Improving the Oral Bioavailability of an Anti-Glioma Prodrug CAT3 Using Novel Solid Lipid Nanoparticles Containing Oleic Acid-CAT3 Conjugates

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Cell Culture
2.1.2. Animals
2.2. Methods
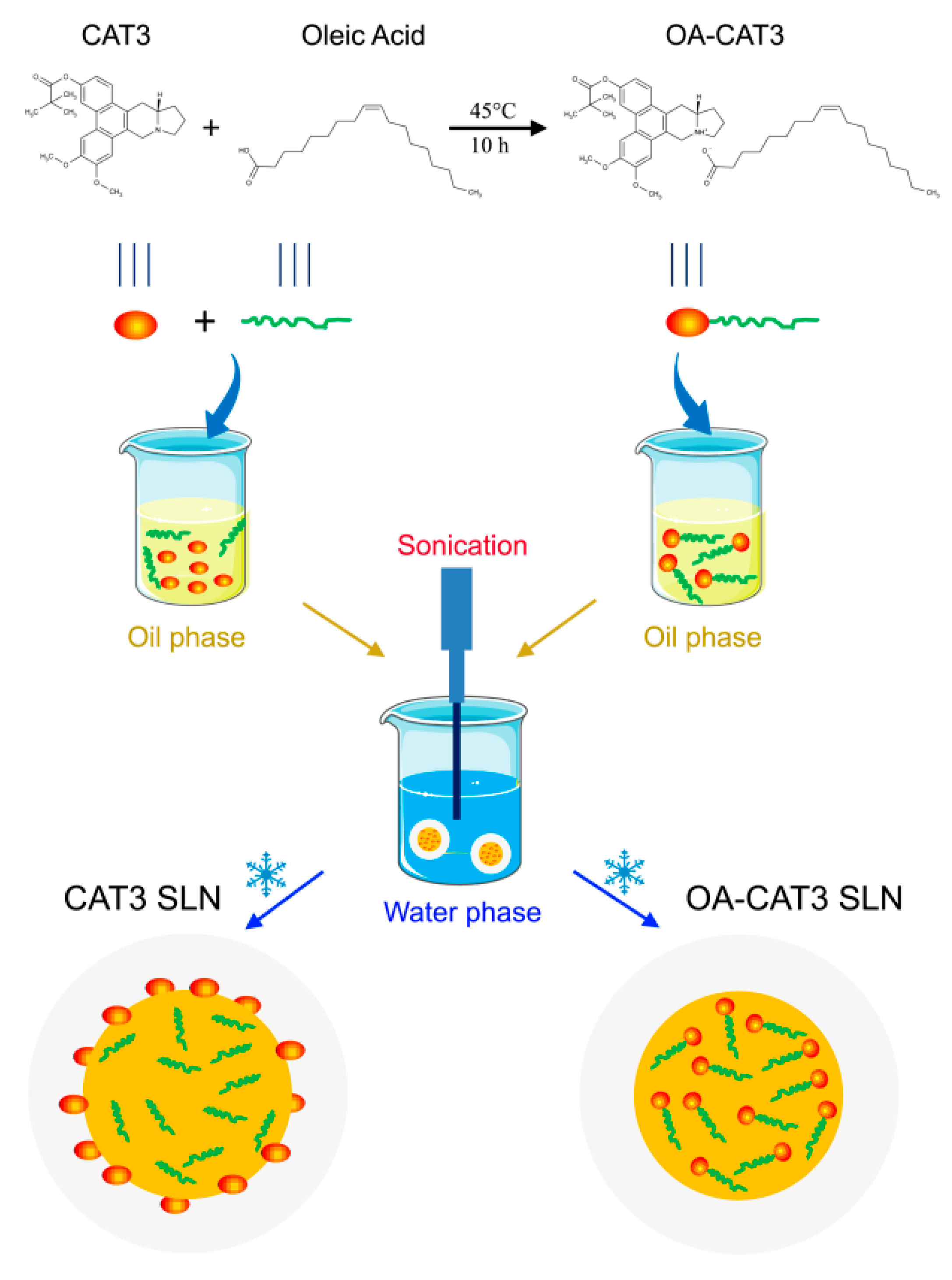
2.2.1. Preparation and Spectral Analysis of OA-CAT3
2.2.2. Preparation of OA-CAT3-SLN and CAT3-SLN
2.2.3. Physicochemical Characterization of OA-CAT3-SLN and CAT3-SLN
Morphology and Particle Size
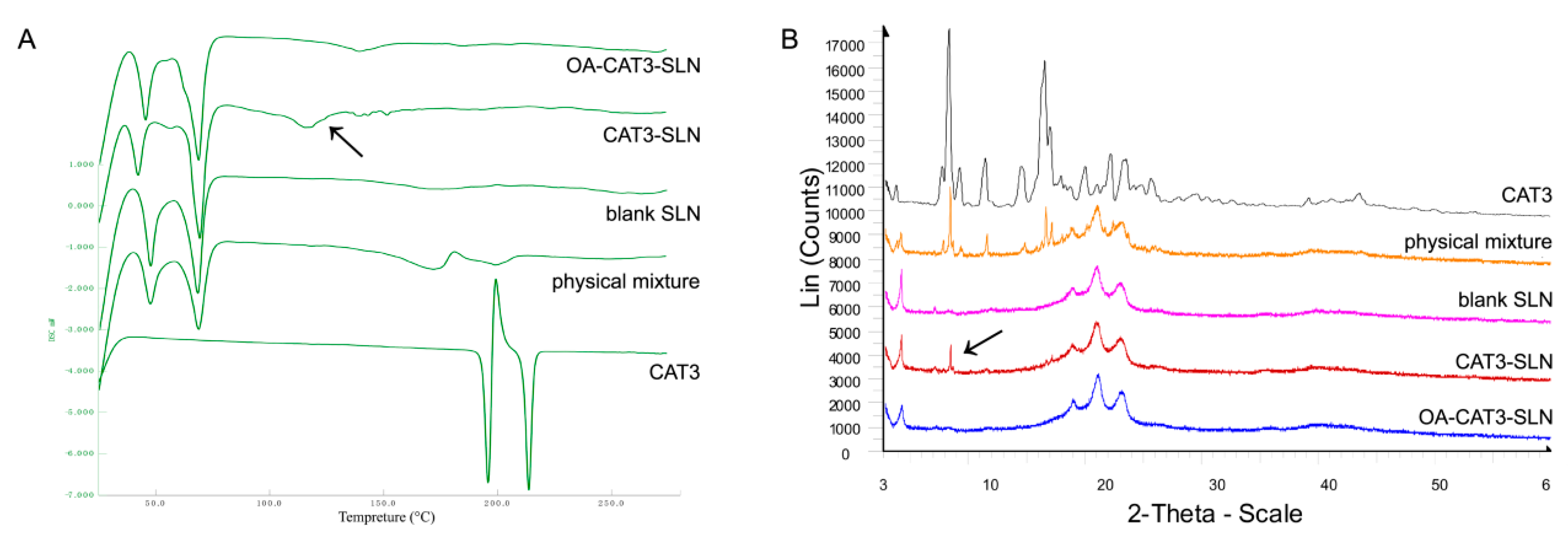
Differential Scanning Calorimetry (DSC) and Powder X-ray Diffractometry (PXRD)
Drug Loading (DL) and Encapsulation Efficiency (EE)
In Vitro Release and Releasing Mechanism
2.2.4. In Vitro and In Vivo Bio Evaluation of OA-CAT3-SLN
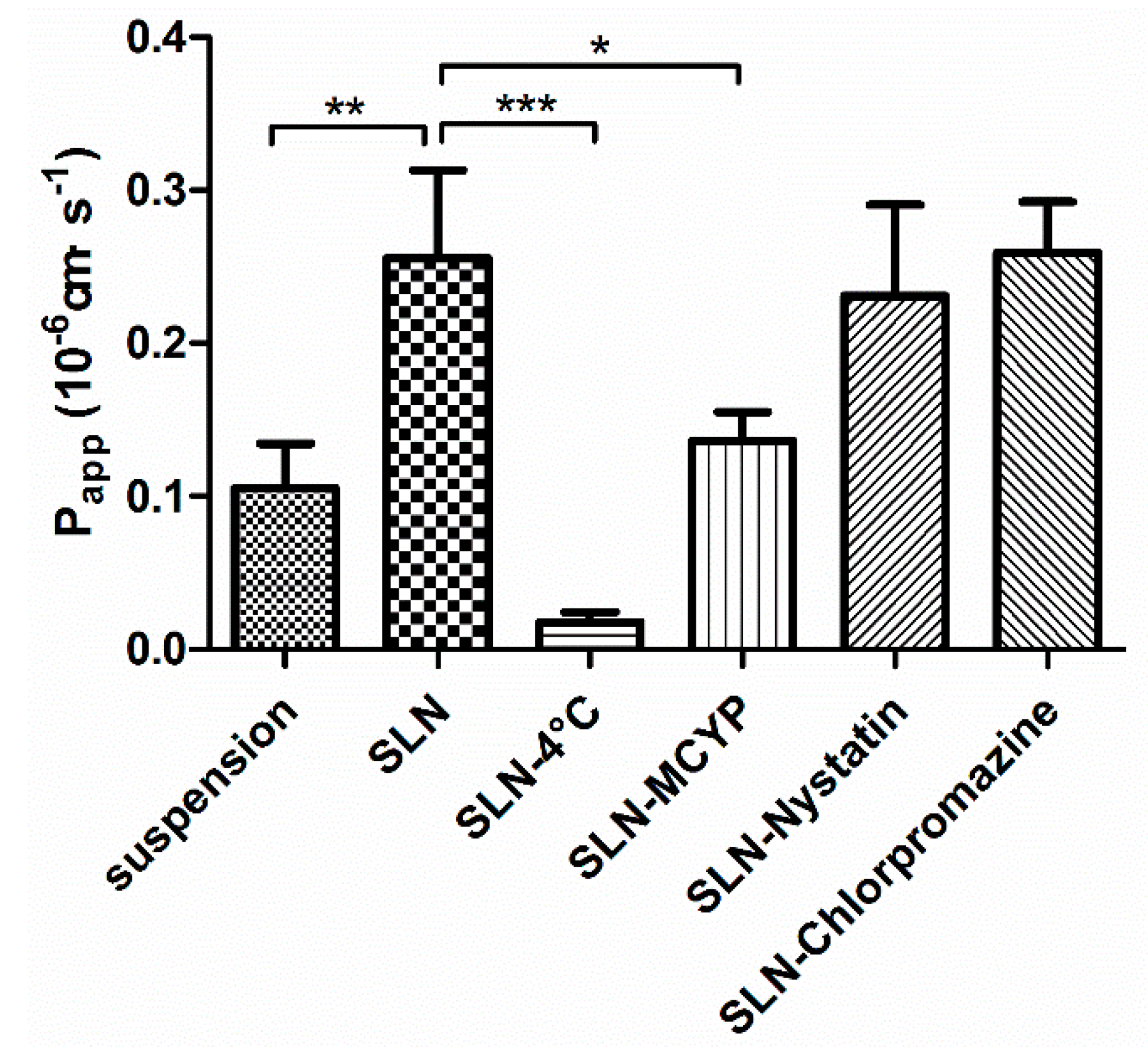
Transepithelial Transport Study
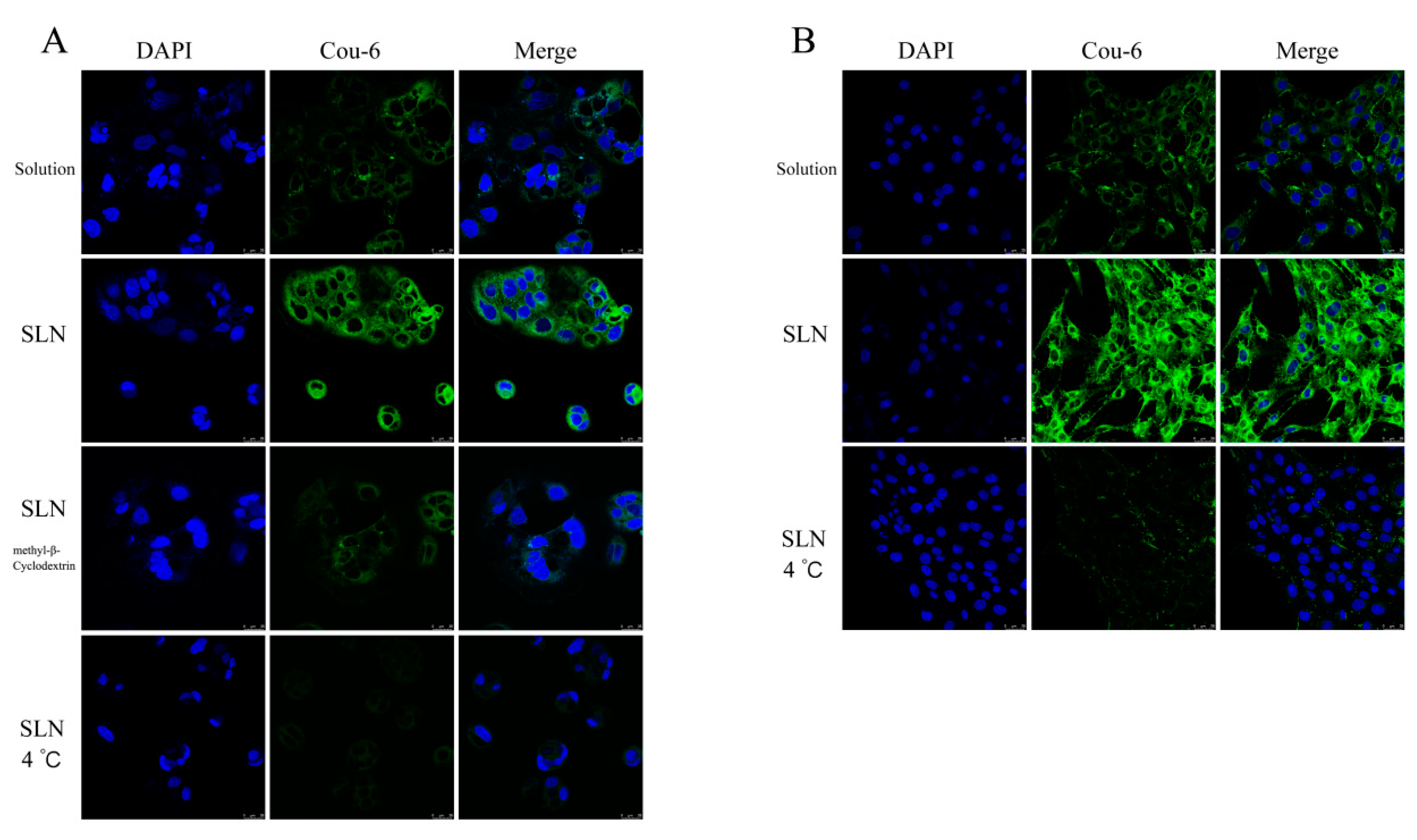
In vitro Cellular Uptake Study
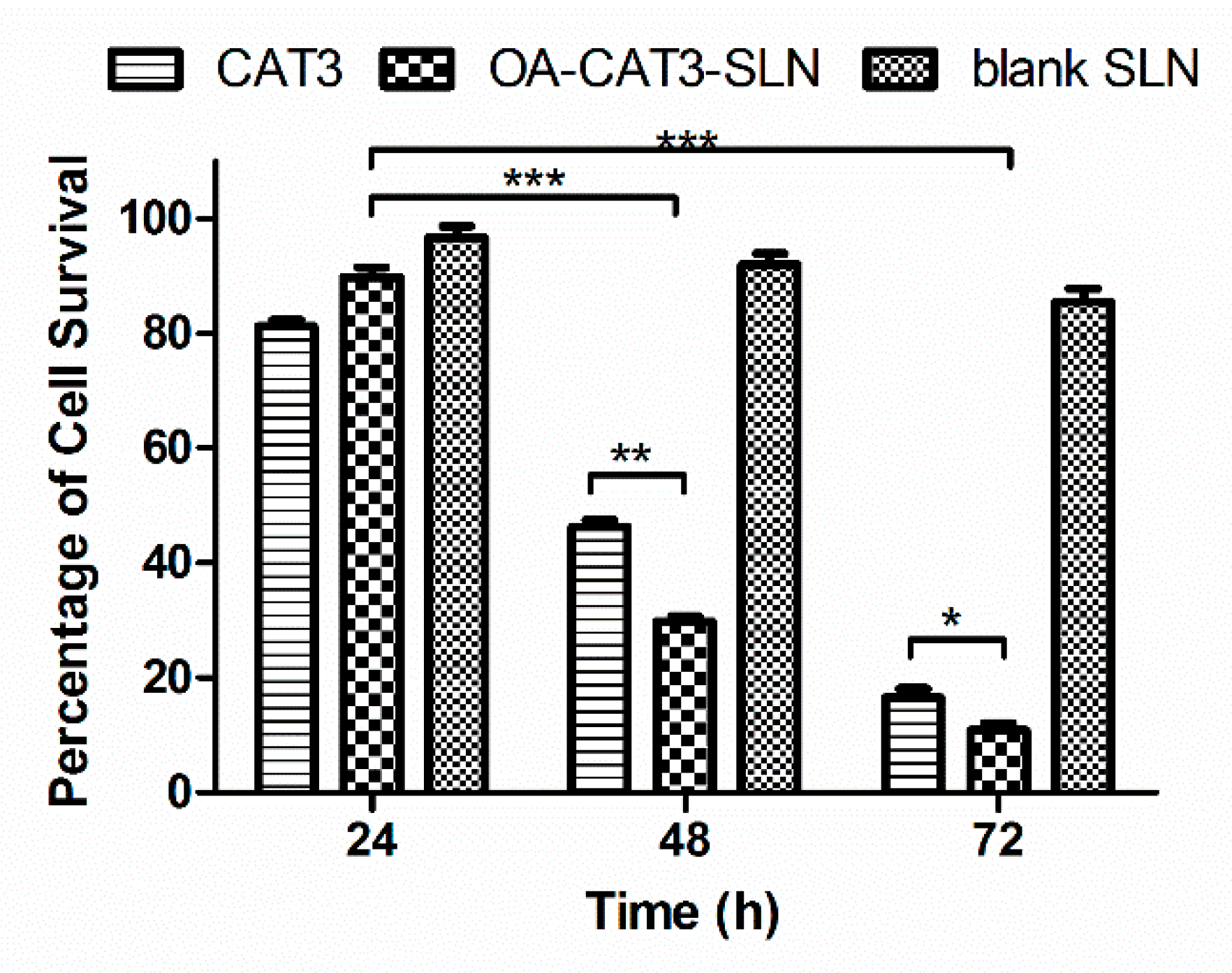
In Vitro Cytotoxicity Analysis
In Vivo Bioavailability Studies
2.2.5. Statistical Analysis
3. Results
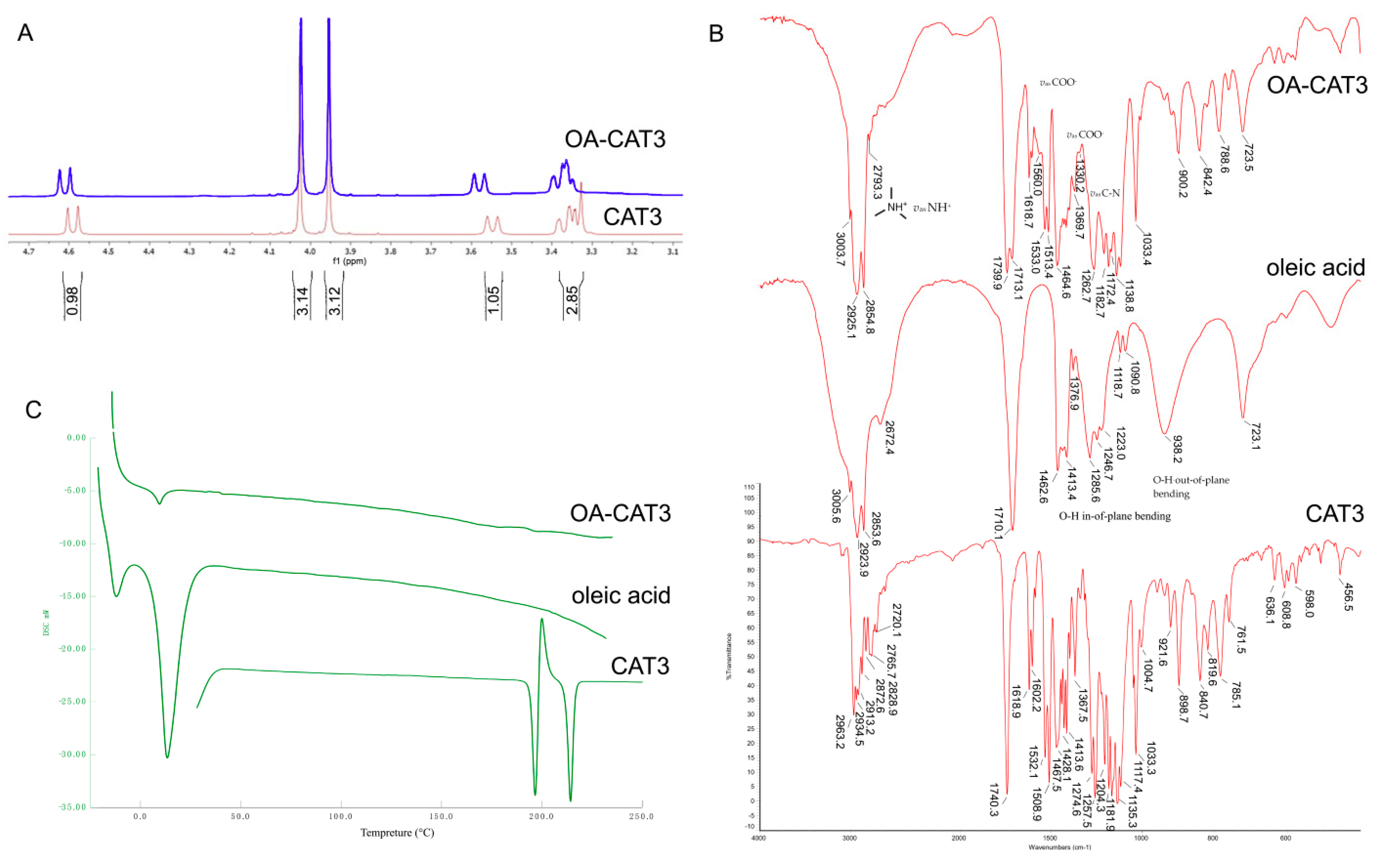
3.1. Characterization of OA-CAT3 by NMR, Fourier Transform Infrared Spectroscopy (FTIR), and Differential Scanning Calorimetry (DSC)
3.2. Characterizations of OA-CAT3-SLN and CAT3-SLN
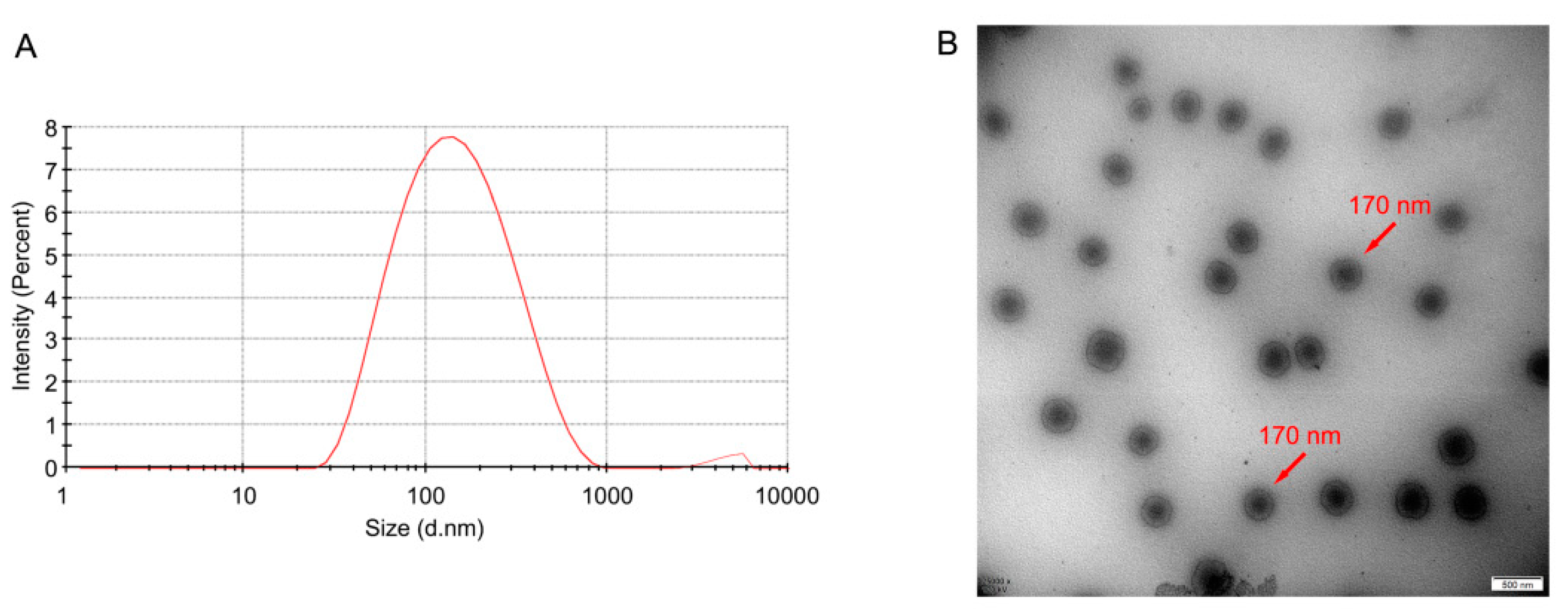
3.2.1. Droplet Size, PDI, Zeta Potential, and Morphology
3.2.2. Drug Loading (DL) and Encapsulation Efficiency (EE)
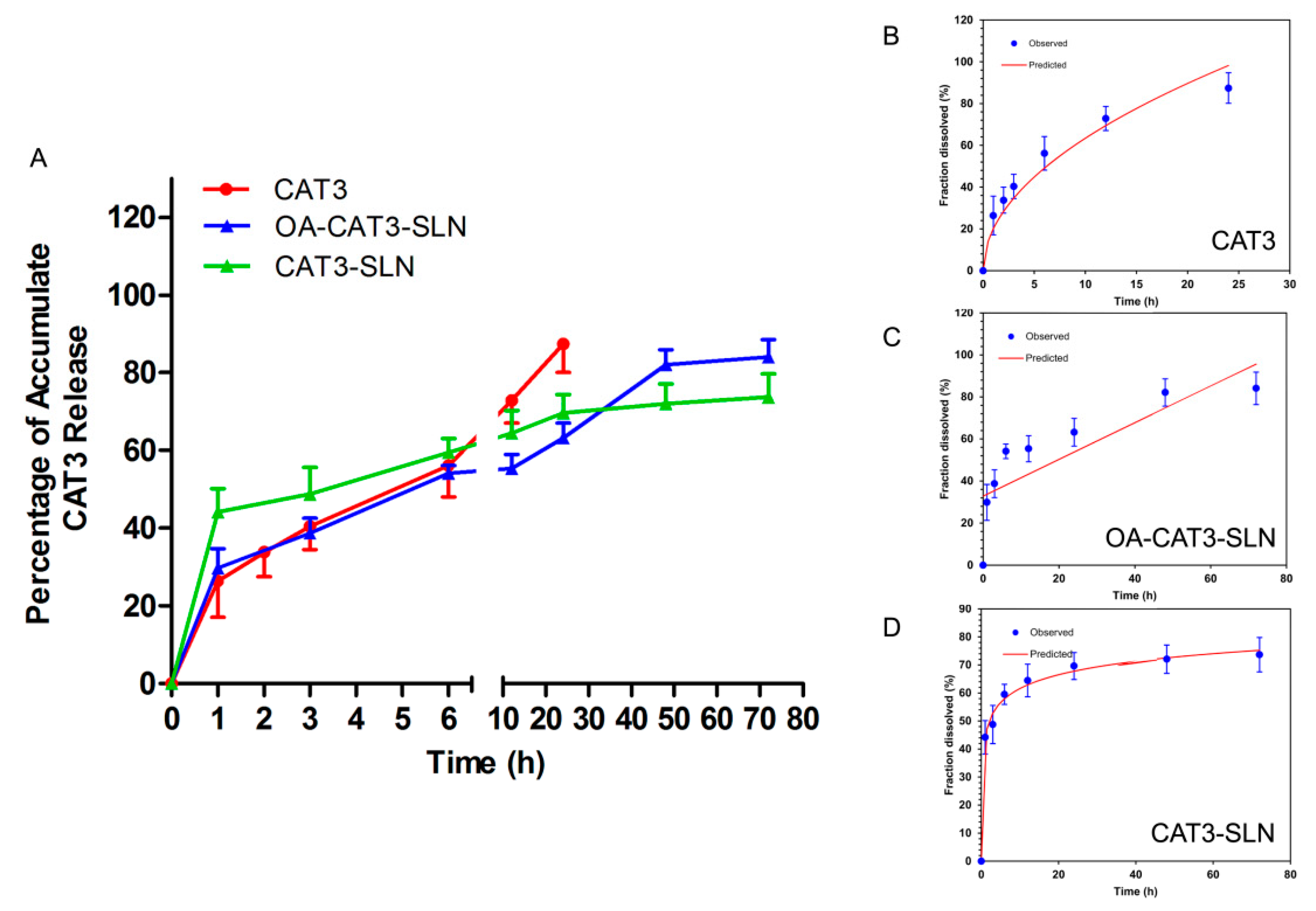
3.2.3. In Vitro Controlled Release Study
3.2.4. Differential Scanning Calorimetry (DSC) and Powder X-ray Diffractometry (PXRD) Studies
3.3. In Vitro and In Vivo Bio Evaluation of OA-CAT3-SLN
3.3.1. Transepithelial Transport Study
3.3.2. In Vitro Cellular Uptake Study
3.3.3. In Vitro Cytotoxicity Analysis
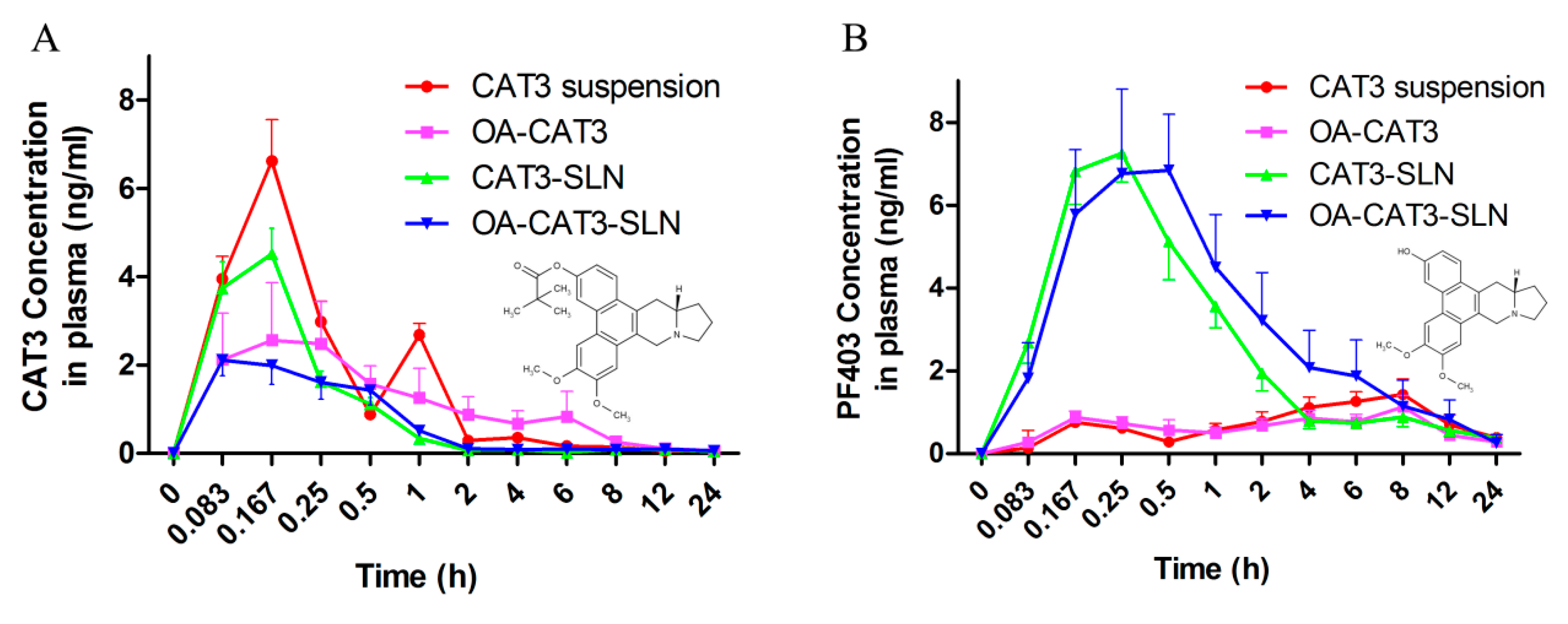
3.3.4. In Vivo Pharmacokinetic Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAT3 | 13a-(S)-3-pivaloyloxyl-6,7-dimethoxyphenanthro(9,10-b)-indolizidine |
| PF403 | 13a-(S)-3-hydroxyl-6,7-dimethoxyphenanthro(9,10-b)-indolizidine |
| GBM | Glioblastoma multiforme |
| TMZ | Temozolomide |
| SLN | Solid lipid nanoparticles |
| MDCK-MDR1 | Madin-Darby canine kidney cells transfected with the human MDR1 gene |
| SD | Sprague-Dawley |
| NMR | Nuclear magnetic resonance |
| TMS | Tetramethylsilane |
| DL | Drug loading |
| EE | Encapsulation efficiency |
| OA | Oleic acid |
| OA-CAT3 | Oleic acid and CAT3 conjugate |
| OA-CAT3-SLN | OA-CAT3 loaded SLN |
| CAT3-SLN | With the same composition and contents to the OA-CAT3-SLN except the preparation of OA-CAT3 in advance |
