Application of Focus Variation Microscopy and Dissolution Imaging in Understanding the Behaviour of Hydrophilic Matrices

 ,
,  ,
,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. True Density
2.2.2. Compact Manufacture
2.2.3. Tablet Dimensions, Hardness and Porosity
2.2.4. Scanning Electron Microscopy (SEM)
2.2.5. Surface Assessment of the Compact Using Focus Variation Microscopy
2.2.6. Dissolution Imaging (Whole Dosage Cell Measurement)
3. Results and Discussion
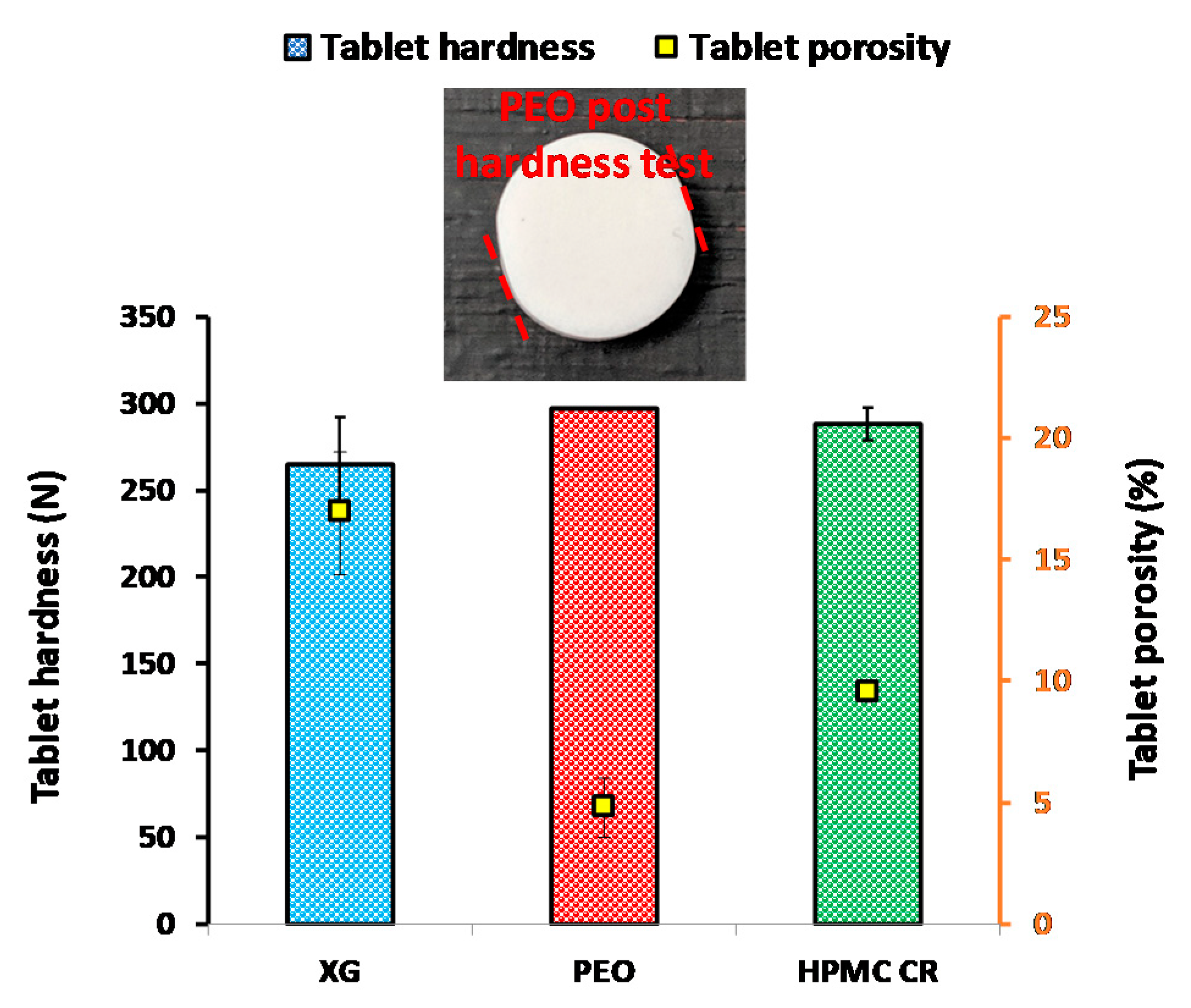
3.1. Micrometric and Tabletting Properties
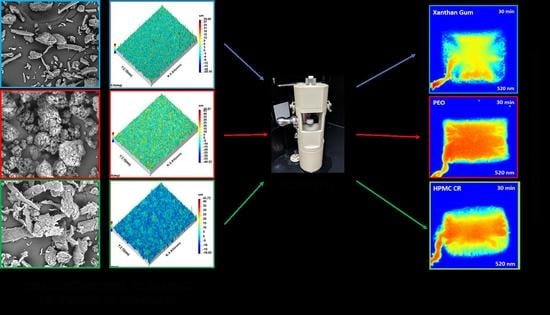
3.2. Focus Variation Microscopy
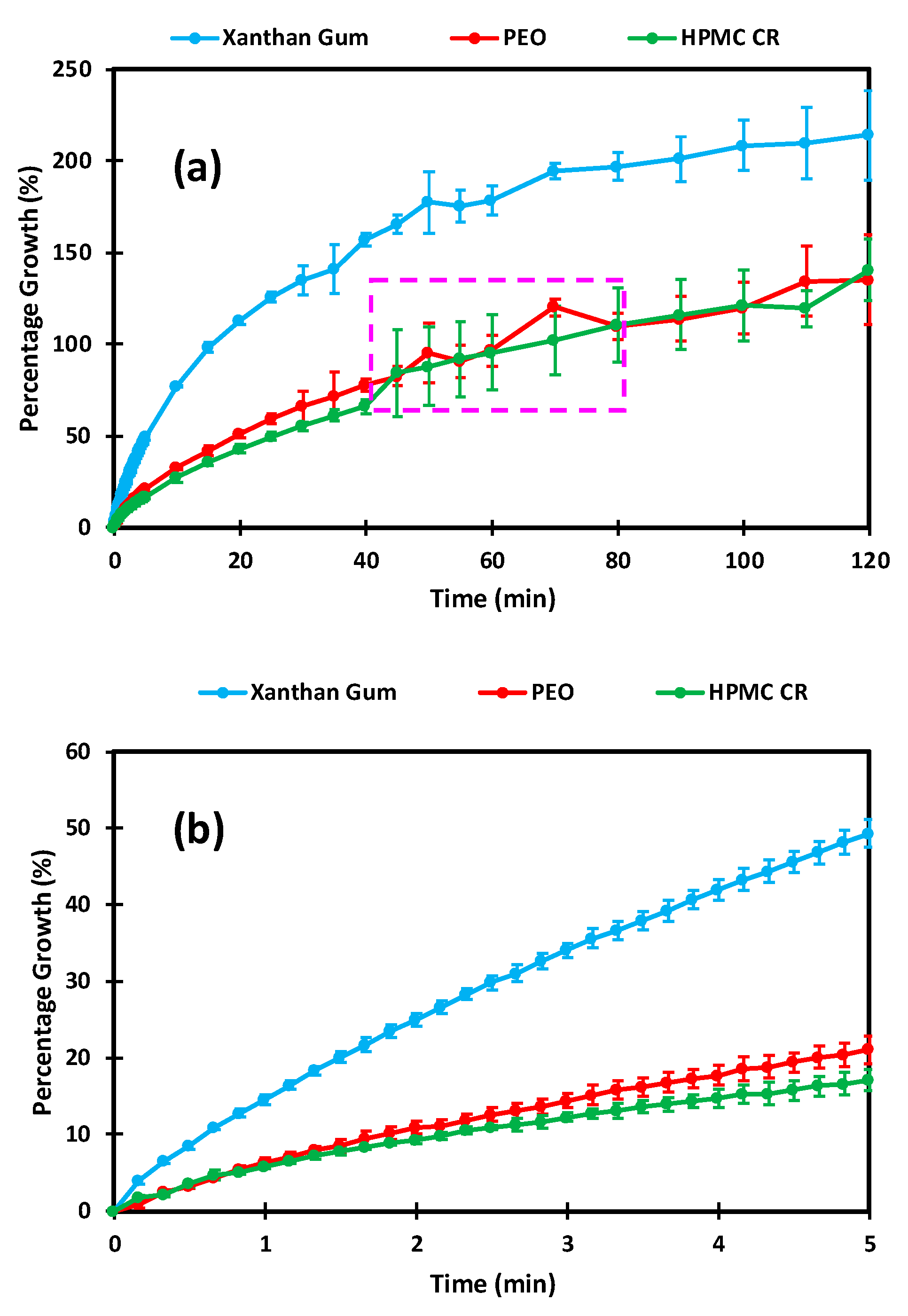
3.3. Dissolution Imaging and Swelling Assessment
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Timmins, P.; Desai, D.; Chen, W.; Wray, P.; Brown, J.; Hanley, S. Advances in mechanistic understanding of release rate control mechanisms of extended-release hydrophilic matrix tablets. Ther. Deliv. 2016, 7, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Alderman, D.A. A review of cellulose ethers in hydrophilic matrices for oral controlled-release dosage forms. Int. J. Pharm. Tech. Prod. Mfr. 1984, 5, 1–9. [Google Scholar]
- Ward, A.; Walton, K.; Mawla, N.; Kaialy, W.; Liu, L.; Timmins, P.; Conway, B.R.; Asare-Addo, K. Development of a novel method utilising dissolution imaging for the measurement of swelling behaviour in hydrophilic matrices. Int. J. Pharm. X 2019, 1, 100013. [Google Scholar] [CrossRef] [PubMed]
- Zahoor, F.D.; Mader, K.T.; Timmins, P.; Brown, J.; Sammon, C. Investigation of within-tablet dynamics for extended release of a poorly soluble basic drug from hydrophilic matrix tablets using ATR–FTIR imaging. Mol. Pharm. 2020, 17, 1090–1099. [Google Scholar] [CrossRef]
- Lazzari, A.; Kleinebudde, P.; Knop, K. Xanthan gum as a rate-controlling polymer for the development of alcohol resistant matrix tablets and mini-tablets. Int. J. Pharm. 2018, 536, 440–449. [Google Scholar] [CrossRef]
- Talukdar, M.M.; Michoel, A.; Rombaut, P.; Kinget, R. Comparative study on xanthan gum and hydroxypropylmethyl cellulose as matrices for controlled-release drug delivery I. Compaction and in vitro drug release behaviour. Int. J. Pharm. 1996, 129, 233–241. [Google Scholar] [CrossRef]
