kaja, A.; Galvan, D.L.; Fletcher, B.S.; Wilson, M.H. PiggyBac transposon-based inducible gene expression in vivo after somatic cell gene transfer. Mol. Ther. 2009, 17, 2115–2120. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Higuchi, Y.; Kawakami, S.; Yamashita, F.; Hashida, M. PiggyBac Transposon-mediated long-term gene expression in mice. Mol. Ther. 2010, 18, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Cooney, A.L.; Singh, B.K.; Sinn, P.L. Hybrid nonviral/viral vector systems for improved piggyBac DNA transposon in vivo delivery. Mol. Ther. 2015, 23, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Li, M.A.; Turner, D.J.; Ning, Z.; Yusa, K.; Liang, Q.; Eckert, S.; Rad, L.; Fitzgerald, T.W.; Craig, N.L.; Bradley, A. Mobilization of giant piggyBac transposons in the mouse genome. Nucleic Acids Res. 2011, 39, e148. [Google Scholar] [CrossRef] [PubMed]
- Katter, K.; Geurts, A.M.; Hoffmann, O.; Mátés, L.; Landa, V.; Hiripi, L.; Moreno, C.; Lazar, J.; Bashir, S.; Zidek, V.; et al. Transposon-mediated transgenesis, transgenic rescue, and tissue-specific gene expression in rodents and rabbits. FASEB J. 2013, 27, 930–941. [Google Scholar] [CrossRef]
- Chapeau, E.A.; Gembarska, A.; Durand, E.Y.; Mandon, E.; Estadieu, C.; Romanet, V.; Wiesmann, M.; Tiedt, R.; Lehar, J.; de Weck, A.; et al. Resistance mechanisms to TP53-MDM2 inhibition identified by in vivo piggyBac transposon mutagenesis screen in an Arf-/- mouse model. Proc. Natl. Acad. Sci. USA 2017, 114, 3151–3156. [Google Scholar] [CrossRef]
- Woodard, L.E.; Wilson, M.H. piggyBac-ing models and new therapeutic strategies. Trends Biotechnol. 2015, 33, 525–533. [Google Scholar] [CrossRef]
- Li, X.; Burnight, E.R.; Cooney, A.L.; Malani, N.; Brady, T.; Sander, J.D.; Staber, J.; Wheelan, S.J.; Joung, J.K.; McCray, P.B., Jr.; et al. piggyBac transposase tools for genome engineering. Proc. Natl. Acad. Sci. USA 2013, 110, E2279–E2287. [Google Scholar] [CrossRef]
- Kesselring, L.; Miskey, C.; Zuliani, C.; Querques, I.; Kapitonov, V.; Laukó, A.; Fehér, A.; Palazzo, A.; Diem, T.; Lustig, J.; et al. A single amino acid switch converts the Sleeping Beauty transposase into an efficient unidirectional excisionase with utility in stem cell reprogramming. Nucleic Acids Res. 2020, 48, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Q.; Yu, X.; Li, Y.; Guo, Y.; Liu, Z.; Sun, F.; Hou, W.; Li, C.; Wu, L.; et al. HIV-1 inhibition in cells with CXCR4 mutant genome created by CRISPR-Cas9 and piggyBac recombinant technologies. Sci. Rep. 2018, 8, 8573. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, Z.R.; Yue, R.; Fu, Y.L.; Liu, Z.Y.; Feng, H.Y.; Li, J.G.; Han, S.Y. PiggyBac transposon system with polymeric gene carrier transfected into human T cells. Am. J. Transl. Res. 2019, 11, 7126–7136. [Google Scholar] [PubMed]
- Alessio, A.P.; Fili, A.E.; Garrels, W.; Forcato, D.O.; Olmos Nicotra, M.F.; Liaudat, A.C.; Bevacqua, R.J.; Savy, V.; Hiriart, M.I.; Talluri, T.R.; et al. Establishment of cell-based transposon-mediated transgenesis in cattle. Theriogenology 2016, 85, 1297–1311.e2. [Google Scholar] [CrossRef]