| DSC | Differential scanning calorimetry |
| FTIR | Fourier transform infrared spectroscopy |
| API | Active pharmaceutical ingredient |
| PVDF | Polyvinylidene fluoride |
| TEER | Transepithelial electrical resistance |
| DI | Deionized |
| PDI | Polydispersity index |
| HBSS | Hanks’ balanced salt solution |
| AP | Apical side |
| BL | Basolateral |
| LC-MS/MS | Liquid chromatography-mass spectrometry |
| Papp | Apparent permeability coefficient |
| HPLC | High-performance liquid chromatography |
| C6-luc | Luciferase-expressing C6 cells |
| DAPI | 4′,6-diamidino-2-phenylindole |
| LSCM | laser scanning confocal microscopy |
| MTT | 3-(4, 5-dimethylthiazol-2-yl)-2,5 diphenyltetrazolium bromide |
| MCYP | Methyl-β-Cyclodextrin |
| PBS | Phosphate-buffered saline |
| Cou-6 | Fluorescent Coumarin 6 |
| ISTD | Internal standard |
| DAS | Drug and Statistics |
References
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef] [PubMed]
- DeAngelis, L.M. Brain tumors. N. Engl. J. Med. 2001, 344, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.; Gong, Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 23. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Portnow, J.; Ammirati, M.; Baehring, J.; Brem, H.; Brown, P.; Butowski, N.; Chamberlain, M.C.; Fenstermaker, R.A.; Friedman, A.; et al. Central nervous system cancers, version 1.2015. J. Natl. Compr. Cancer Netw. 2015, 13, 1191–1202. [Google Scholar] [CrossRef]
- Alonso, M.M.; Gomez-Manzano, C.; Bekele, B.N.; Yung, W.A.; Fueyo, J. Adenovirus-based strategies overcome temozolomide resistance by silencing the O6-methylguanine-DNA methyltransferase promoter. Cancer Res. 2007, 67, 11499–11504. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Banelli, B.; Carra, E.; Barbieri, F.; Würth, R.; Parodi, F.; Pattarozzi, A.; Carosio, R.; Forlani, A.; Allemanni, G.; Marubbi, D. The histone demethylase KDM5A is a key factor for the resistance to temozolomide in glioblastoma. Cell Cycle 2015, 14, 3418–3429. [Google Scholar] [CrossRef]
- Ji, M.; Wang, L.; Chen, J.; Xue, N.; Wang, C.; Lai, F.; Wang, R.; Yu, S.; Jin, J.; Chen, X. CAT3, a prodrug of 13a (S)-3-hydroxyl-6, 7-dimethoxyphenanthro [9, 10-b]-indolizidine, circumvents temozolomide-resistant glioblastoma via the Hedgehog signaling pathway, independently of O6-methylguanine DNA methyltransferase expression. Oncotargets Ther. 2018, 11, 3671. [Google Scholar] [CrossRef]