- Talukdar, M.M.; Vinckier, I.; Moldenaers, P.; Kinget, R. Rheological characterization of xanthan gum and hydroxypropylmethyl cellulose with respect to controlled-release drug delivery. J. Pharm. Sci. 1996, 85, 537–540. [Google Scholar] [CrossRef]
- Crowley, M.M.; Zhang, F.; Koleng, J.J.; McGinity, J.W. Stability of polyethylene oxide in matrix tablets prepared by hot-melt extrusion. Biomaterials 2002, 23, 4241–4248. [Google Scholar] [CrossRef]
- Moroni, A.; Ghebre-Sellassie, I. Application of poly (oxyethylene) homopolymers in sustained release solid formulations. Drug Dev. Ind. Pharm. 1995, 21, 1411–1428. [Google Scholar] [CrossRef]
- Kim, C. Drug release from compressed hydrophilic POLYOX-WSR tablets. J. Pharm. Sci. 1995, 84, 303–306. [Google Scholar] [CrossRef]
- Shojaee, S.; Cumming, I.; Kaialy, W.; Nokhodchi, A. The influence of vitamin E succinate on the stability of polyethylene oxide PEO controlled release matrix tablets. Colloids Surf. B Biointerfaces 2013, 111, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Apicella, A.; Cappello, B.; Del Nobile, M.A.; La Rotonda, M.I.; Mensitieri, G.; Nicolais, L. Poly (ethylene oxide)(PEO) and different molecular weight PEO blends monolithic devices for drug release. Biomaterials 1993, 14, 83–90. [Google Scholar] [CrossRef]
- Stringer, J.L.; Peppas, N.A. Diffusion of small molecular weight drugs in radiation-crosslinked poly (ethylene oxide) hydrogels. J. Control. Release 1996, 42, 195–202. [Google Scholar] [CrossRef]
- Efentakis, M.; Vlachou, M. Evaluation of high molecular weight poly (oxyethylene)(Polyox) polymer: Studies of flow properties and release rates of furosemide and captopril from controlled-release hard gelatin capsules. Pharm. Dev. Technol. 2000, 5, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; McGinity, J.W. Properties of sustained-release tablets prepared by hot-melt extrusion. Pharm. Dev. Technol. 1999, 4, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Asare-Addo, K.; Kaialy, W.; Levina, M.; Rajabi-Siahboomi, A.; Ghori, M.U.; Supuk, E.; Laity, P.R.; Conway, B.R.; Nokhodchi, A. The influence of agitation sequence and ionic strength on in vitro drug release from hypromellose (E4M and K4M) ER matrices—The use of the USP III apparatus. Colloids Surf. B Biointerfaces 2013, 104, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Bajwa, G.S.; Hoebler, K.; Sammon, C.; Timmins, P.; Melia, C.D. Microstructural imaging of early gel layer formation in HPMC matrices. J. Pharm. Sci. 2006, 95, 2145–2157. [Google Scholar] [CrossRef]
