Liposomes for Enhanced Bioavailability of Water-Insoluble Drugs: In Vivo Evidence and Recent Approaches

Abstract
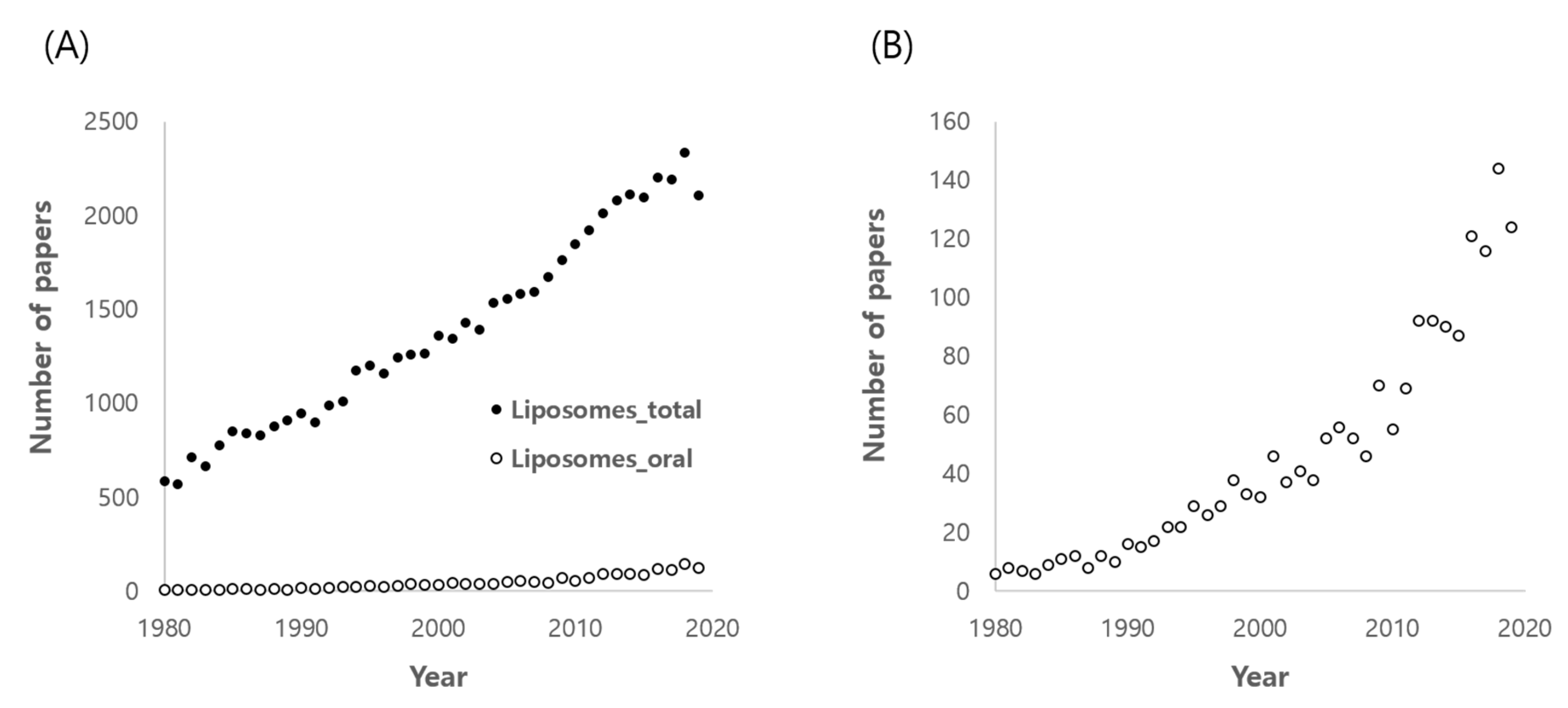
1. Introduction
2. Overview of Liposomes as Drug Delivery System
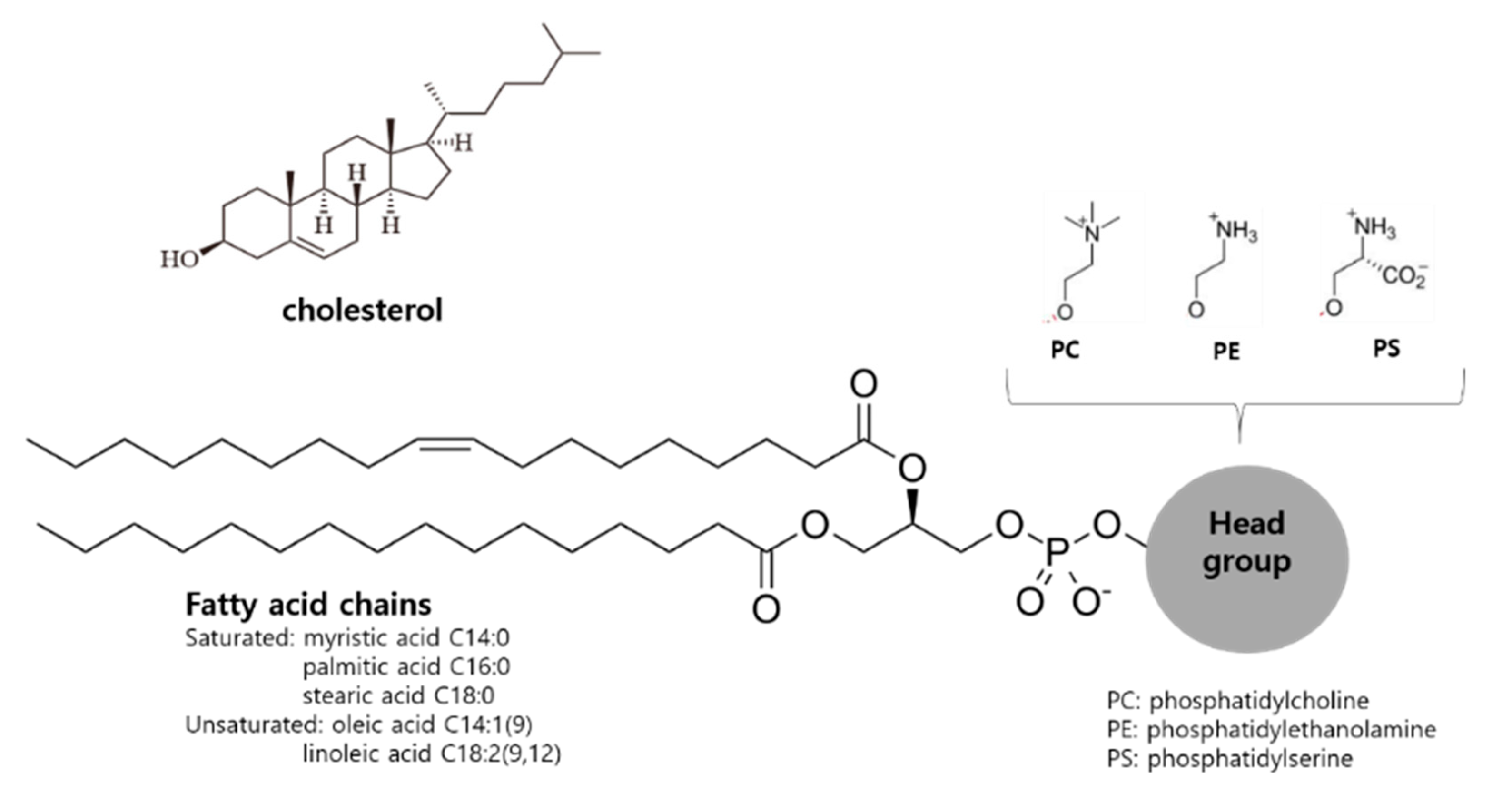
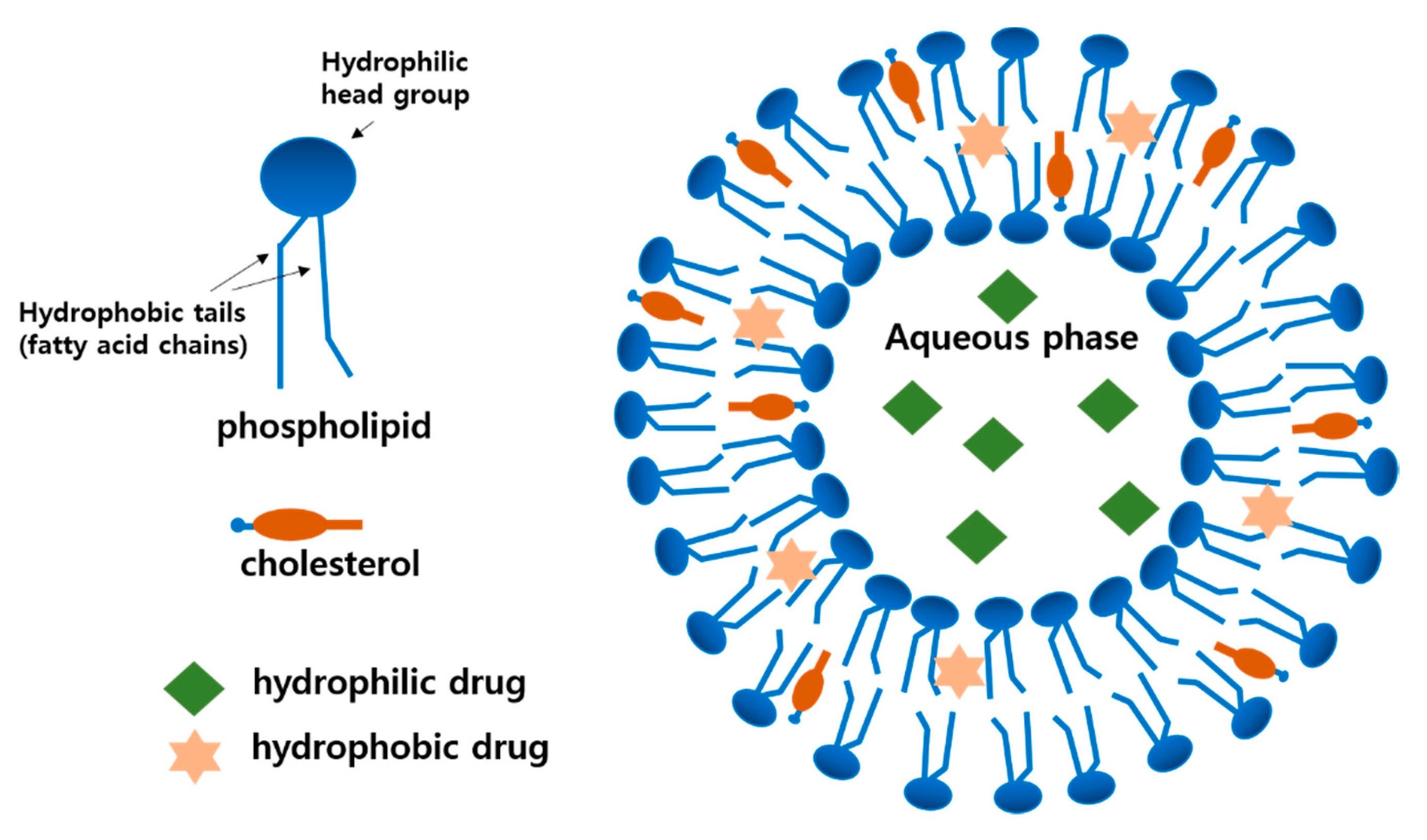
2.1. Basic Composition and Structure
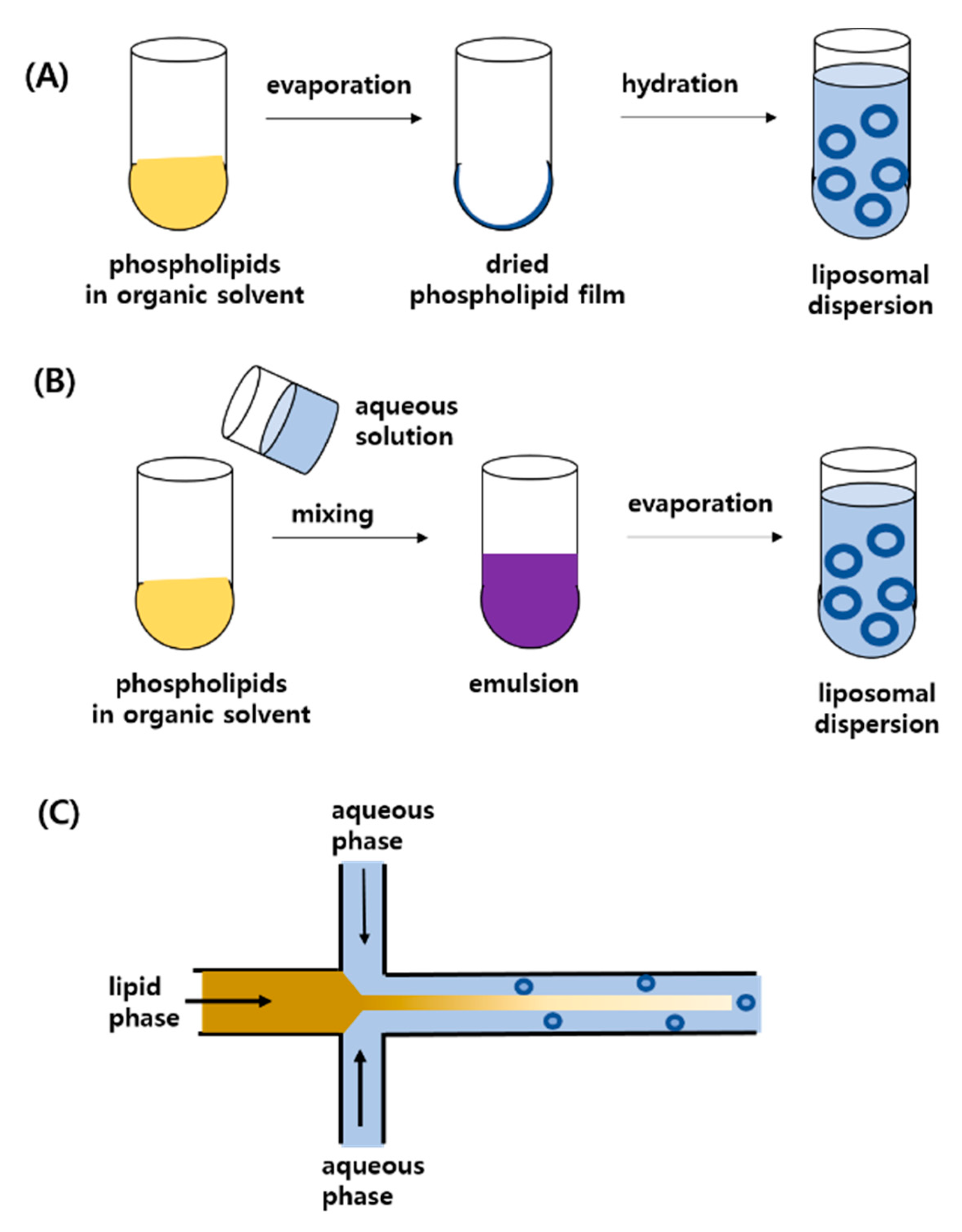
2.2. Preparation Methods
2.3. Evaluation of Liposomal Characteristics
2.4. Advantages and Disadvantages of Liposomes for Oral Delivery
3. Current Approaches Used for In Vivo Studies
3.1. Stabilization
3.1.1. Modulation of Lipid Compositions
3.1.2. Formulation in Solid Forms
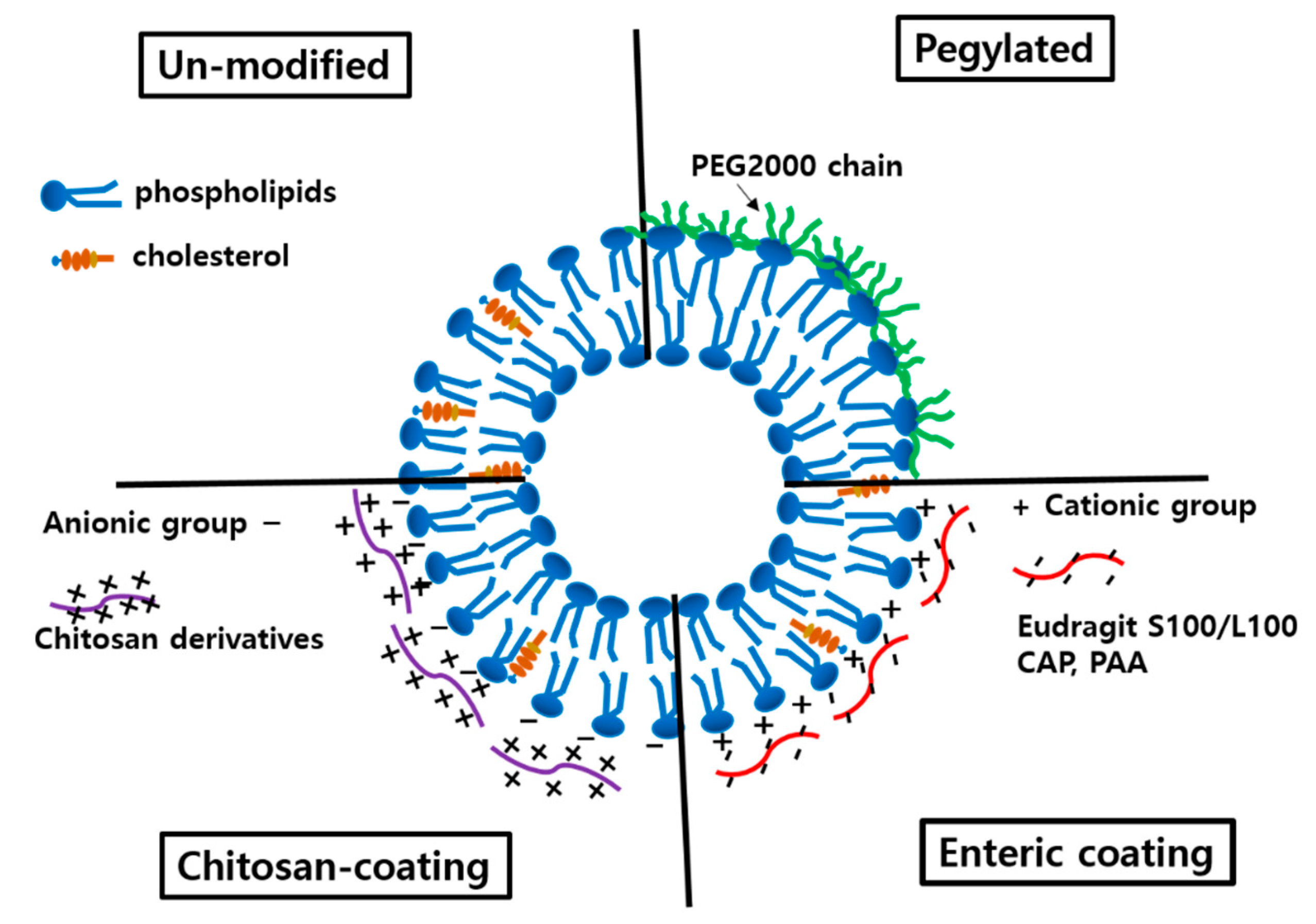
3.1.3. Surface Modification
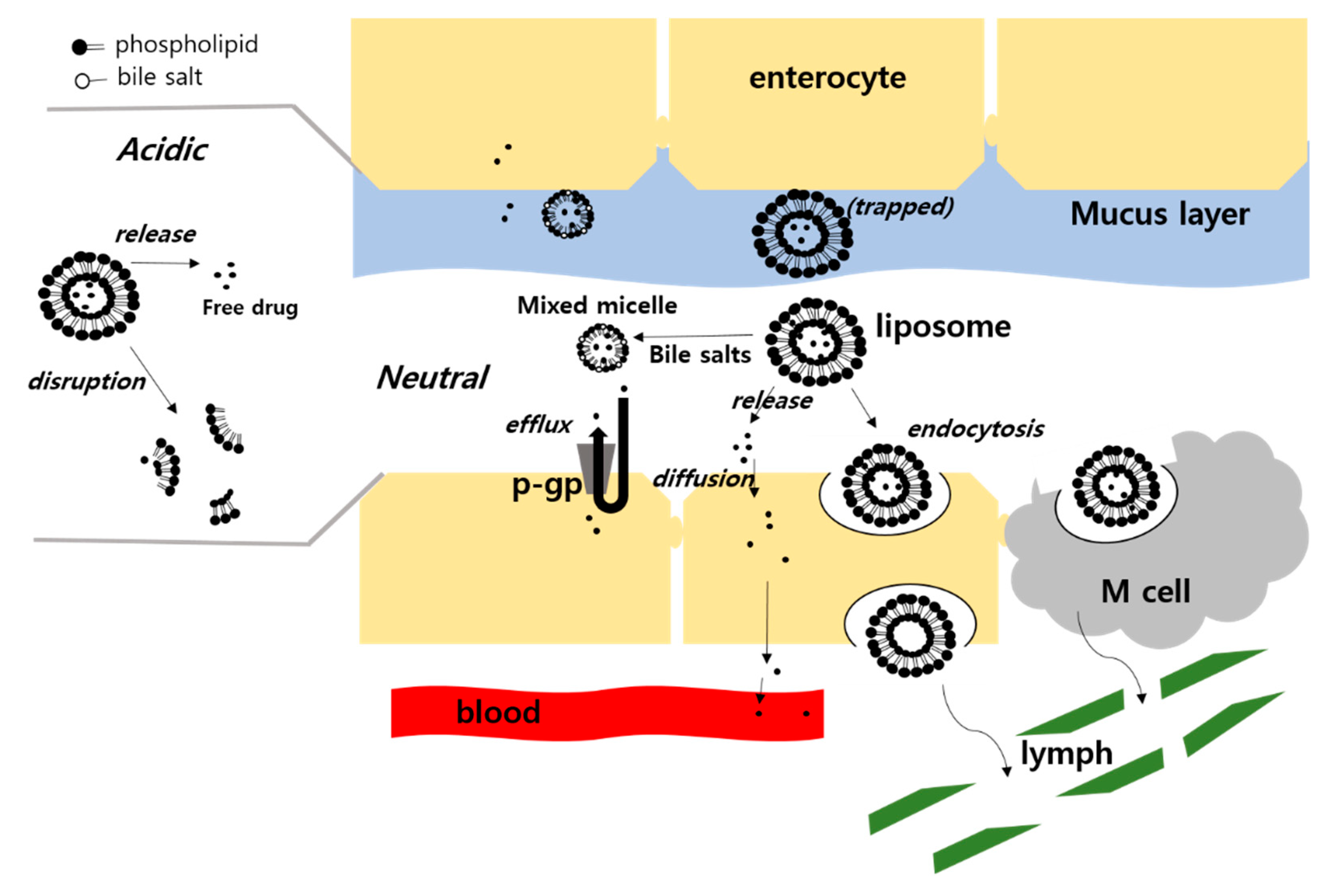
3.2. Enhanced Permeability
3.2.1. Cationic Liposomes
3.2.2. Modification with Chitosan and Its Derivatives
3.2.3. Incorporation of Bile Salts
3.2.4. Modification with Hydrophilic Nonionic Polymers
3.3. Enhanced Dissolution
4. In Vivo Evidence for Enhanced BA by Liposomes
4.1. Apigenin
4.2. Carbamazepine
4.3. Carvedilol
4.4. Curcumin
4.5. Cyclosporine A
4.6. Daidzein
4.7. Docetaxel
4.8. Dronedarone
4.9. Fenofibrate
4.10. Flutamide
4.11. Halofantrine
4.12. Indomethacin
4.13. Isradipine
4.14. Lopinavir
4.15. Lovastatin
4.16. Nisoldipine
4.17. Paclitaxel
4.18. Piroxicam
4.19. Raloxifen
4.20. Sorafenib
4.21. Silymarin and Dehydrosilymarin
4.22. Tacrolimus
4.23. Vinpocetin
4.24. Zaleplon
5. Future Trends and Missions
5.1. Ligand Modification for Active Absorption
5.2. Circumvention of Efflux Pump
5.3. Identification of Absorption Mechanisms
6. Conclusions
Funding
Conflicts of Interest
References
- Ghadi, R.; Dand, N. BCS Class IV Drugs: Highly Notorious Candidates for Formulation Development. J. Control. Release 2017, 248, 71–95. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Nekkanti, V. Insoluble Drug Delivery Strategies: Review of Recent Advances and Business Prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H.; Han, H.K. Nanomedicines: Current Status and Future Perspectives in Aspect of Drug Delivery and Pharmacokinetics. J. Pharm. Investig. 2018, 48, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.S.; Lamprecht, A. Cargoing P-Gp Inhibitors via Nanoparticle Sensitizes Tumor Cells against Doxorubicin. Int. J. Pharm. 2015, 478, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Huang, L. Nanoparticles Containing Insoluble Drug for Cancer Therapy. Biotechnol. Adv. 2014, 32, 778–788. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal Drug Delivery Systems: From Concept to Clinical Applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef]
- Has, C.; Sunthar, P. A Comprehensive Review on Recent Preparation Techniques of Liposomes. J. Liposome Res. 2019, 1–30. [Google Scholar] [CrossRef]
- He, H.; Lu, Y.; Qi, J.; Zhu, Q.; Chen, Z.; Wu, W. Adapting Liposomes for Oral Drug Delivery. Acta Pharm. Sin. B 2019, 9, 36–48. [Google Scholar] [CrossRef]
- Wearley, L.L. Recent Progress in Protein and Peptide Delivery by Noninvasive Routes. Crit. Rev. Ther. Drug Carrier Syst. 1991, 8, 331–394. [Google Scholar]
- Fong, S.Y.; Brandl, M.; Bauer-Brandl, A. Phospholipid-Based Solid Drug Formulations for Oral Bioavailability Enhancement: A Meta-Analysis. Eur. J. Pharm. Sci. 2015, 80, 89–110. [Google Scholar] [CrossRef]
- Abu Lila, A.S.; Ishida, T. Liposomal Delivery Systems: Design Optimization and Current Applications. Biol. Pharm. Bull. 2017, 40, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ran, R.; Wang, H.; Liu, Y.; Hui, Y.; Sun, Q.; Seth, A.; Wibowo, D.; Chen, D.; Zhao, C.X. Microfluidic Self-Assembly of a Combinatorial Library of Single- and Dual-Ligand Liposomes for in Vitro and in Vivo Tumor Targeting. Eur. J. Pharm. Biopharm. 2018, 130, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.; Gupta, N.; Mulla, N.S.; Shukla, S.; Guerrero, Y.A.; Gupta, V. Role of in Vitro Release Methods in Liposomal Formulation Development: Challenges and Regulatory Perspective. AAPS J. 2017, 19, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Lee, S.L.; Tyner, K.M. Liposomal Drug Product Development and Quality: Current US Experience and Perspective. AAPS J. 2017, 19, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Liu, Y.; Wang, H.; Wilson, R.; Hui, Y.; Yu, L.; Wibowo, D.; Zhang, C.; Whittaker, A.K.; Middelberg, A.P.J.; et al. Bioinspired Core-Shell Nanoparticles for Hydrophobic Drug Delivery. Angew. Chem. Int. Ed. 2019, 58, 14357–14364. [Google Scholar] [CrossRef]
- Telange, D.R.; Patil, A.T.; Pethe, A.M.; Fegade, H.; Anand, S.; Dave, V.S. Formulation and Characterization of an Apigenin-Phospholipid Phytosome (APLC) for Improved Solubility, in Vivo Bioavailability, and Antioxidant Potential. Eur. J. Pharm. Sci. 2017, 108, 36–49. [Google Scholar] [CrossRef]