- Liu, Z.; Lv, H.; Li, H.; Zhang, Y.; Zhang, H.; Su, F.; Xu, S.; Li, Y.; Si, Y.; Yu, S.; et al. Interaction studies of an anticancer alkaloid, (+)-(13aS)-deoxytylophorinine, with calf thymus DNA and four repeated double-helical DNAs. Chemotherapy 2011, 57, 310–320. [Google Scholar] [CrossRef]
- Chen, J.; Lv, H.; Hu, J.; Ji, M.; Xue, N.; Li, C.; Ma, S.; Zhou, Q.; Lin, B.; Li, Y. CAT3, a novel agent for medulloblastoma and glioblastoma treatment, inhibits tumor growth by disrupting the Hedgehog signaling pathway. Cancer Lett. 2016, 381, 391–403. [Google Scholar] [CrossRef]
- Lv, H.; Ren, J.; Ma, S.; Xu, S.; Qu, J.; Liu, Z.; Zhou, Q.; Chen, X.; Yu, S. Synthesis, biological evaluation and mechanism studies of deoxytylophorinine and its derivatives as potential anticancer agents. PLoS ONE 2012, 7, e30342. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Y.; Lv, H.; Li, S.; Tang, K.; Zhou, W.; Yu, S.; Chen, X. The novel anti-neuroblastoma agent PF403, inhibits proliferation and invasion in vitro and in brain xenografts. Int. J. Oncol. 2015, 47, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; He, J.; Zhang, R.; Lv, H.; Ma, S.; Chen, Y.; Yu, S.; Chen, X.; Wu, Y.; He, W.; et al. Integrated rapid resolution liquid chromatography-tandem mass spectrometric approach for screening and identification of metabolites of the potential anticancer agent 3,6,7-trimethoxyphenanthroindolizidine in rat urine. Anal. Chim. Acta 2012, 731, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Lv, H.; Li, C.; Ren, J.; Ma, S.; Xu, S.; Chen, X.; Yu, S. Stereospecific Synthesis and Biological Evaluation of Monodesmethyl Metabolites of (+)-13a-(S)-Deoxytylophorinine as Potential Antitumor Agents. Synthesis 2012, 44, 3757–3764. [Google Scholar] [CrossRef]
- Shen, M.-Y.; Liu, T.-I.; Yu, T.-W.; Kv, R.; Chiang, W.-H.; Tsai, Y.-C.; Chen, H.-H.; Lin, S.-C.; Chiu, H.-C. Hierarchically targetable polysaccharide-coated solid lipid nanoparticles as an oral chemo/thermotherapy delivery system for local treatment of colon cancer. Biomaterials 2019, 197, 86–100. [Google Scholar] [CrossRef]
- Naseri, N.; Valizadeh, H.; Zakeri-Milani, P. Solid lipid nanoparticles and nanostructured lipid carriers: Structure, preparation and application. Adv. Pharm. Bull. 2015, 5, 305. [Google Scholar] [CrossRef]
- Muchow, M.; Maincent, P.; Müller, R.H. Lipid nanoparticles with a solid matrix (SLN®, NLC®, LDC®) for oral drug delivery. Drug Dev. Ind. Pharm. 2008, 34, 1394–1405. [Google Scholar] [CrossRef]
- Schwarz, C. Solid lipid nanoparticles (SLN) for controlled drug delivery II. Drug incorporation and physicochemical characterization. J. Microencapsul. 1999, 16, 205–213. [Google Scholar] [CrossRef]