- Avalle, P.; Pygall, S.R.; Pritchard, J.; Jastrzemska, A. Interrogating erosion-based drug liberation phenomena from hydrophilic matrices using near infrared (NIR) spectroscopy. Eur. J. Pharm. Sci. 2013, 48, 72–79. [Google Scholar] [CrossRef]
- Tomer, G.; Mantle, M.D.; Gladden, L.F.; Newton, J.M. Measuring water distribution in extrudates using magnetic resonance imaging (MRI). Int. J. Pharm. 1999, 189, 19–28. [Google Scholar] [CrossRef]
- Kaunisto, E.; Abrahmsen-Alami, S.; Borgquist, P.; Larsson, A.; Nilsson, B.; Axelsson, A. A mechanistic modelling approach to polymer dissolution using magnetic resonance microimaging. J. Control. Release 2010, 147, 232–241. [Google Scholar] [CrossRef]
- Christmann, V.; Rosenberg, J.; Seega, J.; Lehr, C. Simultaneous in vivo visualization and localization of solid oral dosage forms in the rat gastrointestinal tract by magnetic resonance imaging (MRI). Pharm. Res. 1997, 14, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Kulinowski, P.; Dorożyński, P.; Młynarczyk, A.; Węglarz, W.P. Magnetic resonance imaging and image analysis for assessment of HPMC matrix tablets structural evolution in USP apparatus 4. Pharm. Res. 2011, 28, 1065–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashraf, M.; luorno, V.L.; Coffin-Beach, D.; Evans, C.A.; Augsburger, L.L. A novel nuclear magnetic resonance (NMR) imaging method for measuring the water front penetration rate in hydrophilic polymer matrix capsule plugs and its role in drug release. Pharm. Res. 1994, 11, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Rajabi-Siahboomi, A.R.; Bowtell, R.W.; Mansfield, P.; Davies, M.C.; Melia, C.D. Structure and behavior in hydrophilic matrix sustained release dosage forms: 4. Studies of water mobility and diffusion coefficients in the gel layer of HPMC tablets using NMR imaging. Pharm. Res. 1996, 13, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Dinarvand, R.; Wood, B.; D’Emanuele, A. Measurement of the diffusion of 2,2,2-trifluoroacetamide within thermoresponsive hydrogels using NMR imaging. Pharm. Res. 1995, 12, 1376–1379. [Google Scholar] [CrossRef] [PubMed]