- El-Zein, H.; Riad, L.; El-Bary, A.A. Enhancement of Carbamazepine Dissolution: In Vitro and in Vivo Evaluation. Int. J. Pharm. 1998, 168, 209–220. [Google Scholar] [CrossRef]
- Ghassemi, S.; Haeri, A.; Shahhosseini, S.; Dadashzadeh, S. Labrasol-Enriched Nanoliposomal Formulation: Novel Approach to Improve Oral Absorption of Water-Insoluble Drug, Carvedilol. AAPS PharmSciTech 2018, 19, 2961–2970. [Google Scholar] [CrossRef]
- Kim, J.H.; Shin, D.H.; Kim, J.S. Preparation, Characterization, and Pharmacokinetics of Liposomal Docetaxel for Oral Administration. Arch. Pharm. Res. 2018, 41, 765–775. [Google Scholar] [CrossRef]
- Kovvasu, S.P.; Kunamaneni, P.; Yeung, S.; Rueda, J.; Betageri, G.V. Formulation of Dronedarone Hydrochloride-Loaded Proliposomes: In Vitro and in Vivo Evaluation using Caco-2 and Rat Model. AAPS PharmSciTech 2019, 20, 226. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, Y.; Chen, J.; Lai, J.; Sun, J.; Hu, F.; Wu, W. Enhanced Bioavailability of the Poorly Water-Soluble Drug Fenofibrate by using Liposomes Containing a Bile Salt. Int. J. Pharm. 2009, 376, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Youssef, S.F.; Elnaggar, Y.S.; Abdallah, O.Y. Elaboration of Polymersomes versus Conventional Liposomes for Improving Oral Bioavailability of the Anticancer Flutamide. Nanomedicine 2018, 13, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.R.; Betageri, G.V. Enhanced Oral Absorption of Halofantrine Enantiomers after Encapsulation in a Proliposomal Formulation. J. Pharm. Pharmacol. 2002, 54, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Sugihara, H.; Yamamoto, H.; Kawashima, Y.; Takeuchi, H. Effectiveness of Submicronized Chitosan-Coated Liposomes in Oral Absorption of Indomethacin. J. Liposome Res. 2012, 22, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Bobbala, S.K.; Veerareddy, P.R. Formulation, Evaluation, and Pharmacokinetics of Isradipine Proliposomes for Oral Delivery. J. Liposome Res. 2012, 22, 285–294. [Google Scholar] [CrossRef]
- Yanamandra, S.; Venkatesan, N.; Kadajji, V.G.; Wang, Z.; Issar, M.; Betageri, G.V. Proliposomes as a Drug Delivery System to Decrease the Hepatic First-Pass Metabolism: Case Study using a Model Drug. Eur. J. Pharm. Sci. 2014, 64, 26–36. [Google Scholar] [CrossRef]
- Nekkanti, V.; Rueda, J.; Wang, Z.; Betageri, G.V. Comparative Evaluation of Proliposomes and Self Micro-Emulsifying Drug Delivery System for Improved Oral Bioavailability of Nisoldipine. Int. J. Pharm. 2016, 505, 79–88. [Google Scholar] [CrossRef]
- Mirza, S.; Miroshnyk, I.; Habib, M.J.; Brausch, J.F.; Hussain, M.D. Enhanced Dissolution and Oral Bioavailability of Piroxicam Formulations: Modulating Effect of Phospholipids. Pharmaceutics 2010, 2, 339–350. [Google Scholar] [CrossRef]
- Velpula, A.; Jukanti, R.; Janga, K.Y.; Sunkavalli, S.; Bandari, S.; Kandadi, P.; Veerareddy, P.R. Proliposome Powders for Enhanced Intestinal Absorption and Bioavailability of Raloxifene Hydrochloride: Effect of Surface Charge. Drug Dev. Ind. Pharm. 2013, 39, 1895–1906. [Google Scholar] [CrossRef]
- Zhao, M.; Lee, S.H.; Song, J.G.; Kim, H.Y.; Han, H.K. Enhanced Oral Absorption of Sorafenib Via the Layer-by-Layer Deposition of a pH-Sensitive Polymer and Glycol Chitosan on the Liposome. Int. J. Pharm. 2018, 544, 14–20. [Google Scholar] [CrossRef]
- Xiao, Y.-Y.; Song, Y.-M.; Chen, Z.-P.; Ping, Q.-N. Preparation of Silymarin Proliposome: A New Way to Increase Oral Bioavailability of Silymarin in Beagle Dogs. Int. J. Pharm. 2006, 319, 162–168. [Google Scholar]
- Chu, C.; Tong, S.S.; Xu, Y.; Wang, L.; Fu, M.; Ge, Y.R.; Yu, J.N.; Xu, X.M. Proliposomes for Oral Delivery of Dehydrosilymarin: Preparation and Evaluation in Vitro and in Vivo. Acta Pharmacol. Sin. 2011, 32, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Nekkanti, V.; Rueda, J.; Wang, Z.; Betageri, G.V. Design, Characterization, and in Vivo Pharmacokinetics of Tacrolimus Proliposomes. AAPS PharmSciTech 2016, 17, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; He, L.; Nie, S.; Guan, J.; Zhang, X.; Yang, X.; Pan, W. Optimized Preparation of Vinpocetine Proliposomes by a Novel Method and in Vivo Evaluation of its Pharmacokinetics in New Zealand Rabbits. J. Control. Release 2009, 140, 61–68. [Google Scholar] [CrossRef]
- Janga, K.Y.; Jukanti, R.; Velpula, A.; Sunkavalli, S.; Bandari, S.; Kandadi, P.; Veerareddy, P.R. Bioavailability Enhancement of Zaleplon Via Proliposomes: Role of Surface Charge. Eur. J. Pharm. Biopharm. 2012, 80, 347–357. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.; Su, T.; Feng, L.; Long, Y.; Chen, Z. Silica-Coated Flexible Liposomes as a Nanohybrid Delivery System for Enhanced Oral Bioavailability of Curcumin. Int. J. Nanomed. 2012, 7, 5995–6002. [Google Scholar] [CrossRef]