- Dudhipala, N.; Veerabrahma, K. Candesartan cilexetil loaded solid lipid nanoparticles for oral delivery: Characterization, pharmacokinetic and pharmacodynamic evaluation. Drug Deliv. 2016, 23, 395–404. [Google Scholar] [CrossRef]
- Manjunath, K.; Venkateswarlu, V. Pharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administration. J. Control. Release 2005, 107, 215–228. [Google Scholar] [CrossRef]
- Sahu, P.K.; Mishra, D.K.; Jain, N.; Rajoriya, V.; Jain, A.K. Mannosylated solid lipid nanoparticles for lung-targeted delivery of Paclitaxel. Drug Dev. Ind. Pharm. 2015, 41, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, L.; Gu, W.; Gao, Y.; Lin, L.; Zhang, Z.; Xi, Y.; Li, Y. The performance of docetaxel-loaded solid lipid nanoparticles targeted to hepatocellular carcinoma. Biomaterials 2009, 30, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Dudhipala, N.; Janga, K.Y.; Gorre, T. Comparative study of nisoldipine-loaded nanostructured lipid carriers and solid lipid nanoparticles for oral delivery: Preparation, characterization, permeation and pharmacokinetic evaluation. Artif. Cells Nanomed. Biotechnol. 2018, 46, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.; Runge, S.; Ravelli, V.; Mehnert, W.; Thünemann, A.F.; Souto, E. Oral bioavailability of cyclosporine: Solid lipid nanoparticles (SLN®) versus drug nanocrystals. Int. J. Pharm. 2006, 317, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.; Asadullah Madni, A.I.; Khan, W.S.; Khan, M.I.; Mahmood, M.A.; Ashfaq, M.; Bajwa, S.Z.; Shakir, I. Solid and liquid lipid-based binary solid lipid nanoparticles of diacerein: In vitro evaluation of sustained release, simultaneous loading of gold nanoparticles, and potential thermoresponsive behavior. Int. J. Nanomed. 2015, 10, 2805. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Radtke, M.; Wissing, S.A. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv. Drug Deliv. Rev. 2002, 54, S131–S155. [Google Scholar] [CrossRef]
- Almeida, E.D.P.; Dipieri, L.V.; Rossetti, F.C.; Marchetti, J.M.; Bentley, M.V.L.; Nunes, R.D.S.; Sarmento, V.H.V.; Valerio, M.E.G.; Júnior, J.J.R.; Montalvão, M.M. Skin permeation, biocompatibility and antitumor effect of chloroaluminum phthalocyanine associated to oleic acid in lipid nanoparticles. Photodiagnosis Photodyn. Ther. 2018, 24, 262–273. [Google Scholar] [CrossRef]
- Inoue, T.; Hisatsugu, Y.; Yamamoto, R.; Suzuki, M. Solid-liquid phase behavior of binary fatty acid mixtures: 1. Oleic acid/stearic acid and oleic acid/behenic acid mixtures. Chem. Phys. Lipids 2004, 127, 143–152. [Google Scholar] [CrossRef]