- Markl, D.; Wang, P.; Ridgway, C.; Karttunen, A.; Bawuah, P.; Ketolainen, J.; Gane, P.; Peiponen, K.E.; Zeitler, J.A. Resolving the rapid water absorption of porous functionalised calcium carbonate powder compacts by terahertz pulsed imaging. Chem. Eng. Res. Des. 2018, 132, 1082–1090. [Google Scholar] [CrossRef]
- Yassin, S.; Su, K.; Lin, H.; Gladden, L.F.; Axel Zeitler, J. Diffusion and swelling measurements in pharmaceutical powder compacts using terahertz pulsed imaging. J. Pharm. Sci. 2015, 104, 1658–1667. [Google Scholar] [CrossRef]
- Haaser, M.; Karrout, Y.; Velghe, C.; Cuppok, Y.; Gordon, K.C.; Pepper, M.; Siepmann, J.; Rades, T.; Taday, P.F.; Strachan, C.J. Application of terahertz pulsed imaging to analyse film coating characteristics of sustained-release coated pellets. Int. J. Pharm. 2013, 457, 521–526. [Google Scholar] [CrossRef]
- Melia, C.D.; Marshall, P.; Stark, P.; Cunningham, S.; Kinahan, A.; Devane, J. Investigating in vitro drug release mechanisms inside dosage forms. Adv. Exp. Med. Biol. 1997, 423, 129–135. [Google Scholar]
- Healy, A.M.; Corrigan, O.I.; Allan, J. The effect of dissolution on the surface texture of model solid-dosage forms as assessed by non-contact laser profilometry. Pharm. Technol. Eur. 1995, 7, 14. [Google Scholar]
- Pajander, J.; Baldursdottir, S.; Rantanen, J.; Østergaard, J. Behaviour of HPMC compacts investigated using UV-imaging. Int. J. Pharm. 2012, 427, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Timmins, P.; Pygall, S.R.; Melia, C.D. Hydrophilic Matrix Tablets for Oral Controlled Release; Springer: New York, NY, USA, 2014. [Google Scholar]
- Ward, A.; Walton, K.; Box, K.; Østergaard, J.; Gillie, L.J.; Conway, B.R.; Asare-Addo, K. Variable-focus microscopy and UV surface dissolution imaging as complementary techniques in intrinsic dissolution rate determination. Int. J. Pharm. 2017, 530, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Walton, K.; Stoycheva, S.; Wallis, M.; Adebisi, A.; Nep, E.; Ngwuluka, N.C.; Shaboun, S.; Smith, A.M.; Conway, B.R.; et al. The use of visible and UV dissolution imaging for the assessment of propranolol hydrochloride in liquisolid compacts of Sesamum radiatum gum. J. Drug Deliv. Sci. Technol. 2020, 56, 101511. [Google Scholar] [CrossRef]