- Chen, H.; Wu, J.; Sun, M.; Guo, C.; Yu, A.; Cao, F.; Zhao, L.; Tan, Q.; Zhai, G. N-Trimethyl Chitosan Chloride-Coated Liposomes for the Oral Delivery of Curcumin. J. Liposome Res. 2012, 22, 100–109. [Google Scholar] [CrossRef]
- Tian, M.P.; Song, R.X.; Wang, T.; Sun, M.J.; Liu, Y.; Chen, X.G. Inducing Sustained Release and Improving Oral Bioavailability of Curcumin Via Chitosan Derivatives-Coated Liposomes. Int. J. Biol. Macromol. 2018, 120, 702–710. [Google Scholar] [CrossRef]
- Shah, N.M.; Parikh, J.; Namdeo, A.; Subramanian, N.; Bhowmick, S. Preparation, Characterization and in Vivo Studies of Proliposomes Containing Cyclosporine A. J. Nanosci. Nanotechnol. 2006, 6, 2967–2973. [Google Scholar] [CrossRef]
- Guan, P.; Lu, Y.; Qi, J.; Niu, M.; Lian, R.; Hu, F.; Wu, W. Enhanced Oral Bioavailability of Cyclosporine A by Liposomes Containing a Bile Salt. Int. J. Nanomed. 2011, 6, 965–974. [Google Scholar]
- Deng, J.; Zhang, Z.; Liu, C.; Yin, L.; Zhou, J.; Lv, H. The Studies of N-Octyl-N-Arginine-Chitosan Coated Liposome as an Oral Delivery System of Cyclosporine A. J. Pharm. Pharmacol. 2015, 67, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xia, D.; Li, X.; Zhu, Q.; Yu, H.; Zhu, C.; Gan, Y. Comparative Study of Pluronic((R)) F127-Modified Liposomes and Chitosan-Modified Liposomes for Mucus Penetration and Oral Absorption of Cyclosporine A in Rats. Int. J. Pharm. 2013, 449, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, W.; Wang, J.; Liu, H.; Chen, Y. Preparation and Pharmacokinetic Study of Daidzein Long-Circulating Liposomes. Nanoscale Res. Lett. 2019, 14, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.M.; Shelat, P.K.; Lalwani, A.N. QbD Based Development of Proliposome of Lopinavir for Improved Oral Bioavailability. Eur. J. Pharm. Sci. 2017, 108, 50–61. [Google Scholar] [CrossRef]
- Jain, S.; Kumar, D.; Swarnakar, N.K.; Thanki, K. Polyelectrolyte Stabilized Multilayered Liposomes for Oral Delivery of Paclitaxel. Biomaterials 2012, 33, 6758–6768. [Google Scholar] [CrossRef]
- Li, M.; Zou, P.; Tyner, K.; Lee, S. Physiologically Based Pharmacokinetic (PBPK) Modeling of Pharmaceutical Nanoparticles. AAPS J. 2017, 19, 26–42. [Google Scholar] [CrossRef]
- Bibi, S.; Kaur, R.; Henriksen-Lacey, M.; McNeil, S.E.; Wilkhu, J.; Lattmann, E.; Christensen, D.; Mohammed, A.R.; Perrie, Y. Microscopy Imaging of Liposomes: From Coverslips to Environmental SEM. Int. J. Pharm. 2011, 417, 138–150. [Google Scholar] [CrossRef]
- Homayun, B.; Lin, X.; Choi, H.J. Challenges and Recent Progress in Oral Drug Delivery Systems for Biopharmaceuticals. Pharmaceutics 2019, 11, 129. [Google Scholar] [CrossRef]
- Nguyen, T.X.; Huang, L.; Gauthier, M.; Yang, G.; Wang, Q. Recent Advances in Liposome Surface Modification for Oral Drug Delivery. Nanomedicine 2016, 11, 1169–1185. [Google Scholar] [CrossRef]
- Hong, S.S.; Choi, J.Y.; Kim, J.O.; Lee, M.K.; Kim, S.H.; Lim, S.J. Development of Paclitaxel-Loaded Liposomal Nanocarrier Stabilized by Triglyceride Incorporation. Int. J. Nanomed. 2016, 11, 4465–4477. [Google Scholar]
- Chaudhary, S.; Garg, T.; Murthy, R.S.; Rath, G.; Goyal, A.K. Recent Approaches of Lipid-Based Delivery System for Lymphatic Targeting Via Oral Route. J. Drug Target. 2014, 22, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Babadi, D.; Dadashzadeh, S.; Osouli, M.; Daryabari, M.S.; Haeri, A. Nanoformulation Strategies for Improving Intestinal Permeability of Drugs: A More Precise Look at Permeability Assessment Methods and Pharmacokinetic Properties Changes. J. Control. Release 2020, 321, 669–709. [Google Scholar] [CrossRef] [PubMed]
- Kokkona, M.; Kallinteri, P.; Fatouros, D.; Antimisiaris, S.G. Stability of SUV Liposomes in the Presence of Cholate Salts and Pancreatic Lipases: Effect of Lipid Composition. Eur. J. Pharm. Sci. 2000, 9, 245–252. [Google Scholar] [CrossRef]
- Hu, S.; Niu, M.; Hu, F.; Lu, Y.; Qi, J.; Yin, Z.; Wu, W. Integrity and Stability of Oral Liposomes Containing Bile Salts Studied in Simulated and Ex Vivo Gastrointestinal Media. Int. J. Pharm. 2013, 441, 693–700. [Google Scholar] [CrossRef]
- Liu, W.; Ye, A.; Liu, W.; Liu, C.; Han, J.; Singh, H. Behaviour of Liposomes Loaded with Bovine Serum Albumin during in Vitro Digestion. Food Chem. 2015, 175, 16–24. [Google Scholar] [CrossRef]
- Tian, J.N.; Ge, B.Q.; Shen, Y.F.; He, Y.X.; Chen, Z.X. Thermodynamics and Structural Evolution during a Reversible Vesicle-Micelle Transition of a Vitamin-Derived Bolaamphiphile Induced by Sodium Cholate. J. Agric. Food Chem. 2016, 64, 1977–1988. [Google Scholar] [CrossRef]
- Bhatt, P.; Lalani, R.; Vhora, I.; Patil, S.; Amrutiya, J.; Misra, A.; Mashru, R. Liposomes Encapsulating Native and Cyclodextrin Enclosed Paclitaxel: Enhanced Loading Efficiency and its Pharmacokinetic Evaluation. Int. J. Pharm. 2018, 536, 95–107. [Google Scholar] [CrossRef]