- Makwana, V.; Jain, R.; Patel, K.; Nivsarkar, M.; Joshi, A. Solid lipid nanoparticles (SLN) of Efavirenz as lymph targeting drug delivery system: Elucidation of mechanism of uptake using chylomicron flow blocking approach. Int. J. Pharm. 2015, 495, 439–446. [Google Scholar] [CrossRef]
- Venkateswarlu, V.; Manjunath, K. Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J. Control. Release 2004, 95, 627–638. [Google Scholar] [CrossRef]
- Kumar, V.V.; Chandrasekar, D.; Ramakrishna, S.; Kishan, V.; Rao, Y.M.; Diwan, P.V. Development and evaluation of nitrendipine loaded solid lipid nanoparticles: Influence of wax and glyceride lipids on plasma pharmacokinetics. Int. J. Pharm. 2007, 335, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Dolatabadi, J.E.N.; Hamishehkar, H.; Eskandani, M.; Valizadeh, H. Formulation, characterization and cytotoxicity studies of alendronate sodium-loaded solid lipid nanoparticles. Colloids Surf. B Biointerfaces 2014, 117, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Gao, Y.; Bouchacra, N.; Löbenberg, R. Evaluation of the DDSolver Software Applications. BioMed Res. Int. 2014, 2014, 204925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef]
- Yuan, H.; Chen, C.-Y.; Chai, G.-H.; Du, Y.-Z.; Hu, F.-Q. Improved transport and absorption through gastrointestinal tract by PEGylated solid lipid nanoparticles. Mol. Pharm. 2013, 10, 1865–1873. [Google Scholar] [CrossRef]
- Hu, Q.; Luo, Y. Recent advances of polysaccharide-based nanoparticles for oral insulin delivery. Int. J. Biol. Macromol. 2018, 120, 775–782. [Google Scholar] [CrossRef]
- Wu, L.; Liu, M.; Shan, W.; Cui, Y.; Zhang, Z.; Huang, Y. Lipid nanovehicles with adjustable surface properties for overcoming multiple barriers simultaneously in oral administration. Int. J. Pharm. 2017, 520, 216–227. [Google Scholar] [CrossRef]
- Teskač, K.; Kristl, J. The evidence for solid lipid nanoparticles mediated cell uptake of resveratrol. Int. J. Pharm. 2010, 390, 61–69. [Google Scholar] [CrossRef]
- Han, S.; Kwon, S.; Jeong, Y.; Yu, E.; Park, S. Physical characterization and in vitro skin permeation of solid lipid nanoparticles for transdermal delivery of quercetin. Int. J. Cosmet. Sci. 2014, 36, 588–597. [Google Scholar] [CrossRef]
- Kang, B.K.; Lee, J.S.; Chon, S.K.; Jeong, S.Y.; Yuk, S.H.; Khang, G.; Lee, H.B.; Cho, S.H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm. 2004, 274, 65–73. [Google Scholar] [CrossRef]
