Therapeutic Approaches to Treat Mitochondrial Diseases: “One-Size-Fits-All” and “Precision Medicine” Strategies

 ,
, 
 ,
, 
Abstract
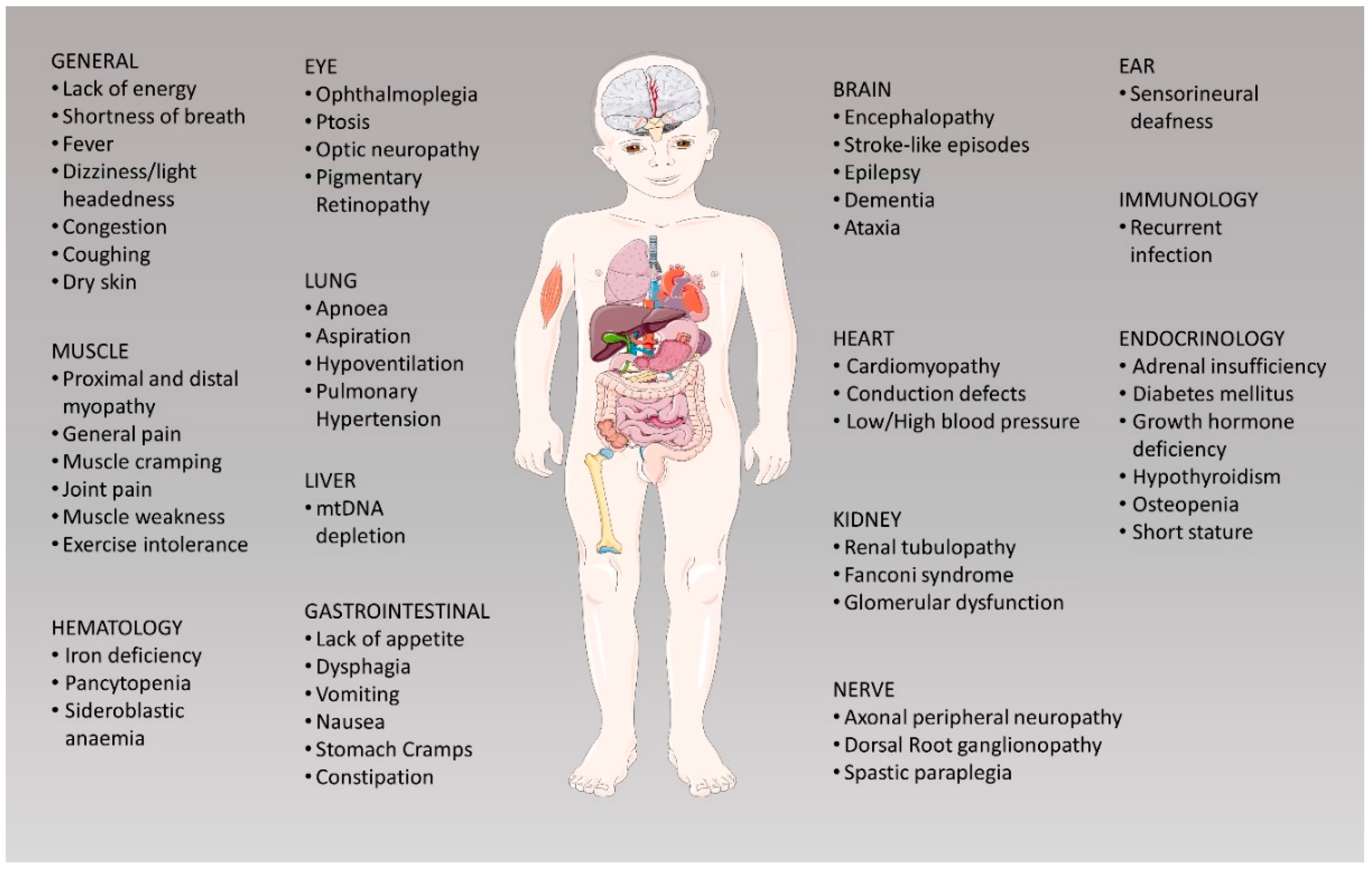
1. Genetics of Mitochondrial Diseases
2. Therapeutic Approaches to Treat Mitochondrial Disorders
3. “One-Size-Fits-All” Approaches
3.1. Physical Exercise
3.2. Dietary Approaches
3.3. Exposure to Hypoxia
3.4. Strategies to Increase ATP Levels
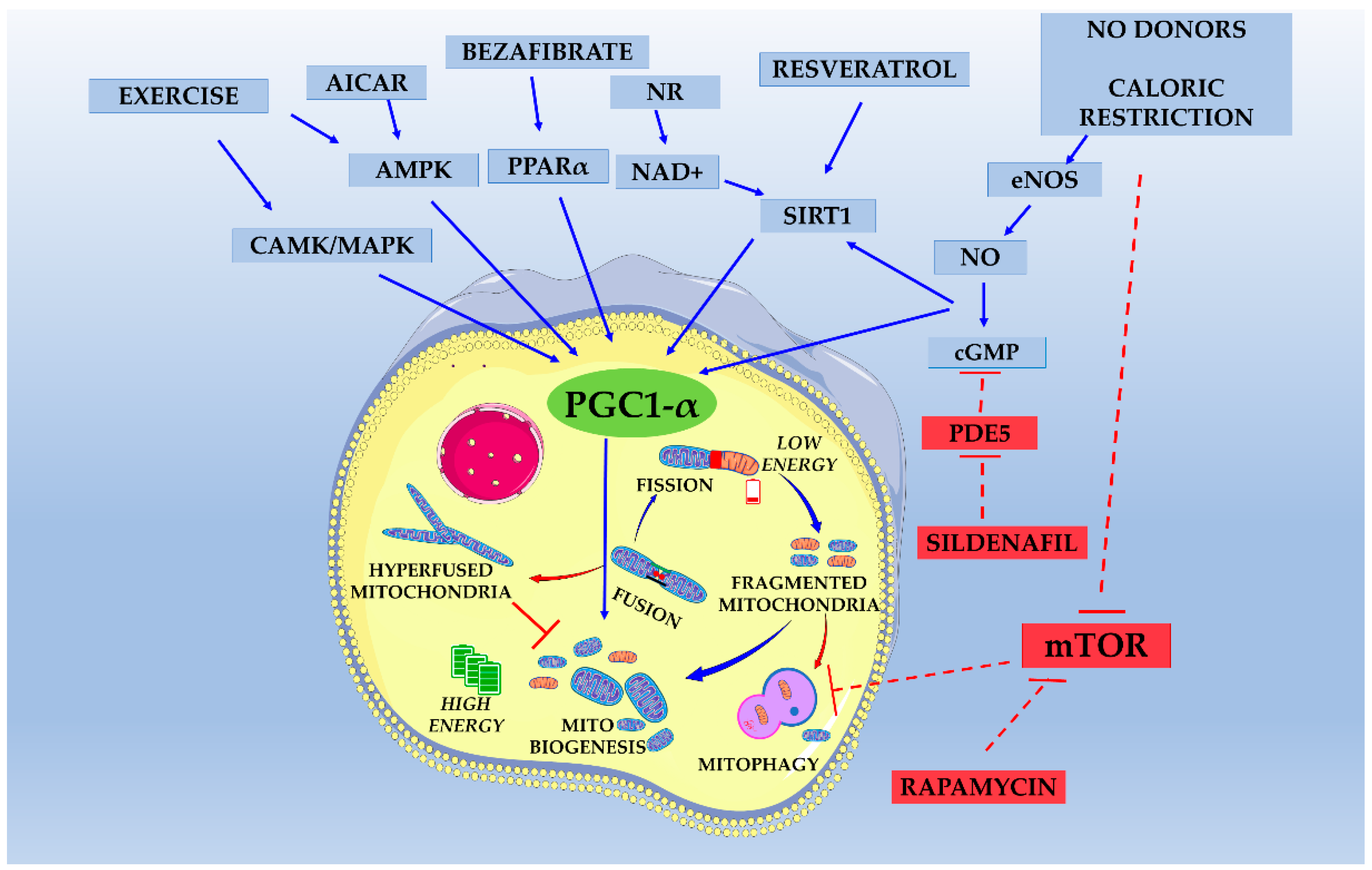
3.5. Pharmacological Stimulation of Mitochondrial Biogenesis
3.5.1. 5-Aminoimidazole-4-Carboxamide Ribonucleoside (AICAR)
3.5.2. Bezafibrate and Other PPAR Agonists
3.5.3. Modulating NAD+ Pool
3.5.4. I-BET 525762A
3.5.5. Polyphenols and Other Pharmacognostic Products
3.6. Pharmacological Modulation of the NO/cGMP/PKG Pathway
3.6.1. L-Arginine and L-Citrulline
3.6.2. Natriuretic Peptides and Cyclic Guanosine Monophosphate
3.6.3. PDE5 Inhibitors
3.7. Antioxidants
3.7.1. Glutathione
3.7.2. Cysteamine
3.7.3. N-Acetylcysteine
3.7.4. Lipoic Acid
3.7.5. Vitamin C
3.7.6. Vitamin E
3.7.7. Coenzyme Q10
3.7.8. Idebenone
3.7.9. MitoQ
3.8. Redox-Active Molecules
3.8.1. EPI-Molecules
3.8.2. JP4-039
3.8.3. KH176
3.8.4. SKQ1
3.9. Pharmacological Modulation of Mitochondrial Dynamics
3.10. Pharmacological Protection of Cardiolipin
3.11. Pharmacological Modulation of Autophagy
3.12. Bypassing cI-cIII-cIV Defects with Alternative Enzymes
4. Precision Medicine Approaches for PMD Caused by mtDNA Defects
4.1. Pre-Implantation Therapies to Prevent the Transmission of mtDNA Mutations
4.1.1. Pre-Implantation Genetic Diagnosis
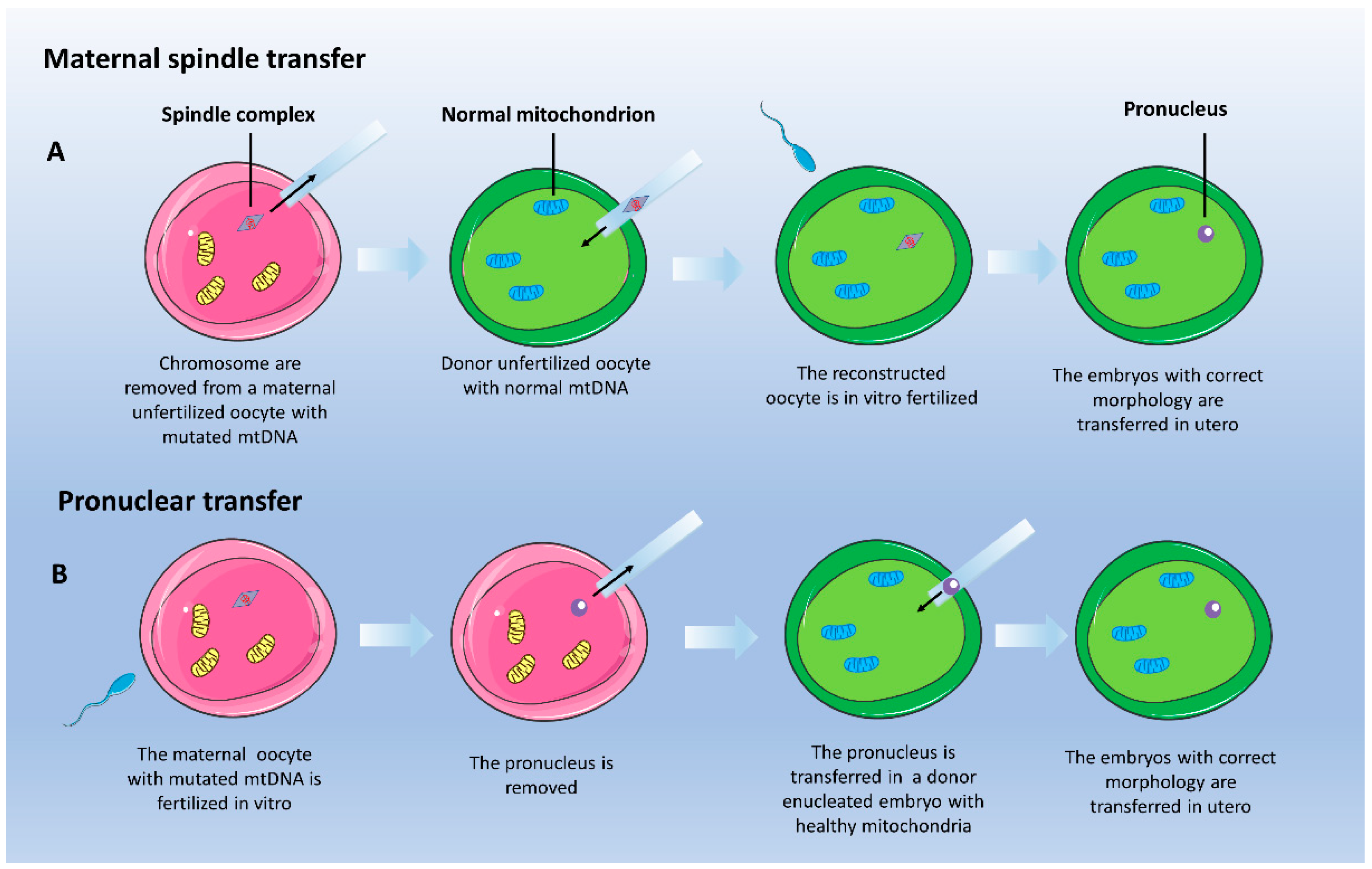
4.1.2. Mitochondrial Donation: Maternal Spindle Transfer
4.1.3. Mitochondrial Donation: Pronuclear Transfer
4.2. Personalized Therapies for mtDNA Disorders
4.2.1. Delivery of Nucleic Acids to the Mitochondria
4.2.2. Heteroplasmic Shift
4.2.3. Allotopic Gene Expression
4.2.4. Mitochondrial Augmentation Therapy
5. Precision Medicine Approaches for PMD Caused by Nuclear Defects
5.1. Gene Therapy Approaches
5.2. Liver Transplantation
5.3. PMD Characterised by Systemic Accumulation of Toxic Compounds
5.3.1. Application of Gene Therapy Protocol
5.3.2. Application of Liver Transplantation Protocol
5.3.3. Cell Replacement
5.4. Molecular Bypass Therapy in Disorders of mtDNA Instability
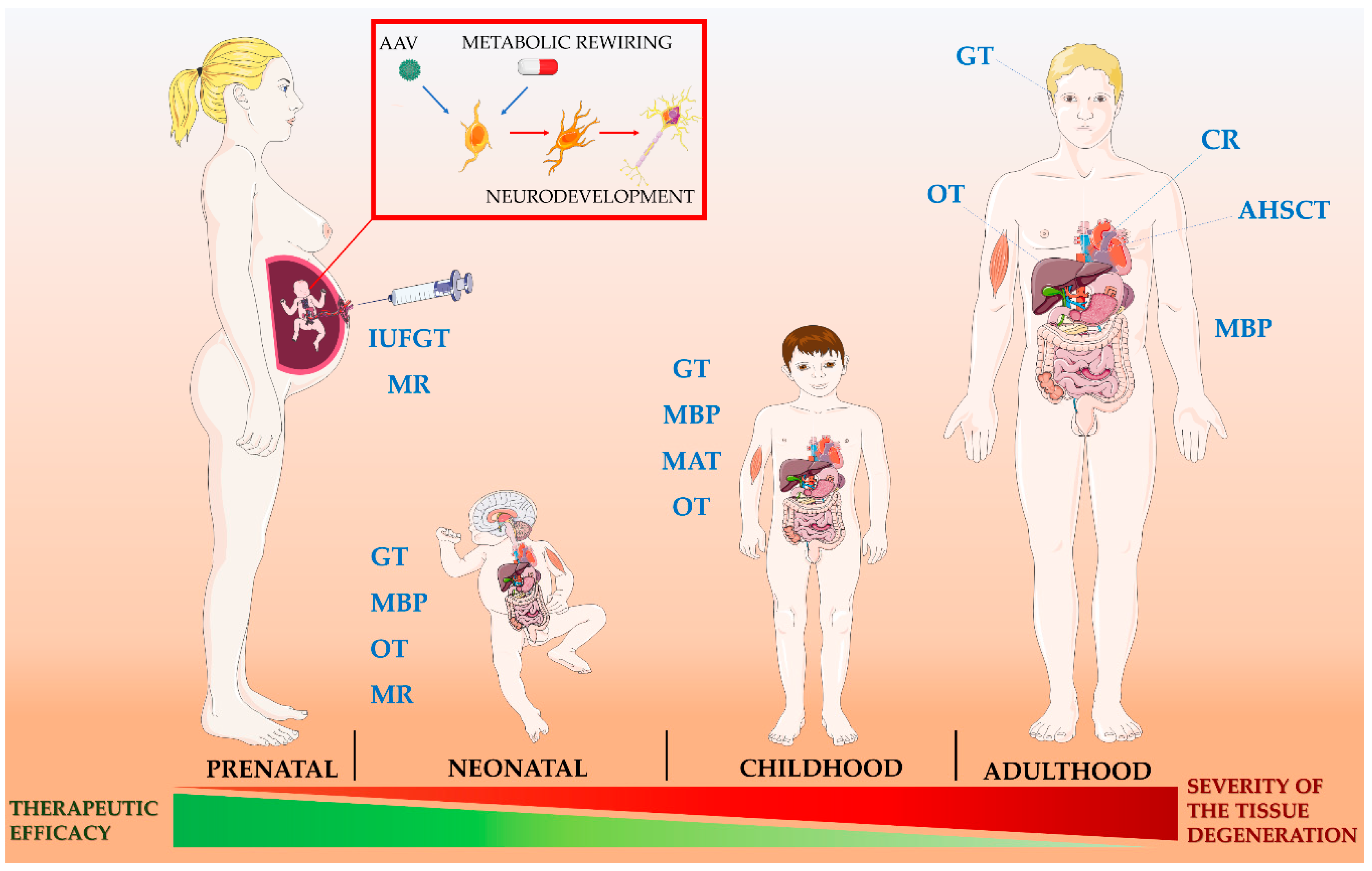
6. Future Perspectives
6.1. Fetal Gene Therapy
6.2. Metabolic Rewiring
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy | Model | Study | Ref. |
|---|---|---|---|
| Strategies to increase ATP levels | |||
| Febuxostat plus inosine | patients with homoplasmic mtDNA mutation | clinical | [65] |
| patient with mitochondrial diabetes with heteroplasmic mutation in tRNA leucine 1 | clinical | [65] | |
| Pharmacological stimulation of mitochondrial biogenesis | |||
| AICAR | mouse models of COX defects | pre-clinical | [74] |
| mouse model Cox10-Mef2c-Cre | pre-clinical | [82] | |
| CI deficient cells (NDUFS2, NDUFS4, NDUFAF4, C20ORF7, FOXRED1, NDUFA12L) | pre-clinical | [83] | |
| Bezafibrate and other PPAR agonists | MRC-deficient patients’ fibroblasts | pre-clinical | [87] |
| SCO2 mutant fibroblasts | pre-clinical | [88] | |
| DNM1L mutant cells | pre-clinical | [89] | |
| mouse models of COX defects | pre-clinical | [74] | |
| Deletor mouse | pre-clinical | [90] | |
| Mutator mouse | pre-clinical | [91] | |
| patients with MM | clinical | [92] | |
| NAD+ precursors | Sco2 ko mouse | pre-clinical | [81] |
| Deletor mouse | pre-clinical | [106] | |
| GBA-PD Drosophila melanogaster | pre-clinical | [107] | |
| Ndufs4 ko mice | pre-clinical | [108] | |
| Niacin | patients with MM | clinical (NCT03973203) | [109] |
| I-BET 525762A | cybrids carrying 3796A>G mutation | pre-clinical | [110] |
| Polyphenols and other Pharmacognostic Products | |||
| Resveratrol | fibroblasts from MT-TL1, MT-TK, MT-ATP6-patients | pre-clinical | [125] |
| Curcumin | LHON patients | clinical (NCT00528151) | unpublished |
| Pharmacological modulation of the NO/cGMP/PKG pathway | |||
| L-arginine and L-citrulline | MELAS patients | clinical (open-label trial) | [162,164,168] |
| MELAS patients | clinical (open-label trial) | [165] | |
| various PMD patients | clinical (NCT02809170) | unpublished | |
| MELAS patients | Phase-1 clinical trial NCT03952234 | unpublished | |
| PDE5 inhibitors | NPCs with homoplasmic mutation in MT-ATP6 | pre-clinical | [176] |
| LHON | clinical (case report) | [179] | |
| Antioxidant | |||
| Glutathione | MM patients | clinical (double-blind cross-over study) | [188] |
| fibroblasts of patients carrying the m.3243A>G and m.8344A>G mutations | pre-clinical | [189] | |
| Cysteamine | C. elegans model of CI defect, FBXL4 mutant human fibroblast, and zebrafish models of pharmacologically induced CI and CIV defects | pre-clinical | [192] |
| Cysteamine bitartrate delayed-release (RP103) | PMD paediatric patients | clinical (NCT02023866) | unpublished |
| N-acetylcysteine | Ethe1 mouse model | pre-clinical | [195] |
| ETHE1 patients | clinical (compassionate use) | [198,199,200] | |
| Lipoic acid and CoQ10 | PMD patients | clinical (randomized, double-blind, placebo-controlled, crossover study) | [206] |
| Vitamin C and K | PMD patients | Clinical | [208,209,210] |
| Vitamin E | fibroblasts from patients with CI defect | pre-clinical | [211,212] |
| Coenzyme Q10 | MERRF cells | pre-clinical | [220] |
| KSS patients | clinical | [221] | |
| KSS patents and other MM with CPEO | clinical | [222,223,224,226,228,230] | |
| patient with mitochondrial encephalomyopathy with COX deficiency | clinical | [225] | |
| patients with mitochondrial cytopathies | clinical | [227] | |
| PMD patients | clinical | [229] | |
| patients with different OXPHOS defects | clinical | [231] | |
| PMD patients | randomized, double-blind, cross-over trial | [232] | |
| patients with different PMD; 15 patients with MM | clinical (multicenter study) | [233] | |
| PMD paediatric patients | clinical (NCT00432744) | [234] | |
| Idebenone | fibroblasts from LHON patients | pre-clinical | [237,238] |
| mouse model of LHON | pre-clinical | [239] | |
| 85 LHON patients with to m.3460G>A, m.11778G>A, and m.14484T>C mutations | clinical (“RHODOS” study, NCT00747487) | [241] | |
| subset of LHON patients from RHODOS study | clinical (RHODOS-OFU, NCT01421381) | [242] | |
| patient with TXN2 mutation | [243] | ||
| Opa1 mutant mice | pre-clinical | [245] | |
| seven DOA patients | clinial | [246] | |
| 87 DOA patients | clinical (retrospective cohort study) | [247] | |
| Redox-Active Molecules | |||
| EPI-743 | PMD patients | clinical | [259] |
| patients with Leigh syndrome | clinical (prospective single-arm subject-controlled trial) | [260] | |