- Armengol, X.; Estelrich, J. Physical Stability of Different Liposome Compositions obtained by Extrusion Method. J. Microencapsul. 1995, 12, 525–535. [Google Scholar] [CrossRef]
- Ugwu, S.; Zhang, A.; Parmar, M.; Miller, B.; Sardone, T.; Peikov, V.; Ahmad, I. Preparation, Characterization, and Stability of Liposome-Based Formulations of Mitoxantrone. Drug Dev. Ind. Pharm. 2005, 31, 223–229. [Google Scholar] [CrossRef]
- Hashemzadeh, H.; Javadi, H.; Darvishi, M.H. Study of Structural Stability and Formation Mechanisms in DSPC and DPSM Liposomes: A Coarse-Grained Molecular Dynamics Simulation. Sci. Rep. 2020, 10, 1837. [Google Scholar] [CrossRef]
- Payne, N.I.; Browning, I.; Hynes, C.A. Characterization of Proliposomes. J. Pharm. Sci. 1986, 75, 330–333. [Google Scholar] [CrossRef]
- Basavaraj, S.; Betageri, G.V. Can Formulation and Drug Delivery Reduce Attrition during Drug Discovery and Development-Review of Feasibility, Benefits and Challenges. Acta Pharm. Sin. B 2014, 4, 3–17. [Google Scholar] [CrossRef]
- Brocks, D.R.; Toni, J.W. Pharmacokinetics of Halofantrine in the Rat: Stereoselectivity and Interspecies Comparisons. Biopharm. Drug Dispos. 1999, 20, 165–169. [Google Scholar] [CrossRef]
- Wu, W.; Lu, Y.; Qi, J. Oral Delivery of Liposomes. Ther. Deliv. 2015, 6, 1239–1241. [Google Scholar] [CrossRef]
- Niu, M.; Lu, Y.; Hovgaard, L.; Guan, P.; Tan, Y.; Lian, R.; Qi, J.; Wu, W. Hypoglycemic Activity and Oral Bioavailability of Insulin-Loaded Liposomes Containing Bile Salts in Rats: The Effect of Cholate Type, Particle Size and Administered Dose. Eur. J. Pharm. Biopharm. 2012, 81, 265–272. [Google Scholar] [CrossRef]
- Lopes, M.A.; Abrahim, B.A.; Cabral, L.M.; Rodrigues, C.R.; Seica, R.M.; de Baptista Veiga, F.J.; Ribeiro, A.J. Intestinal Absorption of Insulin Nanoparticles: Contribution of M Cells. Nanomedicine 2014, 10, 1139–1151. [Google Scholar] [CrossRef]
- Alavi, S.; Haeri, A.; Dadashzadeh, S. Utilization of Chitosan-Caged Liposomes to Push the Boundaries of Therapeutic Delivery. Carbohydr. Polym. 2017, 157, 991–1012. [Google Scholar] [CrossRef]
- Yeh, T.H.; Hsu, L.W.; Tseng, M.T.; Lee, P.L.; Sonjae, K.; Ho, Y.C.; Sung, H.W. Mechanism and Consequence of Chitosan-Mediated Reversible Epithelial Tight Junction Opening. Biomaterials 2011, 32, 6164–6173. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kong, M.; Zhou, Z.; Yan, D.; Yu, X.; Cheng, X.; Feng, C.; Liu, Y.; Chen, X. Mechanism of Surface Charge Triggered Intestinal Epithelial Tight Junction Opening upon Chitosan Nanoparticles for Insulin Oral Delivery. Carbohydr. Polym. 2017, 157, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Crayton, S.H.; Thawani, J.P.; Amirshaghaghi, A.; Tsourkas, A.; Cheng, Z. A pH-Responsive Drug-Delivery Platform Based on Glycol Chitosan-Coated Liposomes. Small 2015, 11, 4870–4874. [Google Scholar] [CrossRef] [PubMed]
- Mertins, O.; Dimova, R. Binding of Chitosan to Phospholipid Vesicles Studied with Isothermal Titration Calorimetry. Langmuir 2011, 27, 5506–5515. [Google Scholar] [CrossRef] [PubMed]
- Elnaggar, Y.S. Multifaceted Applications of Bile Salts in Pharmacy: An Emphasis on Nanomedicine. Int. J. Nanomed. 2015, 10, 3955–3971. [Google Scholar] [CrossRef] [PubMed]
- Huckaby, J.T.; Lai, S.K. PEGylation for Enhancing Nanoparticle Diffusion in Mucus. Adv. Drug Deliv. Rev. 2018, 124, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Frey, A.; Giannasca, K.T.; Weltzin, R.; Giannasca, P.J.; Reggio, H.; Lencer, W.I.; Neutra, M.R. Role of the Glycocalyx in Regulating Access of Microparticles to Apical Plasma Membranes of Intestinal Epithelial Cells: Implications for Microbial Attachment and Oral Vaccine Targeting. J. Exp. Med. 1996, 184, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Netsomboon, K.; Bernkop-Schnurch, A. Mucoadhesive vs. Mucopenetrating Particulate Drug Delivery. Eur. J. Pharm. Biopharm. 2016, 98, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL: The Drawbacks and Advantages of Vehicle Selection for Drug Formulation. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef]
- Ana, R.; Mendes, M.; Sousa, J.; Pais, A.; Falcao, A.; Fortuna, A.; Vitorino, C. Rethinking Carbamazepine Oral Delivery using Polymer-Lipid Hybrid Nanoparticles. Int. J. Pharm. 2019, 554, 352–365. [Google Scholar] [CrossRef]
- Pavan Kumar, M.; Srawan Kumar, G.Y.; Apte, S.; Madhusudan Rao, Y. Review of Solubilization Techniques for a Poorly Water-Soluble Drug: Carbamazepine. PDA J. Pharm. Sci. Technol. 2010, 64, 264–277. [Google Scholar]
- Ogawa, R.; Stachnik, J.M.; Echizen, H. Clinical Pharmacokinetics of Drugs in Patients with Heart Failure: An Update (Part 2, Drugs Administered Orally). Clin. Pharmacokinet. 2014, 53, 1083–1114. [Google Scholar] [CrossRef]
- Hamed, R.; Awadallah, A.; Sunoqrot, S.; Tarawneh, O.; Nazzal, S.; AlBaraghthi, T.; Al Sayyad, J.; Abbas, A. pH-Dependent Solubility and Dissolution Behavior of Carvedilol—Case Example of a Weakly Basic BCS Class II Drug. AAPS PharmSciTech 2016, 17, 418–426. [Google Scholar] [CrossRef]