- Boetker, J.P.; Rantanen, J.; Rades, T.; Müllertz, A.; Østergaard, J.; Jensen, H. A new approach to dissolution testing by UV imaging and finite element simulations. Pharm. Res. 2013, 30, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Asare-Addo, K.; Walton, K.; Ward, A.; Totea, A.; Taheri, S.; Alshafiee, M.; Mawla, N.; Bondi, A.; Evans, W.; Adebisi, A.; et al. Direct imaging of the dissolution of salt forms of a carboxylic acid drug. Int. J. Pharm. 2018, 551, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Asare-Addo, K.; Alshafiee, M.; Walton, K.; Ward, A.; Totea, A.; Taheri, S.; Mawla, N.; Adebisi, A.O.; Elawad, S.; Diza, C.; et al. Effect of preparation method on the surface properties and UV imaging of indomethacin solid dispersions. Eur. J. Pharm. Biopharm. 2019, 137, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Hulse, W.L.; Gray, J.; Forbes, R.T. A discriminatory intrinsic dissolution study using UV area imaging analysis to gain additional insights into the dissolution behaviour of active pharmaceutical ingredients. Int. J. Pharm. 2012, 434, 133–139. [Google Scholar] [CrossRef]
- Østergaard, J.; Meng-Lund, E.; Larsen, S.W.; Larsen, C.; Petersson, K.; Lenke, J.; Jensen, H. Real-time UV imaging of nicotine release from transdermal patch. Pharm. Res. 2010, 27, 2614–2623. [Google Scholar]
- He, S.; Østergaard, J.; Ashna, M.; Nielsen, C.U.; Jacobsen, J.; Mu, H. Microenvironmental pH modifying films for buccal delivery of saquinavir: Effects of organic acids on pH and drug release in vitro. Int. J. Pharm. 2020, 585, 119567. [Google Scholar] [CrossRef]
- Brown, B.; Fazili, Z.; Ward, A.; Walton, K.; Blunt, L.; Østergaard, J.; Asare-Addo, K. An investigation of drug compact topography as relates to intrinsic dissolution rates determined by dissolution imaging. J. Drug Deliv. Sci. Technol. 2020, 102143. [Google Scholar] [CrossRef]