| children with mitochondrial encephalopathy | Clinical | [261] | |
| one patient with Leigh syndrome due to ND3 mutation | Clinical | [262] | |
| patients with LHON | clinical (open-label trial) | [263] | |
| patients with Leigh syndrome | clinical (NCT02352896) | unpublished | |
| PMD paediatric patients | clinical (NCT01642056) | unpublished | |
| JP4-039 | ACAD9- VLCAD-, ETHE1 and MOCS1 mutant fibroblasts | pre-clinical | [266,267] |
| KH176 | cellular models of CI defects | pre-clinical | [268] |
| Ndufs4 ko mouse model | pre-clinical | [269,270] | |
| patients with m.3242A>G mutation | clinical (KHENERGY STUDY, NCT02909400) | [272] | |
| patients with MELAS | clinical (KHENERGYZE Study, NCT04165239) | [273] | |
| SKQ1 | Mutator mouse | pre-clinical | [281] |
| Pharmacological modulation of mitochondrial dynamics | |||
| Cytotoxic Necrotizing Factor 1 (CNF1) | MERRF fibroblasts | pre-clinical | [300] |
| Pharmacological protection of cardiolipin | |||
| Elamipretide | patients with primary mitochondrial myopathy (MM) | clinical (NCT03323749) | [310] |
| Barth syndrome | clinical (NCT03098797) | unpublished | |
| LHON patients | clinical (NCT02693119) | unpublished | |
| age-related macular degeneration (AMD) with non-central geographic atrophy | clinical (NCT03891875) | unpublished | |
| Pharmacological modulation of autophagy | |||
| Rapamycin | Ndufs4 ko mouse | pre-clinical | [313] |
| muscle-specific Cox15 ko mouse | pre-clinical | [314] | |
| ND2-deficient Drosophila model of LS | pre-clinical | [315] | |
| MT-ATP6-mutant, iPSCs-derived neurons | pre-clinical | [316] | |
| gas-1 (fc21) nematodes | pre-clinical | [317] | |
| mice with CoQ10 deficiency | pre-clinical | [320] | |
| TwKOastro mice | pre-clinical | [59] | |
| Everolimus (rapamycin analogue) | MELAS patients | clinical (NCT03747328) | [318] |
| children affected by Leigh disease or MELAS | Clinical | [319] | |
| Bypassing cI-cIII-cIV defects with alternative enzymes | |||
| NDI1 | CI deficiency in Drosophila melanogaster | pre-clinical | [324] |
| mouse model of LHON | pre-clinical | [238] | |
| mouse model of Leigh syndrome | pre-clinical | [239] | |
| AOX, NDI1 | ρ0 mouse cells | pre-clinical | [325] |
| AOX | CIII-IV deficiencies in human cells | pre-clinical | [326] |
| CIV deficient Drosophila melanogaster | pre-clinical | [327] | |
| Bcs1lp.S78G knock-in mice | pre-clinical | [330,331] | |
| Acta-Cox15 ko model | pre-clinical | [332] | |
| NDH-2 | human CI deficient fibroblasts | pre-clinical | [333] |
| Personalized therapies for mtDNA disorders | |||
| Mitochondrial donation: maternal spindle transfer | legally approved for use in the U.K | clinical | [343] |
| rhesus macaques | pre-clinical | [344] | |
| woman carrying mtDNA mutation of Leigh syndrome (8993 T>G) | clinical | [347,348] | |
| Mitochondrial donation: pronuclear transfer | mito-mouse | pre-clinical | [349] |
| Delivery of nucleic acids to the mitochondria | patient’s cell with a G625A heteroplasmic mutation in the tRNAPhe | pre-clinical | [355] |
| ND3 mutant fibroblasts | pre-clinical | [356] | |
| MERFF and KSS cybrids | pre-clinical | [354] | |
| Heteroplasmic shift | various cells with heteroplasmic mutation | pre-clinical | [359,360,361] |
| mouse model of heteroplasmic PMD | pre-clinical | [366,367] | |
| Allotopic gene expression | cybrids with m.8993T>G mutation | pre-clinical | [369] |
| rat model of LHON | pre-clinical | [370] | |
| LHON patients | clinical (NTC01267422; NCT02064569) | [371,372] | |
| Mitochondrial augmentation therapy | children with Pearson syndrome | clinical | Unpublished |
| children with KSS or Pearson Syndrome | clinical (NCT03384420) | unpublished | |
| KSS patient | clinical | [375] | |
| Precision medicine approaches for PMD caused by nuclear defects | |||
| Gene therapy approaches | Ant1 ko mouse model | pre-clinical | [381] |
| Tymp ko mouse model | pre-clinical | [390] | |
| Ethe1 ko mouse model | pre-clinical | [389] | |
| Ndufs4 mouse model | pre-clinical | [391] | |
| Mpv17 ko mouse model | pre-clinical | [58] | |
| mouse model of Leigh syndrome | pre-clinical | [382] | |
| Liver transplantation | PMD patients | clinical | [385] |
| DGUOK-deficient patients | clinical | [386] | |
| 25-year-old MNGIE patient | clinical | [395] | |
| ETHE1 patients | clinical | [396,397] | |
| Cell replacement | MNGIE patients | clinical | [400,401] |
| Molecular bypass therapy in disorders of mtDNA instability | Tk2 mouse model | pre-clinical | [405,406,407] |
| DGUOK mutant fibroblasts | pre-clinical | [404] | |
| RRM2B mutant fibroblasts | pre-clinical | [408] | |
| dguok−/− zebrafish | pre-clinical | [409] | |
| early-onset TK2-patients | clinical (open-label study) | [410] | |
References
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.B.; Chinnery, P.F. Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat. Rev. Genet. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Blok, R.B.; Dahl, H.-H.M.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh syndrome: Clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996, 39, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef]
- Goto, Y.-I.; Nonaka, I.; Horai, S. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nat. Cell Biol. 1990, 348, 651–653. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, A.E.; Petty, R.K.; Morgan-Hughes, J.A. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am. J. Hum. Genet. 1990, 46, 428–433. [Google Scholar]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Lopez-Gallardo, E.; Solano, A.; Herrero-Martin, M.D.; Martinez-Romero, I.; Castano-Perez, M.D.; Andreu, A.L.; Herrera, A.; Lopez-Perez, M.J.; Ruiz-Pesini, E.; Montoya, J. NARP syndrome in a patient harbouring an insertion in the MT-ATP6 gene that results in a truncated protein. J. Med. Genet. 2008, 46, 64–67. [Google Scholar] [CrossRef]
- Pitceathly, R.D.; Murphy, S.M.; Cottenie, E.; Chalasani, A.; Sweeney, M.G.; Woodward, C.; Mudanohwo, E.E.; Hargreaves, I.; Heales, S.; Land, J.; et al. Genetic dysfunction of MT-ATP6 causes axonal Charcot-Marie-Tooth disease. Neurology 2012, 79, 1145–1154. [Google Scholar] [CrossRef]
- Verny, C.; Guegen, N.; Desquiret-Dumas, V.; Chevrollier, A.; Prundean, A.; Dubas, F.; Cassereau, J.; Ferre, M.; Amati-Bonneau, P.; Bonneau, D.; et al. Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. Mitochondrion 2011, 11, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Bugiardini, E.; Bottani, E.; Marchet, S.; Poole, O.V.; Benincá, C.; Horga, A.; Woodward, C.; Lam, A.; Hargreaves, I.; Chalasani, A.; et al. Expanding the molecular and phenotypic spectrum of truncating MT-ATP6 mutations. Neurol. Genet. 2020, 6, e381. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.T.; DiMauro, S.; Zeviani, M.; Lombes, A.; Shanske, S.; Miranda, A.F.; Nakase, H.; Bonilla, E.; Werneck, L.C.; Servidei, S.; et al. Mitochondrial DNA Deletions in Progressive External Ophthalmoplegia and Kearns-Sayre Syndrome. N. Engl. J. Med. 1989, 320, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Pearson, H.A.; Lobel, J.S.; Kocoshis, S.A.; Naiman, J.L.; Windmiller, J.; Lammi, A.T.; Hoffman, R.; Marsh, J.C. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J. Pediatr. 1979, 95, 976–984. [Google Scholar] [CrossRef]