- Stillhart, C.; Durr, D.; Kuentz, M. Toward an Improved Understanding of the Precipitation Behavior of Weakly Basic Drugs from Oral Lipid-Based Formulations. J. Pharm. Sci. 2014, 103, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.Y.; Meng, X.; Li, S.; Gan, R.Y.; Li, Y.; Li, H.B. Bioactivity, Health Benefits, and Related Molecular Mechanisms of Curcumin: Current Progress, Challenges, and Perspectives. Nutrients 2018, 10, 1553. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T. Curcumin as a Functional Food-Derived Factor: Degradation Products, Metabolites, Bioactivity, and Future Perspectives. Food Funct. 2018, 9, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Ipar, V.S.; Dsouza, A.; Devarajan, P.V. Enhancing Curcumin Oral Bioavailability through Nanoformulations. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 459–480. [Google Scholar] [CrossRef]
- Sahariah, P.; Gaware, V.S.; Lieder, R.; Jonsdottir, S.; Hjalmarsdottir, M.A.; Sigurjonsson, O.E.; Masson, M. The Effect of Substituent, Degree of Acetylation and Positioning of the Cationic Charge on the Antibacterial Activity of Quaternary Chitosan Derivatives. Mar. Drugs 2014, 12, 4635–4658. [Google Scholar] [CrossRef]
- Wang, L.; Li, W.; Cheng, D.; Guo, Y.; Wu, R.; Yin, R.; Li, S.; Kuo, H.C.; Hudlikar, R.; Yang, H.; et al. Pharmacokinetics and Pharmacodynamics of Three Oral Formulations of Curcumin in Rats. J. Pharmacokinet. Pharmacodyn. 2020. [Google Scholar] [CrossRef]
- Benet, L.Z. The Drug Transporter-Metabolism Alliance: Uncovering and Defining the Interplay. Mol. Pharm. 2009, 6, 1631–1643. [Google Scholar] [CrossRef]
- Krizova, L.; Dadakova, K.; Kasparovska, J.; Kasparovsky, T. Isoflavones. Molecules 2019, 24, 1076. [Google Scholar] [CrossRef]
- Setchell, K.D.; Faughnan, M.S.; Avades, T.; Zimmer-Nechemias, L.; Brown, N.M.; Wolfe, B.E.; Brashear, W.T.; Desai, P.; Oldfield, M.F.; Botting, N.P.; et al. Comparing the Pharmacokinetics of Daidzein and Genistein with the use of 13C-Labeled Tracers in Premenopausal Women. Am. J. Clin. Nutr. 2003, 77, 411–419. [Google Scholar] [CrossRef]
- Lee, M.K.; Lim, S.J.; Kim, C.K. Preparation, Characterization and in Vitro Cytotoxicity of Paclitaxel-Loaded Sterically Stabilized Solid Lipid Nanoparticles. Biomaterials 2007, 28, 2137–2146. [Google Scholar] [CrossRef]
- Nieuweboer, A.J.; de Morree, E.S.; de Graan, A.J.; Sparreboom, A.; de Wit, R.; Mathijssen, R.H. Inter-Patient Variability in Docetaxel Pharmacokinetics: A Review. Cancer Treat. Rev. 2015, 41, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Dorian, P. Clinical Pharmacology of Dronedarone: Implications for the Therapy of Atrial Fibrillation. J. Cardiovasc. Pharmacol. Ther. 2010, 15, 15S–18S. [Google Scholar] [CrossRef] [PubMed]
- Feeney, O.M.; Crum, M.F.; McEvoy, C.L.; Trevaskis, N.L.; Williams, H.D.; Pouton, C.W.; Charman, W.N.; Bergstrom, C.A.S.; Porter, C.J.H. 50years of Oral Lipid-Based Formulations: Provenance, Progress and Future Perspectives. Adv. Drug Deliv. Rev. 2016, 101, 167–194. [Google Scholar] [CrossRef] [PubMed]
- Posti, J.; Katila, K.; Kostiainen, T. Dissolution Rate Limited Bioavailability of Flutamide, and in Vitro—In Vivo Correlation. Eur. J. Pharm. Biopharm. 2000, 49, 35–39. [Google Scholar] [CrossRef]
- Zarmpi, P.; Flanagan, T.; Meehan, E.; Mann, J.; Fotaki, N. Biopharmaceutical Aspects and Implications of Excipient Variability in Drug Product Performance. Eur. J. Pharm. Biopharm. 2017, 111, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.P.; Srinivas, N.R.; Babu, R.J. Use of Sorbitol as Pharmaceutical Excipient in the Present Day Formulations—Issues and Challenges for Drug Absorption and Bioavailability. Drug Dev. Ind. Pharm. 2019, 45, 1421–1429. [Google Scholar] [CrossRef]
- Karbwang, J.; Na Bangchang, K. Clinical Pharmacokinetics of Halofantrine. Clin. Pharmacokinet. 1994, 27, 104–119. [Google Scholar] [CrossRef]
- Christensen, H.R.; Antonsen, K.; Simonsen, K.; Lindekaer, A.; Bonde, J.; Angelo, H.R.; Kampmann, J.P. Bioavailability and Pharmacokinetics of Isradipine After Oral and Intravenous Administration: Half-Life Shorter than Expected? Pharmacol. Toxicol. 2000, 86, 178–182. [Google Scholar] [CrossRef]
- Maniyar, M.G.; Kokare, C.R. Formulation and Evaluation of Spray Dried Liposomes of Lopinavir for Topical Application. J. Pharm. Investig. 2019, 49, 259–270. [Google Scholar] [CrossRef]
- Schachter, M. Chemical, Pharmacokinetic and Pharmacodynamic Properties of Statins: An Update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef]