- Bai, D.-P.; Yang, M.-M.; Chen, Y.-L. PiggyBac transposon-mediated gene transfer in Cashmere goat fetal fibroblast cells. Biosci. Biotechnol. Biochem. 2012, 76, 933–937. [Google Scholar] [CrossRef]
- Kim, S.J.; Kwon, H.S.; Kwon, D.K.; Koo, O.J.; Moon, J.H.; Park, E.J.; Yum, S.Y.; Lee, B.C.; Jang, G. Production of transgenic porcine embryos reconstructed with induced pluripotent stem-like cells derived from porcine endogenous factors using piggyBac system. Cell. Reprogram. 2019, 21, 26–36. [Google Scholar] [CrossRef]
- Sato, M.; Maeda, K.; Koriyama, M.; Inada, E.; Saitoh, I.; Miura, H.; Ohtsuka, M.; Nakamura, S.; Sakurai, T.; Watanabe, S.; et al. The piggyBac-based gene delivery system can confer successful production of cloned porcine blastocysts with multigene constructs. Int. J. Mol. Sci. 2016, 17, E1424. [Google Scholar] [CrossRef]
- Miura, H.; Inoko, H.; Inoue, I.; Okada, Y.; Tanaka, M.; Sato, M.; Ohtsuka, M. PiggyBac-mediated generation of stable transfectants with surface HLA expression from a small number of cells. Anal. Biochem. 2013, 437, 29–31. [Google Scholar] [CrossRef]
- Behringer, R.; Gertsenstein, M.; Nagy, K.V.; Nagy, A. Integrating piggyBac transposon transgenes into mouse fibroblasts using chemical methods. Cold Spring Harb. Protoc. 2017. [Google Scholar] [CrossRef]
- Wang, G.; Yang, L.; Grishin, D.; Rios, X.; Ye, L.Y.; Hu, Y.; Li, K.; Zhang, D.; Church, G.M.; Pu, W.T. Efficient, footprint-free human iPSC genome editing by consolidation of Cas9/CRISPR and piggyBac technologies. Nat. Protoc. 2017, 12, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. PiggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Woltjen, K.; Kim, S.I.; Nagy, A. The piggyBac transposon as a platform technology for somatic cell reprogramming studies in mouse. Methods Mol. Biol. 2016, 1357, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Oceguera-Yanez, F.; Sakurai, C.; Nakagawa, M.; Yamanaka, S.; Woltjen, K. Inducible transgene expression in human iPS cells using versatile all-in-one piggyBac transposons. Methods Mol. Biol. 2016, 1357, 111–131. [Google Scholar] [CrossRef]
- Ding, S.; Wu, X.; Li, G.; Han, M.; Zhuang, Y.; Xu, T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 2005, 122, 473–483. [Google Scholar] [CrossRef]
- Li, T.; Shuai, L.; Mao, J.; Wang, X.; Wang, M.; Zhang, X.; Wang, L.; Li, Y.; Li, W.; Zhou, Q. Efficient production of fluorescent transgenic rats using the piggyBac transposon. Sci. Rep. 2016, 6, 33225. [Google Scholar] [CrossRef]
- Bai, D.P.; Yang, M.M.; Qu, L.; Chen, Y.L. Generation of a transgenic cashmere goat using the piggyBac transposition system. Theriogenology 2017, 93, 1–6. [Google Scholar] [CrossRef]
- Yum, S.Y.; Lee, S.J.; Park, S.G.; Shin, I.G.; Hahn, S.E.; Choi, W.J.; Kim, H.S.; Kim, H.J.; Bae, S.H.; Lee, J.H.; et al. Long-term health and germline transmission in transgenic cattle following transposon-mediated gene transfer. BMC Genomics 2018, 19, 387. [Google Scholar] [CrossRef]
