Self-Assembled Disulfide Bond Bearing Paclitaxel—Camptothecin Prodrug Nanoparticle for Lung Cancer Therapy


 ,
, 
Abstract
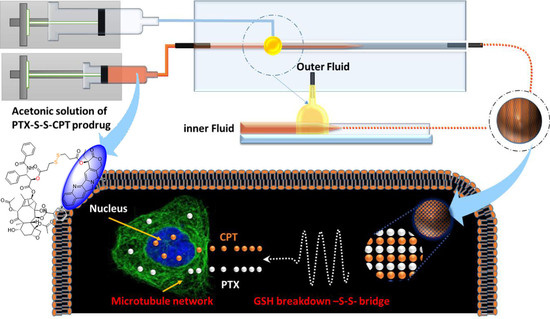
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of PTX-S-S-CPT
2.3. Characterization of CPT-S-S-PTX
2.4. Assembly of Three-Dimensional Microfluidic Chip
2.5. Preparation of Nanoparticles
2.6. Characterization of the Nanoparticles
2.7. In Vitro Release of PTX and CPT from the Nanoparticles
2.8. Cell Assays
2.8.1. Cell Culture and Maintenance
2.8.2. Cytotoxicity Assay
2.8.3. Cellular Uptake Evaluation
3. Results
3.1. Characterization of CPT-S-S-PTX Prodrug
3.2. Characterization of Prodrug-Assembled Nanoparticles
3.3. In Vitro Release of CPT from the CPT-S-S-PTX Nanoparticles
3.4. In Vitro Cell Viability Assay
3.5. Cell Uptake
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, Y.; Borrel, A.; Ghemtio, L.; Regad, L.; Boije af Gennäs, G.; Camproux, A.-C.; Yli-Kauhaluoma, J.; Xhaard, H. Structural Isosteres of Phosphate Groups in the Protein Data Bank. J. Chem. Inf. Model. 2017, 57, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.E.; Krishna, A.; Ma, Y.; Webb, T.E.; Marshall, D.C.; Tooke, C.L.; Spencer, J.; Clarke, T.B.; Armstrong, A.; Edwards, A.M. Exploitation of Antibiotic Resistance as a Novel Drug Target: Development of a β-Lactamase-Activated Antibacterial Prodrug. J. Med. Chem. 2019, 62, 4411–4425. [Google Scholar] [CrossRef] [PubMed]
- Albert, A. Chemical Aspects of Selective Toxicity. Nature 1958, 182, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Subbaiah, M.A.M.; Meanwell, N.A.; Kadow, J.F.; Subramani, L.; Annadurai, M.; Ramar, T.; Desai, S.D.; Sinha, S.; Subramanian, M.; Mandlekar, S.; et al. Coupling of an Acyl Migration Prodrug Strategy with Bio-activation To Improve Oral Delivery of the HIV-1 Protease Inhibitor Atazanavir. J. Med. Chem. 2018, 61, 4176–4188. [Google Scholar] [CrossRef]
- Rautio, J.; Meanwell, N.A.; Di, L.; Hageman, M.J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018, 17, 559–587. [Google Scholar] [CrossRef]
- Andriotis, E.G.; Eleftheriadis, G.K.; Karavasili, C.; Fatouros, D.G. Development of Bio-Active Patches Based on Pectin for the Treatment of Ulcers and Wounds Using 3D-Bioprinting Technology. Pharmaceutics 2020, 12, 56. [Google Scholar] [CrossRef]
- Xiao, H.; Yan, L.; Dempsey, E.M.; Song, W.; Qi, R.; Li, W.; Huang, Y.; Jing, X.; Zhou, D.; Ding, J.; et al. Recent progress in polymer-based platinum drug delivery systems. Prog. Polym. Sci. 2018, 87, 70–106. [Google Scholar] [CrossRef]
- Shamay, Y.; Shah, J.; Işık, M.; Mizrachi, A.; Leibold, J.; Tschaharganeh, D.F.; Roxbury, D.; Budhathoki-Uprety, J.; Nawaly, K.; Sugarman, J.L.; et al. Quantitative self-assembly prediction yields targeted nanomedicines. Nature Mater. 2018, 17, 361–368. [Google Scholar] [CrossRef]
- Ma, X.; Özliseli, E.; Zhang, Y.; Pan, G.; Wang, D.; Zhang, H. Fabrication of redox-responsive doxorubicin and paclitaxel prodrug nanoparticles with microfluidics for selective cancer therapy. Biomater. Sci. 2019, 7, 634–644. [Google Scholar] [CrossRef]
- Bilati, U.; Allémann, E.; Doelker, E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur. J. Pharm. Sci. 2005, 24, 67–75. [Google Scholar] [CrossRef]
- Liu, D.; Cito, S.; Zhang, Y.; Wang, C.F.; Sikanen, T.M.; Santos, H.A. A versatile and robust microfluidic platform toward high throughput synthesis of homogeneous nanoparticles with tunable properties. Adv. Mater 2015, 27, 2298–2304. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-H.; Zhang, Y.-M.; Sheng, X.; Wang, J.; Liu, Y. Enzyme-responsive fluorescent camptothecin prodrug/polysaccharide supramolecular assembly for targeted cellular imaging and in situ controlled drug release. ChemComm 2020. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, Y.; Weisensee, K. Conducting Polymeric Nanocomposites with a Three-Dimensional Co-flow Microfluidics Platform. Micromachines 2019, 10, 383. [Google Scholar] [CrossRef]