- Baerlocher, K.E.; Feldges, A.; Weissert, M.; Simonsz, H.J.; Rötig, A. Mitochondrial DNA deletion in an 8-year-old boy with pearson syndrome. J. Inherit. Metab. Dis. 1992, 15, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.; Jain, M.; Xie, X.; Sheth, S.A.; Chang, B.; Goldberger, O.A.; Spinazzola, A.; Zeviani, M.; Carr, S.A.; Mootha, V.K. Systematic identification of human mitochondrial disease genes through integrative genomics. Nat. Genet. 2006, 38, 576–582. [Google Scholar] [CrossRef]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nat. Cell Biol. 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- La Morgia, C.; Maresca, A.; Caporali, L.; Valentino, M.; Carelli, V. Mitochondrial diseases in adults. J. Intern. Med. 2020, 287, 592–608. [Google Scholar] [CrossRef]
- Ghezzi, D.; Zeviani, M. Human diseases associated with defects in assembly of OXPHOS complexes. Essays Biochem. 2018, 62, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Freyssenet, D.; Berthon, P.; Denis, C. Mitochondrial Biogenesis in Skeletal Muscle in Response to Endurance Exercises. Arch. Physiol. Biochem. 1996, 104, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.L.; Murphy, E.A.; McClellan, J.L.; Carmichael, M.D.; Davis, J.M. Exercise training increases mitochondrial biogenesis in the brain. J. Appl. Physiol. 2011, 111, 1066–1071. [Google Scholar] [CrossRef]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; Zierath, J.R. Exercise Metabolism and the Molecular Regulation of Skeletal Muscle Adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef]
- Vettor, R.; Valerio, A.; Ragni, M.; Trevellin, E.; Granzotto, M.; Olivieri, M.; Tedesco, L.; Ruocco, C.; Fossati, A.; Fabris, R.; et al. Exercise training boosts eNOS-dependent mitochondrial biogenesis in mouse heart: Role in adaptation of glucose metabolism. Am. J. Physiol. Metab. 2014, 306, E519–E528. [Google Scholar] [CrossRef]
- Miller, M.W.; Knaub, L.A.; Olivera-Fragoso, L.F.; Keller, A.C.; Balasubramaniam, V.; Watson, P.A.; Reusch, J.E. Nitric oxide regulates vascular adaptive mitochondrial dynamics. Am. J. Physiol. Circ. Physiol. 2013, 304, H1624–H1633. [Google Scholar] [CrossRef]
- Weber, K.; Wilson, J.N.; Taylor, L.; Brierley, E.; Johnson, M.A.; Turnbull, D.M.; Bindoff, L.A. A new mtDNA mutation showing accumulation with time and restriction to skeletal muscle. Am. J. Hum. Genet. 1997, 60, 373–380. [Google Scholar]
- Fu, K.; Hartlen, R.; Johns, T.; Genge, A.; Karpati, G.; Shoubridge, E.A. A novel heteroplasmic tRNAleu(CUN) mtDNA point mutation in a sporadic patient with mitochondrial encephalomyopathy segregates rapidly in skeletal muscle and suggests an approach to therapy. Hum. Mol. Genet. 1996, 5, 1835–1840. [Google Scholar] [CrossRef]
- Clark, K.M.; Bindoff, L.A.; Chrzanowska-Lightowlers, Z.; Andrews, R.M.; Griffiths, P.G.; Johnson, M.A.; Brierley, E.J.; Turnbull, D.M. Reversal of a mitochondrial DNA defect in human skeletal muscle. Nat. Genet. 1997, 16, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Shoubridge, E.A.; Johns, T.; Karpati, G. Complete restoration of a wild-type mtDNA genotype in regenerating muscle fibres in a patient with a tRNA point mutation and mitochondrial encephalomyopathy. Hum. Mol. Genet. 1997, 6, 2239–2242. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A.; Bourgeois, J.M.; Ogborn, D.I.; Little, J.P.; Hettinga, B.P.; Akhtar, M.; Thompson, J.E.; Melov, S.; Mocellin, N.J.; Kujoth, G.C.; et al. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc. Natl. Acad. Sci. USA 2011, 108, 4135–4140. [Google Scholar] [CrossRef] [PubMed]
- Fiuza-Luces, C.; Valenzuela, P.L.; Laine-Menéndez, S.; La Torre, M.F.-D.; Bermejo-Gómez, V.; Rufián-Vázquez, L.; Arenas, J.; Martín, M.A.; Lucia, A.; Moran, M. Physical Exercise and Mitochondrial Disease: Insights from a Mouse Model. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Greggio, C.; Jha, P.; Kulkarni, S.S.; Lagarrigue, S.; Broskey, N.T.; Boutant, M.; Wang, X.; Alonso, S.C.; Ofori, E.; Auwerx, J.; et al. Enhanced Respiratory Chain Supercomplex Formation in Response to Exercise in Human Skeletal Muscle. Cell Metab. 2017, 25, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Voet, N.; Van Der Kooi, E.L.; Van Engelen, B.G.M.; Geurts, A.C.H. Strength training and aerobic exercise training for muscle disease. Cochrane Database Syst. Rev. 2013, 2013, CD003907. [Google Scholar] [CrossRef]
- Tarnopolsky, M. Exercise as a Therapeutic Strategy for Primary Mitochondrial Cytopathies. J. Child Neurol. 2014, 29, 1225–1234. [Google Scholar] [CrossRef]
- Cejudo, P.; Bautista, J.; Montemayor, T.; Villagómez, R.; Jiménez, L.; Ortega, F.; Campos, Y.; Sánchez, H.; Arenas, J. Exercise training in mitochondrial myopathy: A randomized controlled trial. Muscle Nerve 2005, 32, 342–350. [Google Scholar] [CrossRef]
- Jeppesen, T.D.; Schwartz, M.; Olsen, D.B.; Wibrand, F.; Krag, T.; Duno, M.; Hauerslev, S.; Vissing, J. Aerobic training is safe and improves exercise capacity in patients with mitochondrial myopathy. Brain 2006, 129, 3402–3412. [Google Scholar] [CrossRef]
- Taivassalo, T.; Gardner, J.L.; Taylor, R.W.; Schaefer, A.M.; Newman, J.; Barron, M.J.; Haller, R.G.; Turnbull, D.M. Endurance training and detraining in mitochondrial myopathies due to single large-scale mtDNA deletions. Brain 2006, 129, 3391–3401. [Google Scholar] [CrossRef]
- Taivassalo, T.; Fu, K.; Johns, T.; Arnold, D.; Karpati, G.; Shoubridge, E.A. Gene shifting: A novel therapy for mitochondrial myopathy. Hum. Mol. Genet. 1999, 8, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.L.; Blakely, E.L.; Schaefer, A.M.; He, L.; Wyrick, P.; Haller, R.G.; Taylor, R.W.; Turnbull, U.M.; Taivassalo, T. Resistance training in patients with single, large-scale deletions of mitochondrial DNA. Brain 2008, 131, 2832–2840. [Google Scholar] [CrossRef] [PubMed]
- Taivassalo, T.; Shoubridge, E.A.; Chen, J.; Kennaway, N.G.; DiMauro, S.; Arnold, U.L.; Ørngreen, M.C. Aerobic conditioning in patients with mitochondrial myopathies: Physiological, biochemical, and genetic effects. Ann. Neurol. 2001, 50, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Adhihetty, P.J.; Taivassalo, T.; Haller, R.G.; Walkinshaw, D.R.; Hood, D.A. The effect of training on the expression of mitochondrial biogenesis- and apoptosis-related proteins in skeletal muscle of patients with mtDNA defects. Am. J. Physiol. Metab. 2007, 293, E672–E680. [Google Scholar] [CrossRef]