- Lu, I.L.; Chen, C.; Tung, C.Y.; Chen, H.H.; Pan, J.P.; Chang, C.H.; Cheng, J.S.; Chen, Y.A.; Wang, C.H.; Huang, C.W.; et al. Identification of genes associated with cortical malformation using a transposon-mediated somatic mutagenesis screen in mice. Nat. Commun. 2018, 9, 2498. [Google Scholar] [CrossRef]
- Inada, E.; Saitoh, I.; Kubota, N.; Iwase, Y.; Kiyokawa, Y.; Shibasaki, S.; Noguchi, H.; Yamasaki, Y.; Sato, M. piggyBac transposon-based immortalization of human deciduous tooth dental pulp cells with multipotencyandnon-tumorigenicpotential. Int. J. Mol. Sci. 2019, 20, 4904. [Google Scholar] [CrossRef]
- Sato, M.; Saitoh, I.; Inada, E.; Nakamura, S.; Watanabe, S. Potential for isolation of immortalized hepatocyte cell lines by liver-directed in vivo gene delivery of transposons in mice. Stem Cells Int. 2019, 5129526. [Google Scholar] [CrossRef] [PubMed]
- Woodard, L.E.; Cheng, J.; Welch, R.C.; Williams, F.M.; Luo, W.; Gewin, L.S.; Wilson, M.H. Kidney-specific transposon-mediated gene transfer in vivo. Sci. Rep. 2017, 7, 44904. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Ishihara, M.; Watanabe, S.; Ando, N.; Ohtsuka, M.; Sato, M. Intravenous delivery of piggyBac transposons as a useful tool for liver-specific gene-switching. Int. J. Mol. Sci. 2018, 19, E3452. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Scangos, G.A.; Plotkin, D.J.; Barbosa, J.A.; Ruddle, F.H. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 7380–7384. [Google Scholar] [CrossRef]
- Dunn, D.A.; Pinkert, C.A.; Kooyman, D.L. Foundation Review: Transgenic animals and their impact on the drug discovery industry. Drug Discov. Today 2005, 10, 757–767. [Google Scholar] [CrossRef]
- Houdebine, L.M. Transgenic animal models in biomedical research. Methods Mol. Biol. 2007, 360, 163–202. [Google Scholar] [CrossRef]
- Hammer, R.E.; Pursel, V.G.; Rexroad, C.E., Jr.; Wall, R.J.; Bolt, D.J.; Ebert, K.M.; Palmiter, R.D.; Brinster, R.L. Production of transgenic rabbits, sheep and pigs by microinjection. Nature 1985, 315, 680–683. [Google Scholar] [CrossRef]
- Iqbal, K.; Barg-Kues, B.; Broll, S.; Bode, J.; Niemann, H.; Kues, W.A. Cytoplasmic injection of circular plasmids allows targeted expression in mammalian embryos. BioTechniques 2009, 47, 959–968. [Google Scholar] [CrossRef]
- Dunlap-Brown, M.; Butler, S.P.; Velander, W.H.; Gwazdauskas, F.C. Murine embryo development following cytoplasmic injection of linear and condensed DNA. Open J. Anim. Sci. 2012, 2, 23961. [Google Scholar] [CrossRef]
- Li, Z.; Zeng, F.; Meng, F.; Xu, Z.; Zhang, X.; Huang, X.; Tang, F.; Gao, W.; Shi, J.; He, X.; et al. Generation of transgenic pigs by cytoplasmic injection of piggyBac transposase-based pmGENIE-3 plasmids. Biol. Reprod. 2014, 90, 93. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lin, C.; Lu, D.; Ning, Z.; Cox, T.; Melvin, D.; Wang, X.; Bradley, A.; Liu, P. Chromosomal transposition of PiggyBac in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9290–9295. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Bradley, A.; Huang, Y. A piggyBac transposon-based genome-wide library of insertionally mutated Blm-deficient murine ES cells. Genome Res. 2009, 19, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Kong, J.; Stalker, J.; Bradley, A. Chromosomal mobilization and reintegration of Sleeping Beauty and PiggyBac transposons. Genesis 2009, 47, 404–408. [Google Scholar] [CrossRef]