- Fazili, Z.; Ward, A.; Walton, K.; Blunt, L.; Asare-Addo, K. Design and development of a novel fused filament fabrication (FFF) 3D printed diffusion cell with UV imaging capabilities to characterise permeation in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2020, 152, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, F.; Belton, P.; Ward, A.; Asare-Addo, K.; Qi, S. An investigation into the use of low quantities of functional additives to control drug release from hot melt extruded solid dispersions for poorly soluble drug delivery. Int. J. Pharm. 2020, 579, 119172. [Google Scholar] [CrossRef] [PubMed]
- Blunt, L.; Jiang, X. Advanced Techniques for Assessment Surface Topography: Development of a Basis for 3D Surface Texture Standards Surfstand; Kogan Page Science: Sterling, MA, USA, 2007; p. 355. [Google Scholar]
- Al-Karawi, C.; Kaiser, T.; Leopold, C.S. A novel technique for the visualization of tablet punch surfaces: Characterization of surface modification, wear and sticking. Int. J. Pharm. 2017, 530, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Walton, K.; Blunt, L.; Fleming, L.; Goodhand, M.; Lung, H. Areal parametric characterisation of ex-service compressor blade leading edges. Wear 2014, 321, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Walton, K.; Blunt, L.; Fleming, L. The topographic development and areal parametric characterization of a stratified surface polished by mass finishing. Surf. Topogr. Metrol. Prop. 2015, 3, 035003. [Google Scholar] [CrossRef] [Green Version]
- Danzl, R.; Helmli, F.; Scherer, S. Focus variation—A robust technology for high resolution optical 3D surface metrology. Strojniški vestnik. J. Mech. Eng. 2011, 2011, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Walton, K.; Fleming, L.; Goodhand, M.; Racasan, R.; Zeng, W. High fidelity replication of surface texture and geometric form of a high aspect ratio aerodynamic test component. Surf. Topogr. Metrol. Prop. 2016, 4, 025003. [Google Scholar] [CrossRef]
- International Standards Organisation. BS EN ISO 25178-2:2012: Geometrical Product Specifications (GPS). Surface Texture. Areal. Terms, Definitions and Surface Texture Parameters; International Standards Organisation: Geneva, Switzerland, 30 April 2012. [Google Scholar]
- Kelco, C.P. KELTROL®/KELZAN®—Xanthan Gum Book, 8th ed.; CP Kelco: Atlanta, GA, USA, 2008; pp. 1–28. Available online: http://www.bisi.cz/cmsres.axd/get/cms$7CVwRhc3USVqgzxkKF96gI$2BChNrXcTq$2BOUdiEtz5TfYA$2B1dJUbBlKfluXdoDfiqojVRVU$2FkQ343xA$3D (accessed on 25 November 2020).