- Kossoff, E.; Wang, H.-S.; Eh, K.; Wang, H.-S. Dietary Therapies for Epilepsy. Biomed. J. 2013, 36, 2. [Google Scholar] [CrossRef]
- Kang, H.-C.; Lee, Y.M.; Kim, H.D.; Lee, J.S.; Slama, A. Safe and Effective Use of the Ketogenic Diet in Children with Epilepsy and Mitochondrial Respiratory Chain Complex Defects. Epilepsia 2007, 48, 82–88. [Google Scholar] [CrossRef]
- Lee, Y.M.; Kang, H.-C.; Lee, J.S.; Kim, S.H.; Kim, E.Y.; Lee, S.-K.; Slama, A.; Kim, H.D. Mitochondrial respiratory chain defects: Underlying etiology in various epileptic conditions. Epilepsia 2008, 49, 685–690. [Google Scholar] [CrossRef]
- Wexler, I.D.; Hemalatha, S.G.; McConnell, J.; Buist, N.; Dahl, H.-H.M.; Berry, S.A.; Cederbaum, S.D.; Patel, M.S.; Kerr, D.S. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets: Studies in patients with identical mutations. Neurology 1997, 49, 1655–1661. [Google Scholar] [CrossRef]
- Rahman, S. Mitochondrial disease and epilepsy. Dev. Med. Child Neurol. 2012, 54, 397–406. [Google Scholar] [CrossRef]
- Steriade, C.; Andrade, D.M.; Faghfoury, H.; Tarnopolsky, M.A.; Tai, P. Mitochondrial Encephalopathy With Lactic Acidosis and Stroke-like Episodes (MELAS) May Respond to Adjunctive Ketogenic Diet. Pediatr. Neurol. 2014, 50, 498–502. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Frey, S.; Geffroy, G.; Desquiret-Dumas, V.; Gueguen, N.; Bris, C.; Belal, S.; Amati-Bonneau, P.; Chevrollier, A.; Barth, M.; Henrion, D.; et al. The addition of ketone bodies alleviates mitochondrial dysfunction by restoring complex I assembly in a MELAS cellular model. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Santra, S.; Gilkerson, R.W.; Davidson, M.; Schon, E.A. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann. Neurol. 2004, 56, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Schiff, M.; Bénit, P.; El-Khoury, R.; Schlemmer, D.; Benoist, J.-F.; Rustin, P. Mouse Studies to Shape Clinical Trials for Mitochondrial Diseases: High Fat Diet in Harlequin Mice. PLoS ONE 2011, 6, e28823. [Google Scholar] [CrossRef] [PubMed]
- Ahola-Erkkilä, S.; Carroll, C.J.; Peltola-Mjösund, K.; Tulkki, V.; Mattila, I.; Seppänen-Laakso, T.; Orešič, M.; Tyynismaa, H.; Suomalainen, A. Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum. Mol. Genet. 2010, 19, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Purhonen, J.; Rajendran, J.; Mörgelin, M.; Uusi-Rauva, K.; Katayama, S.; Krjutskov, K.; Einarsdottir, E.; Velagapudi, V.; Kere, J.; Jauhiainen, M.; et al. Ketogenic diet attenuates hepatopathy in mouse model of respiratory chain complex III deficiency caused by a Bcs1l mutation. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Bottani, E.; Giordano, C.; Civiletto, G.; Di Meo, I.; Auricchio, A.; Ciusani, E.; Marchet, S.; Lamperti, C.; D’Amati, G.; Viscomi, C.; et al. AAV-mediated Liver-specific MPV17 Expression Restores mtDNA Levels and Prevents Diet-induced Liver Failure. Mol. Ther. 2014, 22, 10–17. [Google Scholar] [CrossRef]
- Brunetti, D.; Dusi, S.; Giordano, C.; Lamperti, C.; Morbin, M.; Fugnanesi, V.; Marchet, S.; Fagiolari, G.; Sibon, O.; Moggio, M.; et al. Pantethine treatment is effective in recovering the disease phenotype induced by ketogenic diet in a pantothenate kinase-associated neurodegeneration mouse model. Brain 2014, 137, 57–68. [Google Scholar] [CrossRef]
- Ignatenko, O.; Nikkanen, J.; Kononov, A.; Zamboni, N.; Ince-Dunn, G.; Suomalainen, A. Mitochondrial spongiotic brain disease: Astrocytic stress and harmful rapamycin and ketosis effect. Life Sci. Alliance 2020, 3, e202000797. [Google Scholar] [CrossRef]
- Ahola, S.; Auranen, M.; Isohanni, P.; Niemisalo, S.; Urho, N.; Buzkova, J.; Velagapudi, V.; Lundbom, N.; Hakkarainen, A.; Muurinen, T.; et al. Modified Atkins diet induces subacute selective ragged-red-fiber lysis in mitochondrial myopathy patients. EMBO Mol. Med. 2016, 8, 1234–1247. [Google Scholar] [CrossRef]
- Roe, C.R.; Sweetman, L.; Roe, D.S.; David, F.; Brunengraber, H. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J. Clin. Investig. 2002, 110, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M.; Jain, I.H.; Goldberger, O.; Rezoagli, E.; Thoonen, R.; Cheng, K.-H.; Sosnovik, D.E.; Scherrer-Crosbie, M.; Mootha, V.K.; Zapol, W.M. Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, E4241–E4250. [Google Scholar] [CrossRef] [PubMed]
- Kamatani, N.; Hashimoto, M.; Sakurai, K.; Gokita, K.; Yoshihara, J.; Sekine, M.; Mochii, M.-A.; Fukuuchi, T.; Yamaoka, N.; Kaneko, K. Clinical studies on changes in purine compounds in blood and urine by the simultaneous administration of febuxostat and inosine, or by single administration of each. Gout Nucleic Acid Metab. 2017, 41, 171–181. [Google Scholar] [CrossRef][Green Version]
- Kamatani, N.; Kushiyama, A.; Toyo-Oka, L.; Toyo-Oka, T. Treatment of two mitochondrial disease patients with a combination of febuxostat and inosine that enhances cellular ATP. J. Hum. Genet. 2019, 64, 351–353. [Google Scholar] [CrossRef]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.-P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef]
- Lee, H.-C.; Wei, Y.-H. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int. J. Biochem. Cell Biol. 2005, 37, 822–834. [Google Scholar] [CrossRef]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of Mitochondrial Transcription Specificity Factors (TFB1M and TFB2M) by Nuclear Respiratory Factors (NRF-1 and NRF-2) and PGC-1 Family Coactivators. Mol. Cell. Biol. 2005, 25, 1354–1366. [Google Scholar] [CrossRef]
- Schreiber, S.N.; Emter, R.; Hock, M.B.; Knutti, D.; Cardenas, J.; Podvinec, M.; Oakeley, E.J.; Kralli, A. The estrogen-related receptor (ERR) functions in PPAR coactivator 1 (PGC-1)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 6472–6477. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Mullur, R.; Liu, Y.-Y.; Brent, G.A. Thyroid Hormone Regulation of Metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.-Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nat. Cell Biol. 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Viscomi, C.; Bottani, E.; Civiletto, G.; Cerutti, R.; Moggio, M.; Fagiolari, G.; Schon, E.A.; Lamperti, C.; Zeviani, M. In Vivo Correction of COX Deficiency by Activation of the AMPK/PGC-1α Axis. Cell Metab. 2011, 14, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A Cold-Inducible Coactivator of Nuclear Receptors Linked to Adaptive Thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Geng, T.; Li, P.; Okutsu, M.; Yin, X.; Kwek, J.; Zhang, M.; Yan, Z. PGC-1α plays a functional role in exercise-induced mitochondrial biogenesis and angiogenesis but not fiber-type transformation in mouse skeletal muscle. Am. J. Physiol. Physiol. 2010, 298, C572–C579. [Google Scholar] [CrossRef]