- Jiang, M.G.; Li, T.; Feng, C.; Fu, R.; Yuan, Y.; Zhou, Q.; Li, X.; Wan, H.; Wang, L.; Li, W.; et al. Generation of transgenic rats through induced pluripotent stem cells. J. Biol. Chem. 2013, 288, 27150–27158. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Xu, Z.; Zou, X.; Zeng, F.; Shi, J.; Liu, D.; Urschitz, J.; Moisyadi, S.; Li, Z. Pig transgenesis by piggyBac transposition in combination with somatic cell nuclear transfer. Transgenic Res. 2013, 22, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Inada, E.; Saitoh, I.; Nakamura, S.; Watanabe, S. In Vivo piggyBac-based gene delivery towards murine pancreatic parenchyma confers sustained expression of gene of interest. Int. J. Mol. Sci. 2019, 20, 3116. [Google Scholar] [CrossRef]
- Tupin, E.; Poirier, B.; Bureau, M.F.; Khallou-Laschet, J.; Vranckx, R.; Caligiuri, G.; Gaston, A.T.; Duong Van Huyen, J.P.; Scherman, D.; Bariéty, J.; et al. Non-viral gene transfer of murine spleen cells achieved by in vivo electroporation. Gene Ther. 2003, 10, 569–579. [Google Scholar] [CrossRef][Green Version]
- Pillai, V.V.; Weber, D.M.; Phinney, B.S.; Selvaraj, V. Profiling of proteins secreted in the bovine oviduct reveals diverse functions of this luminal microenvironment. PLoS ONE 2017, 12, e0188105. [Google Scholar] [CrossRef]
- Relloso, M.; Esponda, P. In-vivo transfection of the female reproductive tract epithelium. Mol. Hum. Reprod. 2000, 6, 1099–1105. [Google Scholar] [CrossRef][Green Version]
- Sato, M. Intraoviductal introduction of plasmid DNA and subsequent electroporation for efficient in vivo gene transfer to murine oviductal epithelium. Mol. Reprod. Dev. 2005, 71, 321–330. [Google Scholar] [CrossRef]
- Loperfido, M.; Jarmin, S.; Dastidar, S.; Di Matteo, M.; Perini, I.; Moore, M.; Nair, N.; Samara-Kuko, E.; Athanasopoulos, T.; Tedesco, F.S.; et al. piggyBac transposons expressing full-length human dystrophin enable genetic correction of dystrophic mesoangioblasts. Nucleic Acids Res. 2016, 44, 744–760. [Google Scholar] [CrossRef]
- Iyer, P.S.; Mavoungou, L.O.; Ronzoni, F.; Zemla, J.; Schmid-Siegert, E.; Antonini, S.; Neff, L.A.; Dorchies, O.M.; Jaconi, M.; Lekka, M.; et al. Autologous cell therapy approach for Duchenne muscular dystrophy using piggyBac transposons and mesoangioblasts. Mol. Ther. 2018, 26, 1093–1108. [Google Scholar] [CrossRef]
- Ley, D.; Van Zwieten, R.; Puttini, S.; Iyer, P.; Cochard, A.; Mermod, N. A piggyBac-mediated approach for muscle gene transfer or cell therapy. Stem Cell Res. 2014, 13, 390–403. [Google Scholar] [CrossRef]
- Troyanovsky, B.; Bitko, V.; Pastukh, V.; Fouty, B.; Solodushko, V. The Functionality of minimal piggyBac transposons in mammalian cells. Mol. Ther. Nucleic Acids 2016, 5, e369. [Google Scholar] [CrossRef]