- Acosta, E. Bioavailability of nanoparticles in nutrient and nutraceutical delivery. Curr. Opin. Colloid Interface Sci. 2009, 14, 3–15. [Google Scholar] [CrossRef]
- Lingling, G.; Yuan, Z.; Weigen, L. Preparation, optimization, characterization and in vivo pharmacokinetic study of asiatic acid tromethamine salt-loaded solid lipid nanoparticles. Drug Dev. Ind. Pharm. 2016, 42, 1325–1333. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Chaudhury, A. Recent advances in lipid nanoparticle formulations with solid matrix for oral drug delivery. AAPS PharmSciTech 2011, 12, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Gaumet, M.; Vargas, A.; Gurny, R.; Delie, F. Nanoparticles for drug delivery: The need for precision in reporting particle size parameters. Eur. J. Pharm. Biopharm. 2008, 69, 1–9. [Google Scholar] [CrossRef]
- Gupta, M.; Vyas, S.P. Development, characterization and in vivo assessment of effective lipidic nanoparticles for dermal delivery of fluconazole against cutaneous candidiasis. Chem. Phys. Lipids 2012, 165, 454–461. [Google Scholar] [CrossRef]
- You, J.; Wan, F.; de Cui, F.; Sun, Y.; Du, Y.-Z.; qiang Hu, F. Preparation and characteristic of vinorelbine bitartrate-loaded solid lipid nanoparticles. Int. J. Pharm. 2007, 343, 270–276. [Google Scholar] [CrossRef]
- Hu, F.-Q.; Jiang, S.-P.; Du, Y.-Z.; Yuan, H.; Ye, Y.-Q.; Zeng, S. Preparation and characterization of stearic acid nanostructured lipid carriers by solvent diffusion method in an aqueous system. Colloids Surf. B Biointerfaces 2005, 45, 167–173. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, X.; Jin, Y.; Shan, W.; Huang, Y. Mechanism study of cellular uptake and tight junction opening mediated by goblet cell-specific trimethyl chitosan nanoparticles. Mol. Pharm. 2014, 11, 1520–1532. [Google Scholar] [CrossRef]
- Chai, G.-H.; Hu, F.-Q.; Sun, J.; Du, Y.-Z.; You, J.; Yuan, H. Transport pathways of solid lipid nanoparticles across Madin–Darby canine kidney epithelial cell monolayer. Mol. Pharm. 2014, 11, 3716–3726. [Google Scholar] [CrossRef]
- Wang, D.; Zhao, P.; Cui, F.; Li, X. Preparation and characterization of solid lipid nanoparticles loaded with total flavones of Hippophae rhamnoides (TFH). PDA J. Pharm. Sci. Technol. 2007, 61, 110–120. [Google Scholar]
- Freitas, C.; Müller, R.H. Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLN™) dispersions. Int. J. Pharm. 1998, 168, 221–229. [Google Scholar] [CrossRef]
- Alexander, A.; Khichariya, A.; Gupta, S.; Patel, R.J.; Giri, T.K.; Tripathi, D.K. Recent expansions in an emergent novel drug delivery technology: Emulgel. J. Control. Release 2013, 171, 122–132. [Google Scholar]