- Heinig, R. Clinical Pharmacokinetics of Nisoldipine Coat-Core. Clin. Pharmacokinet. 1998, 35, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.C.; Choi, J.S.; Han, H.K. Altered Pharmacokinetics of Paclitaxel by the Concomitant use of Morin in Rats. Int. J. Pharm. 2006, 323, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.K.; Ryu, M.H.; Park, S.H.; Kim, J.G.; Kim, J.W.; Cho, S.H.; Park, Y.I.; Park, S.R.; Rha, S.Y.; Kang, M.J.; et al. Efficacy and Safety Findings from DREAM: A Phase III Study of DHP107 (Oral Paclitaxel) Versus i.V. Paclitaxel in Patients with Advanced Gastric Cancer After Failure of First-Line Chemotherapy. Ann. Oncol. 2018, 29, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Chung, H.J.; Hong, J.W.; Yun, C.W.; Chung, H. Absorption Mechanism of DHP107, an Oral Paclitaxel Formulation that Forms a Hydrated Lipidic Sponge Phase. Acta Pharmacol. Sin. 2017, 38, 133–145. [Google Scholar] [CrossRef]
- Li, R.; Eun, J.S.; Lee, M.K. Pharmacokinetics and Biodistribution of Paclitaxel Loaded in Pegylated Solid Lipid Nanoparticles after Intravenous Administration. Arch. Pharm. Res. 2011, 34, 331–337. [Google Scholar] [CrossRef]
- Hong, J.W.; Lee, I.H.; Kwak, Y.H.; Park, Y.T.; Sung, H.C.; Kwon, I.C.; Chung, H. Efficacy and Tissue Distribution of DHP107, an Oral Paclitaxel Formulation. Mol. Cancer. Ther. 2007, 6, 3239–3247. [Google Scholar] [CrossRef]
- Morello, K.C.; Wurz, G.T.; DeGregorio, M.W. Pharmacokinetics of Selective Estrogen Receptor Modulators. Clin. Pharmacokinet. 2003, 42, 361–372. [Google Scholar] [CrossRef]
- Wang, X.Q.; Fan, J.M.; Liu, Y.O.; Zhao, B.; Jia, Z.R.; Zhang, Q. Bioavailability and Pharmacokinetics of Sorafenib Suspension, Nanoparticles and Nanomatrix for Oral Administration to Rat. Int. J. Pharm. 2011, 419, 339–346. [Google Scholar] [CrossRef]
- Granito, A.; Marinelli, S.; Negrini, G.; Menetti, S.; Benevento, F.; Bolondi, L. Prognostic Significance of Adverse Events in Patients with Hepatocellular Carcinoma Treated with Sorafenib. Therap. Adv. Gastroenterol. 2016, 9, 240–249. [Google Scholar] [CrossRef]
- Van Erp, N.P.; Gelderblom, H.; Guchelaar, H.J. Clinical Pharmacokinetics of Tyrosine Kinase Inhibitors. Cancer Treat. Rev. 2009, 35, 692–706. [Google Scholar] [CrossRef]
- Strumberg, D.; Richly, H.; Hilger, R.A.; Schleucher, N.; Korfee, S.; Tewes, M.; Faghih, M.; Brendel, E.; Voliotis, D.; Haase, C.G.; et al. Phase I Clinical and Pharmacokinetic Study of the Novel Raf Kinase and Vascular Endothelial Growth Factor Receptor Inhibitor BAY 43-9006 in Patients with Advanced Refractory Solid Tumors. J. Clin. Oncol. 2005, 23, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Dallio, M.; Loguercio, C. Silymarin/Silybin and Chronic Liver Disease: A Marriage of Many Years. Molecules 2017, 22, 191. [Google Scholar] [CrossRef]
- Tong, S.; Chu, C.; Wei, Y.; Wang, L.; Gao, X.; Xu, X.; Yu, J. Preparation and Effects of 2,3-Dehydrosilymarin, a Promising and Potent Antioxidant and Free Radical Scavenger. J. Pharm. Pharmacol. 2011, 63, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, A.; Angelico, R. Formulation Strategies for Enhancing the Bioavailability of Silymarin: The State of the Art. Molecules 2019, 24, 2155. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Lin, L.C.; Hung, S.C.; Chi, C.W.; Tsai, T.H. Analysis of Silibinin in Rat Plasma and Bile for Hepatobiliary Excretion and Oral Bioavailability Application. J. Pharm. Biomed. Anal. 2007, 45, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Dheer, D.; Jyoti; Gupta, P.N.; Shankar, R. Tacrolimus: An Updated Review on Delivering Strategies for Multifarious Diseases. Eur. J. Pharm. Sci. 2018, 114, 217–227. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Li, J.D.; Yan, C. An Update on Vinpocetine: New Discoveries and Clinical Implications. Eur. J. Pharmacol. 2018, 819, 30–34. [Google Scholar] [CrossRef]
- Pudleiner, P.; Vereczkey, L. Study on the Absorption of Vinpocetine and Apovincaminic Acid. Eur. J. Drug Metab. Pharmacokinet. 1993, 18, 317–321. [Google Scholar] [CrossRef]
- Drover, D.R. Comparative Pharmacokinetics and Pharmacodynamics of Short-Acting Hypnosedatives: Zaleplon, Zolpidem and Zopiclone. Clin. Pharmacokinet. 2004, 43, 227–238. [Google Scholar] [CrossRef]
- Choi, M.S.; Kim, Y.C.; Maeng, H.J. Therapeutic Targets of Vitamin D Receptor Ligands and their Pharmacokinetic Effects by Modulation of Transporters and Metabolic Enzymes. J. Pharm. Investig. 2020, 50, 1–16. [Google Scholar] [CrossRef]
- Russell-Jones, G.J. The Potential use of Receptor-Mediated Endocytosis for Oral Drug Delivery. Adv. Drug Deliv. Rev. 2001, 46, 59–73. [Google Scholar] [CrossRef]