- Yu, C.; Stefanson, O.; Liu, Y.; Wang, Z.A. Novel method of plasmid DNA delivery to mouse bladder urothelium by electroporation. J. Vis. Exp. 2018, 57649. [Google Scholar] [CrossRef]
- Akhtar, A.A.; Gowing, G.; Kobritz, N.; Savinoff, S.E.; Garcia, L.; Saxon, D.; Cho, N.; Kim, G.; Tom, C.M.; Park, H.; et al. Inducible expression of GDNF in transplanted iPSC-derived neural progenitor cells. Stem Cell Rep. 2018, 10, 1696–1704. [Google Scholar] [CrossRef]
- Liang, M.; Woodard, L.E.; Liang, A.; Luo, J.; Wilson, M.H.; Mitch, W.E.; Cheng, J. Protective role of insulin-like growth factor-1 receptor in endothelial cells against unilateral ureteral obstruction-induced renal fibrosis. Am. J. Pathol. 2015, 185, 1234–1250. [Google Scholar] [CrossRef]
- Tagaya, H.; Ishikawa, K.; Hosokawa, Y.; Kobayashi, S.; Ueoka, Y.; Shimada, M.; Ohashi, Y.; Mikami, H.; Yamamoto, M.; Ihara, T.; et al. A method of producing genetically manipulated mouse mammary gland. Breast Cancer Res. 2019, 21, 1–12. [Google Scholar] [CrossRef]
- Smith, T.T.; Stephan, S.B.; Moffett, H.F.; McKnight, L.E.; Ji, W.; Reiman, D.; Bonagofski, E.; Wohlfahrt, M.E.; Pillai, S.P.S.; Stephan, M.T. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017, 12, 813–820. [Google Scholar] [CrossRef]
- O’Neil, R.T.; Saha, S.; Veach, R.A.; Welch, R.C.; Woodard, L.E.; Rooney, C.M.; Wilson, M.H. Transposon-modified antigen-specific T lymphocytes for sustained therapeutic protein delivery in vivo. Nat. Commun. 2018, 9, 1325. [Google Scholar] [CrossRef]
- Saito, T.; Nakatsuji, N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol. 2001, 240, 237–246. [Google Scholar] [CrossRef]
- Tabata, H.; Nakajima, K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: Visualization of neuronal migration in the developing cortex. Neuroscience 2001, 103, 865–872. [Google Scholar] [CrossRef]
- Szczurkowska, J.; Cwetsch, A.W.; dal Maschio, M.; Ghezzi, D.; Ratto, G.M.; Cancedda, L. Targeted in vivo genetic manipulation of the mouse or rat brain by in utero electroporation with a triple-electrode probe. Nat. Protoc. 2016, 11, 399–412. [Google Scholar] [CrossRef]
- Rosin, J.M.; Kurrasch, D.M. In utero electroporation induces cell death and alters embryonic microglia morphology and expression signatures in the developing hypothalamus. J. Neuroinflamm. 2018, 15, 181. [Google Scholar] [CrossRef]
- Huang, C.C.; Carcagno, A. Electroporation of postimplantation mouse embryos in utero. Cold Spring Harb. Protoc. 2018, 2018. [Google Scholar] [CrossRef]
- Sato, M.; Tanigawa, M.; Kikuchi, N. Non-viral gene transfer to surface skin of mid-gestational murine embryos by intraamniotic injection and subsequent electroporation. Mol. Reprod. Dev. 2004, 69, 268–277. [Google Scholar] [CrossRef]
- Henriques-Coelho, T.; Gonzaga, S.; Endo, M.; Zoltick, P.W.; Davey, M.; Leite-Moreira, A.F.; Correia-Pinto, J.; Flake, A.W. Targeted gene transfer to fetal rat lung interstitium by ultrasound-guided intrapulmonary injection. Mol. Ther. 2007, 15, 340–347. [Google Scholar] [CrossRef]