- Hua, S. Comparison of in vitro dialysis release methods of loperamide-encapsulated liposomal gel for topical drug delivery. Int. J. Nanomed. 2014, 9, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhai, Y.; Ye, H.; Lv, Q.; Sun, B.; Luo, C.; Jiang, Q.; Zhang, H.; Xu, Y.; Jing, Y.; et al. High Co-loading Capacity and Stimuli-Responsive Release Based on Cascade Reaction of Self-Destructive Polymer for Improved Chemo-Photodynamic Therapy. ACS Nano 2019, 13, 7010–7023. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Horwitz, S.B. Taxol(®): The First Microtubule Stabilizing Agent. Int. J. Mol. Sci. 2017, 18, 1733. [Google Scholar] [CrossRef]
- Kundranda, M.N.; Niu, J. Albumin-bound paclitaxel in solid tumors: Clinical development and future directions. Drug Des. Devel. Ther. 2015, 9, 3767–3777. [Google Scholar] [CrossRef]
- Pei, Q.; Hu, X.; Zheng, X.; Xia, R.; Liu, S.; Xie, Z.; Jing, X. Albumin-bound paclitaxel dimeric prodrug nanoparticles with tumor redox heterogeneity-triggered drug release for synergistic photothermal/ chemotherapy. Nano Res. 2019, 12, 877. [Google Scholar] [CrossRef]
- Gokduman, K. Strategies Targeting DNA Topoisomerase I in Cancer Chemotherapy: Camptothecins, Nanocarriers for Camptothecins, Organic Non-Camptothecin Compounds and Metal Complexes. Curr. Drug Targets 2016, 17, 1928–1939. [Google Scholar] [CrossRef] [PubMed]
- Dvoranová, D.; Bobeničová, M.; Šoralová, S.; Breza, M. On UV–Vis spectra and structure of the anticancer drug camptothecin in solutions. Chem. Phys. Lett. 2013, 580, 141–144. [Google Scholar] [CrossRef]
- Chao, P.; Deshmukh, M.; Kutscher, H.L.; Gao, D.; Rajan, S.S.; Hu, P.; Laskin, D.L.; Stein, S.; Sinko, P.J. Pulmonary targeting microparticulate camptothecin delivery system: Anticancer evaluation in a rat orthotopic lung cancer model. Anticancer Drugs 2010, 21, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Kotz, F.; Arnold, K.; Bauer, W.; Schild, D.; Keller, N.; Sachsenheimer, K.; Nargang, T.M.; Richter, C.; Helmer, D.; Rapp, B.E. Three-dimensional printing of transparent fused silica glass. Nature 2017, 544, 337–339. [Google Scholar] [CrossRef]
- Kotz, F.; Risch, P.; Arnold, K.; Sevim, S.; Puigmartí-Luis, J.; Quick, A.; Thiel, M.; Hrynevich, A.; Dalton, P.D.; Helmer, D.; et al. Fabrication of arbitrary three-dimensional suspended hollow microstructures in transparent fused silica glass. Nat. Commun. 2019, 10, 1439. [Google Scholar] [CrossRef]
- Sindhwani, S.; Syed, A.M.; Ngai, J.; Kingston, B.R.; Maiorino, L.; Rothschild, J.; MacMillan, P.; Zhang, Y.; Rajesh, N.U.; Hoang, T.; et al. The entry of nanoparticles into solid tumours. Nat. Mater. 2020, 19, 566–575. [Google Scholar] [CrossRef]
- Ding, Y.; Xu, Y.; Yang, W.; Niu, P.; Li, X.; Chen, Y.; Li, Z.; Liu, Y.; An, Y.; Liu, Y.; et al. Investigating the EPR effect of nanomedicines in human renal tumors via ex vivo perfusion strategy. Nano Today 2020, 35, 100970. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Ma, X.; Zhang, L.; Yan, J.; Cui, H.; Zhang, Y.; Wang, D.; Zhang, H. Self-Assembled Disulfide Bond Bearing Paclitaxel—Camptothecin Prodrug Nanoparticle for Lung Cancer Therapy. Pharmaceutics 2020, 12, 1169. https://doi.org/10.3390/pharmaceutics12121169
Gao J, Ma X, Zhang L, Yan J, Cui H, Zhang Y, Wang D, Zhang H. Self-Assembled Disulfide Bond Bearing Paclitaxel—Camptothecin Prodrug Nanoparticle for Lung Cancer Therapy. Pharmaceutics. 2020; 12(12):1169. https://doi.org/10.3390/pharmaceutics12121169
Chicago/Turabian StyleGao, Jingyan, Xiaodong Ma, Lirong Zhang, Jiaqi Yan, Huaguang Cui, Yuezhou Zhang, Dongqing Wang, and Hongbo Zhang. 2020. "Self-Assembled Disulfide Bond Bearing Paclitaxel—Camptothecin Prodrug Nanoparticle for Lung Cancer Therapy" Pharmaceutics 12, no. 12: 1169. https://doi.org/10.3390/pharmaceutics12121169
APA StyleGao, J., Ma, X., Zhang, L., Yan, J., Cui, H., Zhang, Y., Wang, D., & Zhang, H. (2020). Self-Assembled Disulfide Bond Bearing Paclitaxel—Camptothecin Prodrug Nanoparticle for Lung Cancer Therapy. Pharmaceutics, 12(12), 1169. https://doi.org/10.3390/pharmaceutics12121169