- Elmeliegy, M.; Vourvahis, M.; Guo, C.; Wang, D.D. Effect of P-Glycoprotein (P-Gp) Inducers on Exposure of P-Gp Substrates: Review of Clinical Drug-Drug Interaction Studies. Clin. Pharmacokinet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Weinheimer, M.; Fricker, G.; Burhenne, J.; Mylius, P.; Schubert, R. The Application of P-Gp Inhibiting Phospholipids as Novel Oral Bioavailability Enhancers—An in Vitro and in Vivo Comparison. Eur. J. Pharm. Sci. 2017, 108, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Gurjar, R.; Chan, C.Y.S.; Curley, P.; Sharp, J.; Chiong, J.; Rannard, S.; Siccardi, M.; Owen, A. Inhibitory Effects of Commonly used Excipients on P-Glycoprotein in Vitro. Mol. Pharm. 2018, 15, 4835–4842. [Google Scholar] [CrossRef]
- Liu, W.; Ye, A.; Han, F.; Han, J. Advances and Challenges in Liposome Digestion: Surface Interaction, Biological Fate, and GIT Modeling. Adv. Colloid Interface Sci. 2019, 263, 52–67. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs (Therapeutic Category) | Liposome Composition | Encapsulation Efficiency (%) | Physical Forms | Study Subject | Relative BA (fold) | Comparator | Reference |
|---|---|---|---|---|---|---|---|
| BCS Class II drugs | |||||||
| Apigenin (herbal supplement) | Phospholipid 90H | 93.3% | Solid: proliposome (mannitol) | Rat | 1.5 | Free drug suspension | [16] |
| Carbamazepine (antiepilectic) | Drug:DMPG (1:1) | ND | Solid: co-precipitate | Rabbitt | 1.2 (NS) | Tegretol suspenstion | [17] |
| Carvedilol (cardiovascular) | EPC:CH:Labrasol (65:15:20) | 79.8% | Liquid: liposome dispersion | Rat | 2.3 | Free drug suspension | [18] |
| Docetaxel (anticancer) | EPC:SA (1:0.2) with SDC and coating with Eudragit L100/S100 (4:1) | 33.6% | Solid: Freeze-dried liposomes (trehalose, mannitol) | Rat | 3.1 | Free drug solution in polysorbate 80/ethanol/saline (20:13:67) | [19] |
| Dronedarone (antiarrhythmic) | DMPG Na:CH (1:2) | 84% | Solid: proliposomes (MCC) | Rat | 1.5 | Free drug suspension | [20] |
| Fenofibrate (antilipidemic) | SPC:SDC (4:1) | 88.2% | Liquid: liposomal dispersion | Dog | 5.1 | Micronized fenofibrate in capsule | [21] |
| Flutamide (antiandrogen) | SPC:CH (4:1 w/w) | 70.6% | Liquid: liposomal dispersion | Rat | 0.9 | Free drug suspension | [22] |
| Halofantrine (antimalarial) | DSPC:Drug (3:1) Coating with CAP | ND | Solid: proliposomes (enteric coating) | Rat | 1.4 | Free drug suspension | [23] |
| Indomethacin (NSAID) | DSPC:DCP:CH (8:2:1) coating with chitosan | ND | Liquid: liposomal dispersion | Rat | 1.8 | Free drug solution | [24] |
| Isradipine (calcium antagonist) | HSPC:CH (1:1) | 96.8% | Solid: proliposomes (mannitol) | Rat | 2.0 | Free drug suspension | [25] |
| Lovastatin (antilipidemic) | SPC:CH (9:1) | 85.8% | Solid: proliposomes (silicified MCC) | Rat | 1.6 | Free drug suspension | [26] |
| Nisoldipine (calcium channel blocker) | DMPC:CH (4:1) | 85.6% | Solid: proliposome (MCC) | Rat | 3.0 | Free drug suspension | [27] |
| Piroxicam (NSAID) | DMPG | ND | Solid: solid dispersion | Rat | 1.3 (NS) | Free drug suspension | [28] |
| Raloxifen (estrogen receptor modulator) | HSPC:CH with DCP or SA | 94.2% (cationic) 93.2% (anionic) 93.9% (neutral) | Solid: proliposomes (mannitol) | Rat | 3.4 (cationic); 2.6 (anionic); 2.4 (neutral) | Free drug suspension (processed without lipids) | [29] |
| Sorafenib tosylate (anticancer) | DPPC:DPPG:TPGS:CH (8:1:2:4) Coating with Glycol chitosan & Eudragit S100 | 94.6% (uncoated) 96.6% (glycol chitosan-coated) 89.7% (double layer coated) | Liquid: liposome dispersion | Rat | 2.9 (uncoated); 3.0 (glycol chitosan-coated); 5.1 (EudragitS100/glycol chitosan coated) | Free drug | [30] |
| Silymarin (hepatoprotective) | Phospholipid (82% PC) | 92.6% | Solid: proliposomes (mannitol) | Dog | 3.4 | Powder | [31] |
| Dehydrosilymarin (hepatoprotective) | SPC 0.3 g CH 0.075 g IPM 0.2 g Sodium cholate 0.2 g | 70–80% | Solid: proliposomes (mannitol) | Rabbit | 2.2 | Free drug suspension | [32] |
| Tacrolimus (immunosuppressant) | DSPC:CH (4:1) | approx. 70–80% | Solid: proliposomes | Rat | 1.9 | Free drug suspension | [33] |
| Vinpocetine (Cardiovascular) | SPC:CH (9:1, w/w) | 86.3% | Solid: proliposomes (sorbitol) | Rabbit | 3.5 | Free drug suspension | [34] |
| Zaleplon (hypnotic) | HSPC:CH (1:1) with DCP or SA | 93.8% (cationic) 92.5% (anionic) 94.6% (neutral) | Solid: proliposomes (mannitol) | Rat | 4.6 (cationic) 3.0 (anionic) 2.0 (neutral) | Free drug suspension (processed without lipids) | [35] |
| BCS class IV drugs | |||||||
| Curcumin (herbal supplement) | SPC:SDC (85:15 w/w) Coating with Silica | 93.3% | Liquid: liposome dispersion | Rat | 2.3 (uncoated); 3.3 (silica-coated) | Free drug suspension | [36] |
| SPC:CH:TPGS:drug (20:2:12:1) Coating with TMC | 86.7% | Liquid: liposome dispersion | Rat | 6.7 (uncoated); 10.6 (TMC-coated) | Free drug suspension | [37] | |
| SPC:SDC (70:25 w/w) Coating with TMC and CMCS | ND | Liquid: liposome dispersion | Rat | 6.3 (CMCS/TMC-coated); 2 (TMC-coated) | Uncoated liposomes | [38] | |
| Cyclosporine A (immunosuppressant) | ePC:Cremophor EL (10:0.5) | 96.3% | Solid: proliposomes (lactose) | Rat | 9.6 | Free drug suspension | [39] |