- Garcia-Frigola, C.; Carreres, M.I.; Vegar, C.; Herrera, E. Gene delivery into mouse retinal ganglion cells by in utero electroporation. BMC Dev. Biol. 2007, 7, 103. [Google Scholar] [CrossRef]
- Chen, F.; LoTurco, J. A method for stable transgenesis of radial glia lineage in rat neocortex by piggyBac mediated transposition. J. Neurosci. Methods 2012, 207, 172–180. [Google Scholar] [CrossRef]
- Chen, F.; Maher, B.J.; LoTurco, J.J. piggyBac transposon-mediated cellular transgenesis in mammalian forebrain by in utero electroporation. Cold Spring Harb. Protoc. 2014, 7, 741–749. [Google Scholar] [CrossRef]
- Matsui, H.; Fujimoto, N.; Sasakawa, N.; Ohinata, Y.; Shima, M.; Yamanaka, S.; Sugimoto, M.; Hotta, A. Delivery of full length factor VIII using a piggyBac transposon vector to correct a mouse model of hemophilia A. PLoS ONE 2014, 9, e104957. [Google Scholar] [CrossRef]
- Staber, J.M.; Pollpeter, M.J.; Arensdorf, A.; Sinn, P.L.; Rutkowski, D.T.; McCray, P.B., Jr. piggyBac-mediated phenotypic correction of factor VIII deficiency. Mol. Ther. Methods Clin. Dev. 2014, 1, 14042. [Google Scholar] [CrossRef]
- Di Matteo, M.; Samara-Kuko, E.; Ward, N.J.; Waddington, S.N.; McVey, J.H.; Chuah, M.K.; VandenDriessche, T. Hyperactive piggyBac transposons for sustained and robust liver-targeted gene therapy. Mol. Ther. 2014, 22, 1614–1624. [Google Scholar] [CrossRef]
- Puttini, S.; van Zwieten, R.W.; Saugy, D.; Lekka, M.; Hogger, F.; Ley, D.; Kulik, A.J.; Mermod, N. MAR-mediated integration of plasmid vectors for in vivo gene transfer and regulation. BMC Mol. Biol. 2013, 14, 26. [Google Scholar] [CrossRef]
- Cadinanos, J.; Bradley, A. Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res. 2007, 35, e87. [Google Scholar] [CrossRef]
- Lacoste, A.; Berenshteyn, F.; Brivanlou, A.H. An efficient and reversible transposable system for gene delivery and lineage-specific differentiation in human embryonic stem cells. Cell Stem Cell 2009, 5, 332–342. [Google Scholar] [CrossRef]
- Yusa, K.; Zhou, L.; Li, M.A.; Bradley, A.; Craig, N.L. A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. USA 2011, 108, 1531–1536. [Google Scholar] [CrossRef]
- Doherty, J.E.; Huye, L.E.; Yusa, K.; Zhou, L.; Craig, N.L.; Wilson, M.H. Hyperactive piggyBac gene transfer in human cells and in vivo. Hum. Gene Ther. 2012, 23, 311–320. [Google Scholar] [CrossRef]
- Bire, S.; Ley, D.; Casteret, S.; Mermod, N.; Bigot, Y.; Rouleux-Bonnin, F. Optimization of the piggyBac transposon using mRNA and insulators: Toward a more reliable gene delivery system. PLoS ONE 2013, 8, e82559. [Google Scholar] [CrossRef]
- Bire, S.; Gosset, D.; Jégot, G.; Midoux, P.; Pichon, C.; Rouleux-Bonnin, F. Exogenous mRNA delivery and bioavailability in gene transfer mediated by piggyBac transposition. BMC Biotechnol. 2013, 13, 75. [Google Scholar] [CrossRef]
- Bire, S.; Ishac, N.; Rouleux-Bonnin, F. In Vitro synthesis, delivery, and bioavailability of exogenous mRNA in gene transfer mediated by piggyBac transposition. Methods Mol. Biol. 2016, 1428, 187–217. [Google Scholar] [CrossRef]