- Ferstl, M.; Drechsler, M.; Rachel, R.; Rischer, M.; Engel, J.; Backofen, M.; Goepferich, A. The impact of polyelectrolyte structure on the shape of nanoassemblies with cationic peptides. J. Pharm. Sci. 2013, 102, 2599–2607. [Google Scholar] [CrossRef]
- Mawla, N.; Hanley, S.; Walton, K.; Kaialy, W.; Hussain, T.; Ward, A.; Brown, J.; Conway, B.R.; Timmins, P.; Asare-Addo, K. Imaging of the effect of alcohol-containing media on the performance of hypromellose hydrophilic matrix tablets: Comparison of direct compression and regular grades of polymer. Pharmaceutics 2020, 12, 889. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Profile Standards | Areal Standards (ISO 25178) |
|---|---|
| Nominal characteristics of contact (stylus) instruments. (ISO 3274) | Part 1: Areal surface texture drawing indications. |
| Rules and procedures for the assessment of surface texture (ISO 4288) | Part 2: Terms, definitions and surface texture parameters. |
| Metrological characteristics of phase correct filters (ISO 11562) | Part 3: Specification operators. |
| Motif Parameters (ISO 12085) | Part 4: Comparison rules |
| Surfaces having stratified functional properties—Part 1 (ISO 13565 part 1) | Part 5: Verification Operators |
| Surfaces having stratified functional properties—Part 2 (ISO 13565 part 2) | Part 6: Classification of methods for measuring surface texture. |
| Terms definitions and surface texture parameters (ISO 4287) | Part 601: Nominal characteristics of contact stylus instruments |
| Measurement Standards—material measures (ISO 5436 part 1) | Part 602: Nominal characteristics of non-contact (confocal chromatic probe) instruments. |
| Software measurement standards (ISO 5436 part 2) | Part 603: Nominal characteristics of non-contact (phase shifting interferometric microscopy) instruments. |
| Calibration of contact stylus instruments (ISO 12179) | Part 604: Nominal characteristics of non-contact (coherence scanning interferometry) instruments. |
| Surfaces having stratified functional properties—Part 3 (ISO 13565 part 3) | Part 605: Nominal characteristics of non-contact (point autofocus) instruments |
| Indication of surface texture in technical product documentation (ISO 1302) | Part 606: Nominal characteristics of non-contact (variable focus) instruments |
| Surface Parameters | |||
|---|---|---|---|
| 10× Magnification | |||
| Parameter | Xanthan Gum (XG) | Polyethylene oxide (PEO) | Hydroxypropyl methylcellulose (HPMC CR) |
| Str | 0.96 ± 0.26 | 0.93 ± 0.03 | 0.90 ± 0.04 |
| Sal (mm) | 0.03 ± 0.02 | 0.07 ± 0.01 | 0.06 ± 0.00 |
| Sdr (%) | 3.63 ± 0.28 | 2.38 ± 0.11 | 4.38 ± 0.33 |
| Smr2 (%) | 89.82 ± 0.26 | 90.96 ± 0.48 | 91.50 ± 0.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ward, A.; Brown, B.; Walton, K.; Timmins, P.; Conway, B.R.; Asare-Addo, K. Application of Focus Variation Microscopy and Dissolution Imaging in Understanding the Behaviour of Hydrophilic Matrices. Pharmaceutics 2020, 12, 1162. https://doi.org/10.3390/pharmaceutics12121162
Ward A, Brown B, Walton K, Timmins P, Conway BR, Asare-Addo K. Application of Focus Variation Microscopy and Dissolution Imaging in Understanding the Behaviour of Hydrophilic Matrices. Pharmaceutics. 2020; 12(12):1162. https://doi.org/10.3390/pharmaceutics12121162
Chicago/Turabian StyleWard, Adam, Benedict Brown, Karl Walton, Peter Timmins, Barbara R. Conway, and Kofi Asare-Addo. 2020. "Application of Focus Variation Microscopy and Dissolution Imaging in Understanding the Behaviour of Hydrophilic Matrices" Pharmaceutics 12, no. 12: 1162. https://doi.org/10.3390/pharmaceutics12121162
APA StyleWard, A., Brown, B., Walton, K., Timmins, P., Conway, B. R., & Asare-Addo, K. (2020). Application of Focus Variation Microscopy and Dissolution Imaging in Understanding the Behaviour of Hydrophilic Matrices. Pharmaceutics, 12(12), 1162. https://doi.org/10.3390/pharmaceutics12121162