- Jäger, S.; Handschin, C.; St.-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.-H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase--an energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef]
- Cerutti, R.; Pirinen, E.; Lamperti, C.; Marchet, S.; Sauve, A.A.; Li, W.; Leoni, V.; Schon, E.A.; Dantzer, F.; Auwerx, J.; et al. NAD+-Dependent Activation of Sirt1 Corrects the Phenotype in a Mouse Model of Mitochondrial Disease. Cell Metab. 2014, 19, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Peralta, S.; Garcia, S.; Yin, H.Y.; Arguello, T.; Diaz, F.; Moraes, C.T. Sustained AMPK activation improves muscle function in a mitochondrial myopathy mouse model by promoting muscle fiber regeneration. Hum. Mol. Genet. 2016, 25, 3178–3191. [Google Scholar] [CrossRef] [PubMed]
- Saada, A.; Dan, P.; Weissman, S.; Link, G.; Wikstrom, J.D.; Saada, A. Screening for Active Small Molecules in Mitochondrial Complex I Deficient Patient’s Fibroblasts, Reveals AICAR as the Most Beneficial Compound. PLoS ONE 2011, 6, e26883. [Google Scholar] [CrossRef]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.-C.; Staels, B. Sorting out the roles of PPAR in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef]
- Kersten, S. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef]
- Djouadi, F.; Bastin, J. Mitochondrial Genetic Disorders: Cell Signaling and Pharmacological Therapies. Cells 2019, 8, 289. [Google Scholar] [CrossRef]
- Bastin, J.; Aubey, F.; Rötig, A.; Munnich, A.; Djouadi, F. Activation of Peroxisome Proliferator-Activated Receptor Pathway Stimulates the Mitochondrial Respiratory Chain and Can Correct Deficiencies in Patients’ Cells Lacking Its Components. J. Clin. Endocrinol. Metab. 2008, 93, 1433–1441. [Google Scholar] [CrossRef]
- Casarin, A.; Giorgi, G.; Pertegato, V.; Siviero, R.; Cerqua, C.; Doimo, M.; Basso, G.; Sacconi, S.; Cassina, M.; Rizzuto, R.; et al. Copper and bezafibrate cooperate to rescue cytochrome c oxidase deficiency in cells of patients with sco2 mutations. Orphanet J. Rare Dis. 2012, 7, 21. [Google Scholar] [CrossRef]
- Douiev, L.; Sheffer, R.; Horvath, G.A.; Saada, A. Bezafibrate Improves Mitochondrial Fission and Function in DNM1L-Deficient Patient Cells. Cells 2020, 9, 301. [Google Scholar] [CrossRef]
- Yatsuga, S.; Suomalainen, A. Effect of bezafibrate treatment on late-onset mitochondrial myopathy in mice. Hum. Mol. Genet. 2011, 21, 526–535. [Google Scholar] [CrossRef]
- Dillon, L.M.; Hida, A.; Garcia, S.; Prolla, T.A.; Moraes, C.T. Long-Term Bezafibrate Treatment Improves Skin and Spleen Phenotypes of the mtDNA Mutator Mouse. PLoS ONE 2012, 7, e44335. [Google Scholar] [CrossRef] [PubMed]
- Steele, H.; Gomez-Duran, A.; Pyle, A.; Hopton, S.; Newman, J.; Stefanetti, R.J.; Charman, S.J.; Parikh, J.D.; He, L.; Viscomi, C.; et al. Metabolic effects of bezafibrate in mitochondrial disease. EMBO Mol. Med. 2020, 12, e11589. [Google Scholar] [CrossRef] [PubMed]
- Hondares, E.; Mora, O.; Yubero, P.; De La Concepción, M.R.; Iglesias, R.; Giralt, M.; Villarroya, F. Thiazolidinediones and Rexinoids Induce Peroxisome Proliferator-Activated Receptor-Coactivator (PGC)-1α Gene Transcription: An Autoregulatory Loop Controls PGC-1α Expression in Adipocytes via Peroxisome Proliferator-Activated Receptor-γ Coactivation. Endocrinology 2006, 147, 2829–2838. [Google Scholar] [CrossRef] [PubMed]
- Miglio, G.; Rosa, A.C.; Rattazzi, L.; Collino, M.; Lombardi, G.; Fantozzi, R. PPARγ stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss. Neurochem. Int. 2009, 55, 496–504. [Google Scholar] [CrossRef]
- Wilson-Fritch, L.; Burkart, A.; Bell, G.; Mendelson, K.; Leszyk, J.D.; Nicoloro, S.M.; Czech, M.P.; Corvera, S. Mitochondrial Biogenesis and Remodeling during Adipogenesis and in Response to the Insulin Sensitizer Rosiglitazone. Mol. Cell. Biol. 2003, 23, 1085–1094. [Google Scholar] [CrossRef]
- Rong, J.X.; Klein, J.-L.D.; Qiu, Y.; Xie, M.; Johnson, J.H.; Waters, K.M.; Zhang, V.; Kashatus, J.A.; Remlinger, K.S.; Bing, N.; et al. Rosiglitazone Induces Mitochondrial Biogenesis in Differentiated Murine 3T3-L1 and C3H/10T1/2 Adipocytes. PPAR Res. 2011, 2011, 1–11. [Google Scholar] [CrossRef]
- Strum, J.C.; Shehee, R.; Virley, D.; Richardson, J.; Mattie, M.; Selley, P.; Ghosh, S.; Nock, C.; Saunders, A.; Roses, A. Rosiglitazone Induces Mitochondrial Biogenesis in Mouse Brain. J. Alzheimers Dis. 2007, 11, 45–51. [Google Scholar] [CrossRef]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef]
- Bieganowski, P.; Brenner, C. Discoveries of Nicotinamide Riboside as a Nutrient and Conserved NRK Genes Establish a Preiss-Handler Independent Route to NAD+ in Fungi and Humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 Inhibition Increases Mitochondrial Metabolism through SIRT1 Activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Pirinen, E.; Cantó, C.; Jo, Y.S.; Morato, L.; Zhang, H.; Menzies, K.J.; Williams, E.G.; Mouchiroud, L.; Moullan, N.; Hagberg, C.; et al. Pharmacological Inhibition of Poly(ADP-Ribose) Polymerases Improves Fitness and Mitochondrial Function in Skeletal Muscle. Cell Metab. 2014, 19, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; Van Der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD+ synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef]
- Pirinen, E.; Auranen, M.; Khan, N.A.; Brilhante, V.; Urho, N.; Pessia, A.; Hakkarainen, A.; Kuula, J.; Heinonen, U.; Schmidt, M.S.; et al. Niacin Cures Systemic NAD+ Deficiency and Improves Muscle Performance in Adult-Onset Mitochondrial Myopathy. Cell Metab. 2020, 31, 1078–1090.e5. [Google Scholar] [CrossRef]
- Khan, N.A.; Auranen, M.; Paetau, I.; Pirinen, E.; Euro, L.; Forsström, S.; Pasila, L.; Velagapudi, V.; Carroll, C.J.; Auwerx, J.; et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B 3. EMBO Mol. Med. 2014, 6, 721–731. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef]
- Lee, C.F.; Caudal, A.; Abell, L.; Gowda, G.A.N.; Tian, R. Targeting NAD+ Metabolism as Interventions for Mitochondrial Disease. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef]
- Airhart, S.E.; Shireman, L.M.; Risler, L.J.; Anderson, G.D.; Gowda, G.A.N.; Raftery, D.; Tian, R.; Shen, D.D.; O’Brien, K.D. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE 2017, 12, e0186459. [Google Scholar] [CrossRef]
- Barrow, J.J.; Balsa, E.; Verdeguer, F.; Tavares, C.D.J.; Soustek, M.S.; Hollingsworth, L.R.; Jedrychowski, M.; Vogel, R.; Paulo, J.A.; Smeitink, J.; et al. Bromodomain Inhibitors Correct Bioenergetic Deficiency Caused by Mitochondrial Disease Complex I Mutations. Mol. Cell 2016, 64, 163–175. [Google Scholar] [CrossRef]