- Zhang, G.; Budker, V.; Williams, P.; Subbotin, V.; Wolff, J.A. Efficient expression of naked DNA delivered intraarterially to limb muscles of nonhuman primates. Hum. Gene Ther. 2001, 12, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Perez, J.L.; Widmann, T.J.; Adams, I.R. The impact of transposable elements on mammalian development. Development 2016, 143, 4101–4114. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, A.; Valeri, A.; Nollmann, M. Roles of chromatin insulators in the formation of long-range contacts. Nucleus 2015, 6, 118–122. [Google Scholar] [CrossRef]
- Narwade, N.; Patel, S.; Alam, A.; Chattopadhyay, S.; Mittal, S.; Kulkarni, A. Mapping of scaffold/matrix attachment regions in human genome: A data mining exercise. Nucleic Acids Res. 2019, 47, 7247–7261. [Google Scholar] [CrossRef]
- Ley, D.; Harraghy, N.; Le Fourn, V.; Bire, S.; Girod, P.A.; Regamey, A.; Rouleux-Bonnin, F.; Bigot, Y.; Mermod, N. MAR elements and transposons for improved transgene integration and expression. PLoS ONE 2013, 8, e62784. [Google Scholar] [CrossRef]
- Zhao, C.-P.; Guo, X.; Chen, S.-J.; Li, C.-Z.; Yang, Y.; Zhang, J.-H.; Chen, S.-N.; Jia, Y.-L.; Wang, T.-Y. Matrix attachment region combinations increase transgene expression in transfected Chinese hamster ovary cells. Sci. Rep. 2017, 7, 2805. [Google Scholar] [CrossRef]
- Cunningham, S.C.; Siew, S.M.; Hallwirth, C.V.; Bolitho, C.; Sasaki, N.; Garg, G.; Michael, I.P.; Hetherington, N.A.; Carpenter, K.; de Alencastro, G.; et al. Modeling correction of severe urea cycle defects in the growing murine liver using a hybrid recombinant adeno-associated virus/piggyBac transposase gene delivery system. Hepatology 2015, 62, 417–428. [Google Scholar] [CrossRef]
- Siew, S.M.; Cunningham, S.C.; Zhu, E.; Tay, S.S.; Venuti, E.; Bolitho, C.; Alexander, I.E. Prevention of cholestatic liver disease and reduced tumorigenicity in a murine model of PFIC type 3 using hybrid AAV-piggyBac gene therapy. Hepatology 2019, 70, 2047–2061. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, M.; Inada, E.; Saitoh, I.; Watanabe, S.; Nakamura, S. piggyBac-Based Non-Viral In Vivo Gene Delivery Useful for Production of Genetically Modified Animals and Organs. Pharmaceutics 2020, 12, 277. https://doi.org/10.3390/pharmaceutics12030277
Sato M, Inada E, Saitoh I, Watanabe S, Nakamura S. piggyBac-Based Non-Viral In Vivo Gene Delivery Useful for Production of Genetically Modified Animals and Organs. Pharmaceutics. 2020; 12(3):277. https://doi.org/10.3390/pharmaceutics12030277
Chicago/Turabian StyleSato, Masahiro, Emi Inada, Issei Saitoh, Satoshi Watanabe, and Shingo Nakamura. 2020. "piggyBac-Based Non-Viral In Vivo Gene Delivery Useful for Production of Genetically Modified Animals and Organs" Pharmaceutics 12, no. 3: 277. https://doi.org/10.3390/pharmaceutics12030277
APA StyleSato, M., Inada, E., Saitoh, I., Watanabe, S., & Nakamura, S. (2020). piggyBac-Based Non-Viral In Vivo Gene Delivery Useful for Production of Genetically Modified Animals and Organs. Pharmaceutics, 12(3), 277. https://doi.org/10.3390/pharmaceutics12030277