- Aburahma, M.H.; Badr-Eldin, S.M. Compritol 888 ATO: A multifunctional lipid excipient in drug delivery systems and nanopharmaceuticals. Expert Opin. Drug Deliv. 2014, 11, 1865–1883. [Google Scholar] [CrossRef] [PubMed]
- Doijad, R.; Manvi, F.; Godhwani, D.; Joseph, R.; Deshmukh, N. Formulation and targeting efficiency of cisplatin engineered solid lipid nanoparticles. Indian J. Pharm. Sci. 2008, 70, 203. [Google Scholar] [CrossRef]
- Das, S.; Ng, W.K.; Tan, R.B. Are nanostructured lipid carriers (NLCs) better than solid lipid nanoparticles (SLNs): Development, characterizations and comparative evaluations of clotrimazole-loaded SLNs and NLCs? Eur. J. Pharm. Sci. 2012, 47, 139–151. [Google Scholar] [CrossRef]
- zur Mühlen, A.; Schwarz, C.; Mehnert, W. Solid lipid nanoparticles (SLN) for controlled drug delivery–drug release and release mechanism. Eur. J. Pharm. Biopharm. 1998, 45, 149–155. [Google Scholar] [CrossRef]
- Gordillo-Galeano, A.; Mora-Huertas, C.E. Solid lipid nanoparticles and nanostructured lipid carriers: A review emphasizing on particle structure and drug release. Eur. J. Pharm. Biopharm. 2018, 133, 285–308. [Google Scholar] [CrossRef]
- Wang, R.-B.; Lv, H.-N.; Zhu, S.-S.; Ren, X.-D.; Xu, S.; Ma, S.-G.; Liu, Y.-B.; Qu, J.; Yu, S.-S. A novel and practical synthesis of CAT3: A phenanthroindolizidine alkaloid with potential in treating glioblastoma. RSC Adv. 2018, 8, 29301–29308. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Particle Size (nm) | PDI | Zeta Potential (mV) | DL (%) | EE (%) |
|---|---|---|---|---|---|
| Blank SLN | 139.2 ± 2.47 | 0.257 ± 0.003 | −41.7 ± 0.80 ** | / | / |
| OA-CAT3-SLN | 151.3 ± 17.51 | 0.230 ± 0.018 | −26.7 ± 0.46 | 5.478 ± 0.346 | 80.65 ± 6.79 |
| CAT3-SLN | 155.7 ± 3.03 | 0.228 ± 0.007 | −7.9 ± 0.15 ** | 4.067 ± 0.163 ** | 58.48 ± 3.35 ** |
| Parameters | CAT3 | OA-CAT3 | CAT3-SLN | OA-CAT3-SLN | |
|---|---|---|---|---|---|
| AUC(0–t) | ng·mL−1·h | 6.39 ± 0.645 ** | 8.632 ± 3.843 * | 3.093 ± 0.133 | 3.284 ± 1.254 |
| AUC(0–∞) | ng·mL−1·h | 7.23 ± 0.476 ** | 8.954 ± 3.716 * | 3.807 ± 0.441 | 4.356 ± 1.562 |
| MRT(0–t) | H | 3.887 ± 0.805 | 5.113 ± 0.616 | 5.433 ± 0.82 | 6.158 ± 0.704 |
| MRT(0–∞) | H | 8.779 ± 6.346 | 6.48 ± 2.444 | 13.954 ± 6.912 | 16.604 ± 2.597 |
| t1/2z | H | 10.824 ± 6.769 | 4.754 ± 3.951 | 16.742 ± 11.915 | 16.902 ± 1.578 |
| Tmax | H | 0.167 ± 0 | 0.25 ± 0.129 | 0.153 ± 0.034 | 0.125 ± 0.07 |