- Baratta, M.G.; Schinzel, A.C.; Zwang, Y.; Bandopadhayay, P.; Bowman-Colin, C.; Kutt, J.; Curtis, J.; Piao, H.; Wong, L.C.; Kung, A.L.; et al. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.-L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nat. Cell Biol. 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 Is Required for AMPK Activation and the Beneficial Effects of Resveratrol on Mitochondrial Function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nat. Cell Biol. 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Milbrandt, J. Resveratrol stimulates AMP kinase activity in neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 7217–7222. [Google Scholar] [CrossRef]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of Human SIRT1 Activation by Resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef]
- Beher, D.; Wu, J.; Cumine, S.; Kim, K.W.; Lu, S.-C.; Atangan, L.; Wang, M. Resveratrol is Not a Direct Activator of SIRT1 Enzyme Activity. Chem. Biol. Drug Des. 2009, 74, 619–624. [Google Scholar] [CrossRef]
- Kaeberlein, M.; McDonagh, T.; Heltweg, B.; Hixon, J.; Westman, E.A.; Caldwell, S.D.; Napper, A.; Curtis, R.; Distefano, P.S.; Fields, S.; et al. Substrate-specific Activation of Sirtuins by Resveratrol. J. Biol. Chem. 2005, 280, 17038–17045. [Google Scholar] [CrossRef]
- Um, J.-H.; Park, S.-J.; Kang, H.; Yang, S.; Foretz, M.; McBurney, M.W.; Kim, M.K.; Viollet, B.; Chung, J.H. AMP-Activated Protein Kinase-Deficient Mice Are Resistant to the Metabolic Effects of Resveratrol. Diabetes 2010, 59, 554–563. [Google Scholar] [CrossRef]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 Regulates Hepatocyte Lipid Metabolism through Activating AMP-activated Protein Kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 Modulation of the Acetylation Status, Cytosolic Localization, and Activity of LKB1. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef] [PubMed]
- Akyuva, Y.; Nazıroğlu, M. Resveratrol attenuates hypoxia-induced neuronal cell death, inflammation and mitochondrial oxidative stress by modulation of TRPM2 channel. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Revin, V.V.; Pinyaev, S.; Parchaykina, M.V.; Revina, E.S.; Maksimov, G.V.; Kuzmenko, T.P. The Effect of Resveratrol on the Composition and State of Lipids and the Activity of Phospholipase A2 During the Excitation and Regeneration of Somatic Nerves. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef]
- De Paepe, B.; Van Coster, R. A Critical Assessment of the Therapeutic Potential of Resveratrol Supplements for Treating Mitochondrial Disorders. Nutrients 2017, 9, 1017. [Google Scholar] [CrossRef]
- Davis, J.M.; Murphy, E.A.; Carmichael, M.D.; Davis, B. Quercetin increases brain and muscle mitochondrial biogenesis and exercise tolerance. Am. J. Physiol. Integr. Comp. Physiol. 2009, 296, R1071–R1077. [Google Scholar] [CrossRef]
- Koshinaka, K.; Honda, A.; Masuda, H.; Sato, A. Effect of Quercetin Treatment on Mitochondrial Biogenesis and Exercise-Induced AMP-Activated Protein Kinase Activation in Rat Skeletal Muscle. Nutrients 2020, 12, 729. [Google Scholar] [CrossRef]
- Wang, D.-M.; Li, S.-Q.; Wu, W.-L.; Zhu, X.-Y.; Wang, Y.; Yuan, H.-Y. Effects of Long-Term Treatment with Quercetin on Cognition and Mitochondrial Function in a Mouse Model of Alzheimer’s Disease. Neurochem. Res. 2014, 39, 1533–1543. [Google Scholar] [CrossRef]
- Karuppagounder, S.; Madathil, S.; Pandey, M.; Haobam, R.; Rajamma, U.; Mohanakumar, K. Quercetin up-regulates mitochondrial complex-I activity to protect against programmed cell death in rotenone model of Parkinson’s disease in rats. Neuroscience 2013, 236, 136–148. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, Z.; Feng, Z.; Hao, J.; Shen, W.; Li, X.; Sun, L.; Sharman, E.; Wang, Y.; Wertz, K.; et al. Hydroxytyrosol protects against oxidative damage by simultaneous activation of mitochondrial biogenesis and phase II detoxifying enzyme systems in retinal pigment epithelial cells. J. Nutr. Biochem. 2010, 21, 1089–1098. [Google Scholar] [CrossRef]
- Feng, Z.; Bai, L.; Yan, J.; Li, Y.; Shen, W.; Wang, Y.; Wertz, K.; Weber, P.; Zhang, Y.; Chen, Y.; et al. Mitochondrial dynamic remodeling in strenuous exercise-induced muscle and mitochondrial dysfunction: Regulatory effects of hydroxytyrosol. Free Radic. Biol. Med. 2011, 50, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Casuso, R.A.; Al-Fazazi, S.; Hidalgo-Gutierrez, A.; López, L.C.; Plaza-Díaz, J.; Rueda-Robles, A.; Huertas, J.R. Hydroxytyrosol influences exercise-induced mitochondrial respiratory complex assembly into supercomplexes in rats. Free Radic. Biol. Med. 2019, 134, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Zheng, A.; Li, H.; Xu, J.; Cao, K.; Li, H.; Pu, W.; Yang, Z.; Peng, Y.; Long, J.; Liu, J.; et al. Hydroxytyrosol improves mitochondrial function and reduces oxidative stress in the brain of db/db mice: Role of AMP-activated protein kinase activation. Br. J. Nutr. 2015, 113, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, G.; Di Rosa, G.; Scuto, M.; Leri, M.; Stefani, M.; Schmitz-Linneweber, C.; Calabrese, V.; Saul, N. Healthspan Maintenance and Prevention of Parkinson’s-like Phenotypes with Hydroxytyrosol and Oleuropein Aglycone in C. elegans. Int. J. Mol. Sci. 2020, 21, 2588. [Google Scholar] [CrossRef]
- Eckert, G.P.; Schiborr, C.; Hagl, S.; Abdel-Kader, R.; Müller, W.E.; Rimbach, G.; Frank, J. Curcumin prevents mitochondrial dysfunction in the brain of the senescence-accelerated mouse-prone 8. Neurochem. Int. 2013, 62, 595–602. [Google Scholar] [CrossRef]
- Chin, D.; Hagl, S.; Hoehn, A.; Huebbe, P.; Pallauf, K.; Grune, T.; Frank, J.; Eckert, G.P.; Rimbach, G. Adenosine triphosphate concentrations are higher in the brain of APOE3- compared to APOE4-targeted replacement mice and can be modulated by curcumin. Genes Nutr. 2014, 9, 397. [Google Scholar] [CrossRef]
- Kalpravidh, R.W.; Siritanaratkul, N.; Insain, P.; Charoensakdi, R.; Panichkul, N.; Hatairaktham, S.; Srichairatanakool, S.; Phisalaphong, C.; Rachmilewitz, E.; Fucharoen, S. Improvement in oxidative stress and antioxidant parameters in β-thalassemia/Hb E patients treated with curcuminoids. Clin. Biochem. 2010, 43, 424–429. [Google Scholar] [CrossRef]
- Nasseri, E.; Mohammadi, E.; Tamaddoni, A.; Qujeq, D.; Zayeri, F.; Zand, H. Benefits of Curcumin Supplementation on Antioxidant Status in β-Thalassemia Major Patients: A Double-Blind Randomized Controlled Clinical Trial. Ann. Nutr. Metab. 2017, 71, 136–144. [Google Scholar] [CrossRef]
- Musi, N.; Goodyear, L.J. Targeting the AMP-activated protein kinase for the treatment of type 2 diabetes. Curr. Drug Targets Immune Endocr. Metab. Disord. 2002, 2, 119–127. [Google Scholar] [CrossRef]