| SPC:SDC (3:1) | 94.0% | Liquid: liposome dispersion | Rat | 1.2 (NS) | Sandimmune Neoral®® | [40] | |
| SPC:CH (20:1) Coating with OACS | 98.0% | Liquid: liposome dispersion | Rat | 1.7 (uncoated); 3.4 (OACS-coated) | Free drug suspension | [41] | |
| EPC:CH (28:5) with Pluronic F127 | 90.0% | Liquid: liposome dispersion | Rat | 1.8 | Unmodified liposomes | [42] | |
| Daidzein (natural compound) | SPC:CH:DSPEPEG2000 (55:40:5) | 80.2% | Solid: freeze dried liposomes with 3% sucrose | Rat | 2.5 | Free drug suspension | [43] |
| Lopinavir (antiviral) | HSPC, CH (7:3) | Approx. 89% | Solid: proliposome (mannitol) | Rat | 2.2 | Free drug suspension | [44] |
| Paclitaxel (anticancer) | SPC:CH:SA (24.5:11.5:2 w/w) Coating with PAA and then PAH | 81.3% | Solid: freeze dried liposomes with mannitol | Rat | 4.0 (double-layer coated) | Free drug suspension | [45] |
| Characteristics | Representative Techniques |
|---|---|
| Particle size and size distribution | Dynamic light scattering (DLS), Electron microscopy |
| Morphology, lamellarity | Electron microscopy |
| Surface charge | Zeta potential analysis |
| Encapsulation efficiency | Separation of free drug (dialysis, ultrafiltration, size exclusion chromatography) and drug analysis (HPLC etc.) |
| Release rate | Release in physiological media or storage buffer |
| Physical stability | Particle size change in physiological media or storage buffer |
| Advantages | Disadvantages |
|---|---|
| Biocompatibility | Physical instability in liquid state |
| Versatility for drug encapsulation | Lysolipid formation by chemical degradation |
| Flexibility of membrane components | Drug leakage |
| Capability of surface modification | Disruption in the stomach |
| Proposed enhanced permeation | Low permeability of intact liposome in the GI tract |
| Modifiable pharmacokinetic behavior | Difficulty in mass production and quality control |
| Researchers | Formulations | Dose | F | AUC0–∞ (ng·h/L) (* AUC0–12h) | Cmax (ng/L) |
|---|---|---|---|---|---|
| Li et al., 2012 | Free drug suspension | 50 mg/kg (oral) | - | 86.65 * | 71.35 |
| Liposomes (SPC:SDC) | 50 mg/kg (oral) | - | 203.64 * | 128.78 | |
| Silica-coated liposome | 50 mg/kg (oral) | - | 673.79 * | 446.66 | |
| Chen et al., 2012 | Free drug suspension | 250 mg/kg (oral) | - | 244,770 | 46,130 |
| Liposomes (SPC:CH:TPGS) | 40 mg/kg (oral) | - | 263,770 | 32,120 | |
| TMC-coated liposomes | 40 mg/kg (oral) | - | 416,580 | 35,460 | |
| Tian et al., 2018 | Liposomes (SPC:SDC) | 10 mg/kg (oral) | 6% | 528,900 * | 48,200 |
| TMC-coated liposomes | 10 mg/kg (oral) | 12% | 1,218,200 * | 78,300 | |
| CMCS/TMC-coated liposomes | 10 mg/kg (oral) | 38% | 3,021,200 * | 167,800 | |
| Wang et al., 2020 | Intravenous | 40 mg/kg (i.v.) | - | 268,900 | - |
| Commercial product 1 (tablet) | 250 mg/kg (oral) | 0.9% | 20,000 | 12,600 | |
| Commercial product 2 (capsule) | 250 mg/kg (oral) | 0.6% | 10,740 | 9920 | |
| Powder (Sigma) | 250 mg/kg (oral) | 3.1% | 45,600 | 17,800 |
| Researchers | Formulations | Mean Diameter | Dose | AUC0–∞ (µg·h/mL) | Cmax (µg/L) |
|---|---|---|---|---|---|
| Shah et al., 2006 | drug suspension | - | 10 mg/kg | 0.2253 | 0.09 |
| EPC/CreEL-proLip | 10.34 µm | 10 mg/kg | 2.155 | 0.3 | |
| Guan et al., 2011 | Microemulsion | - | 15 mg/kg | 65.41 ± 29.55 | 2.57 ± 0.20 |
| SPC/SDC Lip | 85.6 nm | 15 mg/kg | 73.90 ± 6.63 | 2.65 ± 0.70 | |
| SPC/CH Lip | 98.1 nm | 15 mg/kg | 60.49 ± 10.79 | 2.67 ± 0.69 | |
| Chen et al., 2013 | EPC/CH Lip | 165.25 nm | 10 mg/kg | 9.18 ± 1.06 * | 1.14 ± 0.23 |
| PF127-Lip | 172.82 nm | 10 mg/kg | 11.59 ± 0.70 * | 1.37 ± 0.15 | |
| CS-Lip | 207.81 nm | 10 mg/kg | 6.30 ± 0.97 * | 0.79 ± 0.10 | |
| Deng et al., 2015 | drug suspension | - | 15 mg/kg | 31.14 ± 1.30 | 1.10 ± 0.14 |
| Microemulsion | - | 15 mg/kg | 69.34 ± 7.93 | 3.40 ± 0.24 | |
| SPC/CH Lip | 58.94 nm | 15 mg/kg | 53.29 ± 4.59 | 2.85 ± 0.16 | |
| OACS-Lip | 69.12 nm | 15 mg/kg | 100.98 ± 13.08 | 4.14 ± 0.26 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.-K. Liposomes for Enhanced Bioavailability of Water-Insoluble Drugs: In Vivo Evidence and Recent Approaches. Pharmaceutics 2020, 12, 264. https://doi.org/10.3390/pharmaceutics12030264
Lee M-K. Liposomes for Enhanced Bioavailability of Water-Insoluble Drugs: In Vivo Evidence and Recent Approaches. Pharmaceutics. 2020; 12(3):264. https://doi.org/10.3390/pharmaceutics12030264
Chicago/Turabian StyleLee, Mi-Kyung. 2020. "Liposomes for Enhanced Bioavailability of Water-Insoluble Drugs: In Vivo Evidence and Recent Approaches" Pharmaceutics 12, no. 3: 264. https://doi.org/10.3390/pharmaceutics12030264
APA StyleLee, M.-K. (2020). Liposomes for Enhanced Bioavailability of Water-Insoluble Drugs: In Vivo Evidence and Recent Approaches. Pharmaceutics, 12(3), 264. https://doi.org/10.3390/pharmaceutics12030264