| Vz/F | L/kg | 21,169.855 ± 12,087.293 | 8806.657 ± 8572.706 | 62,090.614 ± 42,268.807 | 61,307.763 ± 18,523.656 |
| CLz/F | L/h/kg | 1388.185 ± 91.525 | 1268.605 ± 450.993 | 2653.874 ± 278.551 | 2576.094 ± 1027.274 |
| Cmax | ng/mL | 6.618 ± 0.942 ** | 2.711 ± 1.201 | 4.539 ± 1.406 ** | 2.245 ± 0.951 |
| Parameters | CAT3 | OA-CAT3 | CAT3-SLN | OA-CAT3-SLN | |
|---|---|---|---|---|---|
| AUC(0–t) | ng·mL−1·h | 18.778 ± 1.953 * | 13.713 ± 5.616 * | 21.723 ± 6.763 * | 32.045 ± 7.425 |
| AUC(0–∞) | ng·mL−1·h | 24.42 ± 2.435 * | 30.861 ± 24.886 | 25.541 ± 9.898 * | 34.73 ± 6.178 |
| MRT(0–t) | H | 9.775 ± 0.104 | 9.677 ± 1.252 | 6.649 ± 1.068 | 5.945 ± 0.889 |
| MRT(0–∞) | H | 16.449 ± 0.87 | 56.077 ± 75.093 | 10.837 ± 4.978 | 7.742 ± 1.978 |
| t1/2z | H | 10.121 ± 0.759 | 39.049 ± 53.163 | 8.94 ± 3.181 | 6.218 ± 1.528 |
| Tmax | H | 8 ± 0 | 4.736 ± 3.834 | 0.264 ± 0.123 | 0.417 ± 0.129 |
| Vz/F | L/kg | 6011.79 ± 535.387 | 15,412.451 ± 13,804.038 | 5298.387 ± 1424.022 | 2807.484 ± 748.904 |
| CLz/F | L/h/kg | 412.919 ± 41.283 | 442.039 ± 193.353 | 439.687 ± 152.452 | 333.068 ± 131.27 |
| Cmax | ng/mL | 1.429 ± 0.171 ** | 1.431 ± 0.315 ** | 7.884 ± 1.621 | 7.36 ± 1.694 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Li, L.; Ye, J.; Wang, R.; Wang, R.; Hu, J.; Wang, Y.; Dong, W.; Xia, X.; Yang, Y.; et al. Improving the Oral Bioavailability of an Anti-Glioma Prodrug CAT3 Using Novel Solid Lipid Nanoparticles Containing Oleic Acid-CAT3 Conjugates. Pharmaceutics 2020, 12, 126. https://doi.org/10.3390/pharmaceutics12020126
Wang H, Li L, Ye J, Wang R, Wang R, Hu J, Wang Y, Dong W, Xia X, Yang Y, et al. Improving the Oral Bioavailability of an Anti-Glioma Prodrug CAT3 Using Novel Solid Lipid Nanoparticles Containing Oleic Acid-CAT3 Conjugates. Pharmaceutics. 2020; 12(2):126. https://doi.org/10.3390/pharmaceutics12020126
Chicago/Turabian StyleWang, Hongliang, Lin Li, Jun Ye, Rubing Wang, Renyun Wang, Jinping Hu, Yanan Wang, Wujun Dong, Xuejun Xia, Yanfang Yang, and et al. 2020. "Improving the Oral Bioavailability of an Anti-Glioma Prodrug CAT3 Using Novel Solid Lipid Nanoparticles Containing Oleic Acid-CAT3 Conjugates" Pharmaceutics 12, no. 2: 126. https://doi.org/10.3390/pharmaceutics12020126
APA StyleWang, H., Li, L., Ye, J., Wang, R., Wang, R., Hu, J., Wang, Y., Dong, W., Xia, X., Yang, Y., Gao, Y., Gao, L., & Liu, Y. (2020). Improving the Oral Bioavailability of an Anti-Glioma Prodrug CAT3 Using Novel Solid Lipid Nanoparticles Containing Oleic Acid-CAT3 Conjugates. Pharmaceutics, 12(2), 126. https://doi.org/10.3390/pharmaceutics12020126