- Goodyear, L.J. The Exercise Pill—Too Good to Be True? N. Engl. J. Med. 2008, 359, 1842–1844. [Google Scholar] [CrossRef]
- Bundred, N.J.; Gardovskis, J.; Jaskiewicz, J.; Eglitis, J.; Paramonov, V.; McCormack, P.; Swaisland, H.; Cavallin, M.; Parry, T.; Carmichael, J.; et al. Evaluation of the pharmacodynamics and pharmacokinetics of the PARP inhibitor olaparib: A Phase I multicentre trial in patients scheduled for elective breast cancer surgery. Investig. New Drugs 2013, 31, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Conze, D.; Brenner, C.; Kruger, C.L. Safety and Metabolism of Long-term Administration of NIAGEN (Nicotinamide Riboside Chloride) in a Randomized, Double-Blind, Placebo-controlled Clinical Trial of Healthy Overweight Adults. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Conze, D.B.; Crespo-Barreto, J.; Kruger, C.L. Safety assessment of nicotinamide riboside, a form of vitamin B3. Hum. Exp. Toxicol. 2016, 35, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Marinescu, A.G.; Chen, J.; Holmes, H.E.; Guarente, L.; Mendes, O.; Morris, M.; Dellinger, R.W. Safety Assessment of High-Purity, Synthetic Nicotinamide Riboside (NR-E) in a 90-Day Repeated Dose Oral Toxicity Study, With a 28-Day Recovery Arm. Int. J. Toxicol. 2020, 39, 307–320. [Google Scholar] [CrossRef]
- Lacza, Z.; Pankotai, E.; Csordás, A.; Gero, D.; Kiss, L.; Horváth, E.M.; Kollai, M.; Busija, D.W.; Szabó, C. Mitochondrial NO and reactive nitrogen species production: Does mtNOS exist? Nitric Oxide 2006, 14, 162–168. [Google Scholar] [CrossRef]
- Leite, A.C.R.; Oliveira, H.C.; Utino, F.L.; Garcia, R.; Alberici, L.C.; Fernandes, M.P.; Castilho, R.F.; Vercesi, A.E. Mitochondria generated nitric oxide protects against permeability transition via formation of membrane protein S-nitrosothiols. Biochim. Biophys. Acta (BBA) Bioenerg. 2010, 1797, 1210–1216. [Google Scholar] [CrossRef]
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci. 2005, 26, 190–195. [Google Scholar] [CrossRef]
- Eqian, J.; Fulton, D.J. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front. Physiol. 2013, 4, 347. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D. cGMP-Dependent Protein Kinases and cGMP Phosphodiesterases in Nitric Oxide and cGMP Action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Kaupp, U.B.; Seifert, R. Cyclic Nucleotide-Gated Ion Channels. Physiol. Rev. 2002, 82, 769–824. [Google Scholar] [CrossRef]
- Fischmeister, R.; Méry, P.-F. Regulation of cardiac Ca2+ channels by cGMP and NO. In Molecular Physiology and Pharmacology of Cardiac Ion Channels and Transporters; Springer: Dordrecht, The Netherlands, 1996; pp. 93–105. [Google Scholar] [CrossRef]
- White, R.E. Cyclic GMP and Ion Channel Regulation. In Adv. Second Messenger and Phosphoprotein Res.; Elsevier: London, UK, 1999; Volume 33, pp. 251–277. [Google Scholar] [CrossRef]
- Brown, G.C. CELL BIOLOGY: Enhanced: NO Says Yes to Mitochondria. Science 2003, 299, 838–839. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef] [PubMed]
- Gutsaeva, D.R.; Carraway, M.S.; Suliman, H.B.; Demchenko, I.T.; Shitara, H.; Yonekawa, H.; Piantadosi, C.A. Transient Hypoxia Stimulates Mitochondrial Biogenesis in Brain Subcortex by a Neuronal Nitric Oxide Synthase-Dependent Mechanism. J. Neurosci. 2008, 28, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Sanders, O. Sildenafil for the Treatment of Alzheimer’s Disease: A Systematic Review. J. Alzheimers Dis. Rep. 2020, 4, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Ohama, E.; Ohara, S.; Ikuta, F.; Tanaka, K.; Nishizawa, M.; Miyatake, T. Mitochondrial angiopathy in cerebral blood vessels of mitochondrial eneephalomyopathy. Acta Neuropathol. 1987, 74, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Vattemi, G.; Mechref, Y.; Marini, M.; Tonin, P.; Minuz, P.; Grigoli, L.; Guglielmi, V.; Klouckova, I.; Chiamulera, C.; Meneguzzi, A.; et al. Increased Protein Nitration in Mitochondrial Diseases: Evidence for Vessel Wall Involvement. Mol. Cell. Proteom. 2010, 10, 110 002964. [Google Scholar] [CrossRef] [PubMed]
- Sarti, P.; Forte, E.; Giuffrè, A.; Mastronicola, D.; Magnifico, M.C.; Arese, M. The Chemical Interplay between Nitric Oxide and Mitochondrial Cytochrome c Oxidase: Reactions, Effectors and Pathophysiology. Int. J. Cell Biol. 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Hsu, J.W.; Emrick, L.T.; Wong, L.-J.C.; Craigen, W.J.; Jahoor, F.; Scaglia, F. Restoration of impaired nitric oxide production in MELAS syndrome with citrulline and arginine supplementation. Mol. Genet. Metab. 2012, 105, 607–614. [Google Scholar] [CrossRef]
- Naini, A.; Kaufmann, P.; Shanske, S.; Engelstad, K.; De Vivo, D.C.; Schon, E.A. Hypocitrullinemia in patients with MELAS: An insight into the “MELAS paradox”. J. Neurol. Sci. 2005, 229, 187–193. [Google Scholar] [CrossRef]
- Koga, Y.; Akita, Y.; Junko, N.; Yatsuga, S.; Povalko, N.; Fukiyama, R.; Ishii, M.; Matsuishi, T. Endothelial dysfunction in MELAS improved by l-arginine supplementation. Neurology 2006, 66, 1766–1769. [Google Scholar] [CrossRef]
- Tengan, C.H.; Kiyomoto, B.H.; Godinho, R.O.; Gamba, J.; Neves, A.C.; Schmidt, B.; Oliveira, A.S.; Gabbai, A.A. The role of nitric oxide in muscle fibers with oxidative phosphorylation defects. Biochem. Biophys. Res. Commun. 2007, 359, 771–777. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Emrick, L.T.; Craigen, W.J.; Scaglia, F. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol. Genet. Metab. 2012, 107, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Akita, Y.; Nishioka, J.; Yatsuga, S.; Povalko, N.; Tanabe, Y.; Fujimoto, S.; Matsuishi, T. L-Arginine improves the symptoms of strokelike episodes in MELAS. Neurology 2005, 64, 710–712. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Emrick, L.T.; Chanprasert, S.; Craigen, W.J.; Scaglia, F. Mitochondria: Role of citrulline and arginine supplementation in MELAS syndrome. Int. J. Biochem. Cell Biol. 2014, 48, 85–91. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Emrick, L.T.; Williamson, K.C.; Craigen, W.J.; Scaglia, F. The effect of citrulline and arginine supplementation on lactic acidemia in MELAS syndrome. Meta Gene 2013, 1, 8–14. [Google Scholar] [CrossRef]
- Koga, Y.; Povalko, N.; Nishioka, J.; Katayama, K.; Kakimoto, N.; Matsuishi, T. MELAS and l-arginine therapy: Pathophysiology of stroke-like episodes. Ann. N. Y. Acad. Sci. 2010, 1201, 104–110. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Emrick, L.T.; Hsu, J.W.; Chanprasert, S.; Almannai, M.; Craigen, W.J.; Jahoor, F.; Scaglia, F. Impaired nitric oxide production in children with MELAS syndrome and the effect of arginine and citrulline supplementation. Mol. Genet. Metab. 2016, 117, 407–412. [Google Scholar] [CrossRef]
- Potter, L.R.; Yoder, A.R.; Flora, D.R.; Antos, L.K.; Dickey, D.M. Natriuretic Peptides: Their Structures, Receptors, Physiologic Functions and Therapeutic Applications. In cGMP: Generators, Effectors and Therapeutic Implications; Schmidt, H.H.H.W., Hofmann, F., Stasch, J.-P., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 341–366. [Google Scholar]
- Miyashita, K.; Itoh, H.; Tsujimoto, H.; Tamura, N.; Fukunaga, Y.; Sone, M.; Yamahara, K.; Taura, D.; Inuzuka, M.; Sonoyama, T.; et al. Natriuretic Peptides/cGMP/cGMP-Dependent Protein Kinase Cascades Promote Muscle Mitochondrial Biogenesis and Prevent Obesity. Diabetes 2009, 58, 2880–2892. [Google Scholar] [CrossRef]
- Engeli, S.; Birkenfeld, A.L.; Badin, P.-M.; Bourlier, V.; Louche, K.; Viguerie, N.; Thalamas, C.; Montastier, E.; Larrouy, D.; Harant, I.; et al. Natriuretic peptides enhance the oxidative capacity of human skeletal muscle. J. Clin. Investig. 2012, 122, 4675–4679. [Google Scholar] [CrossRef]
- Whitaker, R.M.; Wills, L.P.; Stallons, L.J.; Schnellmann, R.G. cGMP-Selective Phosphodiesterase Inhibitors Stimulate Mitochondrial Biogenesis and Promote Recovery from Acute Kidney Injury. J. Pharmacol. Exp. Ther. 2013, 347, 626–634. [Google Scholar] [CrossRef]
- Corbin, J.D. Mechanisms of action of PDE5 inhibition in erectile dysfunction. Int. J. Impot. Res. 2004, 16, S4–S7. [Google Scholar] [CrossRef] [PubMed]
- Mitschke, M.M.; Hoffmann, L.S.; Gnad, T.; Scholz, D.; Kruithoff, K.; Mayer, P.; Haas, B.; Sassmann, A.; Pfeifer, A.; Kilić, A. Increased cGMP promotes healthy expansion and browning of white adipose tissue. FASEB J. 2013, 27, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Lesimple, P.; Bukowiecki, R.; Zink, A.; Inak, G.; Mlody, B.; Singh, M.; Semtner, M.; Mah, N.; Auré, K.; et al. Human iPSC-Derived Neural Progenitors Are an Effective Drug Discovery Model for Neurological mtDNA Disorders. Cell Stem Cell 2017, 20, 659–674.e9. [Google Scholar] [CrossRef] [PubMed]
- Percival, J.M.; Siegel, M.P.; Knowels, G.; Marcinek, D.J. Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum. Mol. Genet. 2013, 22, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Tetsi, L.; Charles, A.-L.; Georg, I.; Goupilleau, F.; Lejay, A.; Talha, S.; Maumy-Bertrand, M.; Lugnier, C.; Geny, B. Effect of the Phosphodiesterase 5 Inhibitor Sildenafil on Ischemia-Reperfusion-Induced Muscle Mitochondrial Dysfunction and Oxidative Stress. Antioxidants 2019, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Cornish, K.S.; Barras, C. Leber’s Hereditary Optic Neuropathy Precipitated by Tadalafil Use for Erectile Dysfunction. Semin. Ophthalmol. 2011, 26, 7–10. [Google Scholar] [CrossRef]
- Choi, M.H.; Lee, I.K.; Kim, G.W.; Kim, B.U.; Han, Y.-H.; Yu, D.-Y.; Park, H.S.; Kim, K.Y.; Lee, J.S.; Choi, C.; et al. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nat. Cell Biol. 2005, 435, 347–353. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, Oxidants, and Aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Wallace, D.C.; Fan, W. The pathophysiology of mitochondrial disease as modeled in the mouse. Genes Dev. 2009, 23, 1714–1736. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [PubMed]
- Polyak, E.; Ostrovsky, J.; Peng, M.; Dingley, S.D.; Tsukikawa, M.; Kwon, Y.J.; McCormack, S.E.; Bennett, M.; Xiao, R.; Seiler, C.; et al. N-acetylcysteine and vitamin E rescue animal longevity and cellular oxidative stress in pre-clinical models of mitochondrial complex I disease. Mol. Genet. Metab. 2018, 123, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M.; Moore, T.; Le, A.; Atkuri, K.; Shah, M.K.; Cusmano-Ozog, K.; Niemi, A.-K.; Cowan, T.M. Degree of Glutathione Deficiency and Redox Imbalance Depend on Subtype of Mitochondrial Disease and Clinical Status. PLoS ONE 2014, 9, e100001. [Google Scholar] [CrossRef] [PubMed]
- Salmi, H.; Leonard, J.V.; Rahman, S.; Lapatto, R. Plasma thiol status is altered in children with mitochondrial diseases. Scand. J. Clin. Lab. Investig. 2012, 72, 152–157. [Google Scholar] [CrossRef]
- Mancuso, M.; Orsucci, D.; LoGerfo, A.; Rocchi, A.; Petrozzi, L.; Nesti, C.; Galetta, F.; Santoro, G.; Murri, L.; Siciliano, G. Oxidative stress biomarkers in mitochondrial myopathies, basally and after cysteine donor supplementation. J. Neurol. 2009, 257, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Bartsakoulia, M.; Mϋller, J.S.; Gomez-Duran, A.; Yu-Wai-Man, P.; Boczonadi, V.; Horváth, H.R. Cysteine Supplementation May be Beneficial in a Subgroup of Mitochondrial Translation Deficiencies. J. Neuromuscul. Dis. 2016, 3, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, G.; Gahl, W.A. Cystinosis. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Besouw, M.; Masereeuw, R.; Heuvel, L.V.D.; Levtchenko, E. Cysteamine: An old drug with new potential. Drug Discov. Today 2013, 18, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Konkwo, C.; Lavorato, M.; Mathew, N.D.; Peng, M.; Ostrovsky, J.; Kwon, Y.-J.; Polyak, E.; Lightfoot, R.; Seiler, C.; et al. Pre-clinical evaluation of cysteamine bitartrate as a therapeutic agent for mitochondrial respiratory chain disease. Hum. Mol. Genet. 2019, 28, 1837–1852. [Google Scholar] [CrossRef]
- Dohil, R.; Rioux, P. Pharmacokinetic Studies of Cysteamine Bitartrate Delayed-Release. Clin. Pharmacol. Drug Dev. 2013, 2, 178–185. [Google Scholar] [CrossRef]
- Ferreira, L.F.; Campbell, K.S.; Reid, M.B. N-acetylcysteine in handgrip exercise: Plasma thiols and adverse reactions. Int. J. Sport Nutr. Exerc. Metab. 2011, 21, 146–154. [Google Scholar] [CrossRef]
- Viscomi, C.; Burlina, A.B.; Dweikat, I.; Savoiardo, M.; Lamperti, C.; Hildebrandt, T.M.; Tiranti, V.; Zeviani, M. Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat. Med. 2010, 16, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Tiranti, V.; Viscomi, C.; Hildebrandt, T.; Di Meo, I.; Mineri, R.; Tiveron, C.; Levitt, M.D.; Prelle, A.; Fagiolari, G.; Rimoldi, M.; et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 2009, 15, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, I.; Fagiolari, G.; Prelle, A.; Viscomi, C.; Zeviani, M.; Tiranti, V. Chronic Exposure to Sulfide Causes Accelerated Degradation of Cytochrome c Oxidase in Ethylmalonic Encephalopathy. Antioxid. Redox Signal. 2011, 15, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Kitzler, T.M.; Gupta, I.R.; Osterman, B.; Poulin, C.; Trakadis, Y.; Waters, P.J.; Buhas, D.C. Acute and Chronic Management in an Atypical Case of Ethylmalonic Encephalopathy. JIMD Rep. 2018, 45, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Kılıç, M.; Dedeoğlu, Ö.; Göçmen, R.; Kesici, S.; Yüksel, D. Successful treatment of a patient with ethylmalonic encephalopathy by intravenous N-acetylcysteine. Metab. Brain Dis. 2017, 32, 293–296. [Google Scholar] [CrossRef]
- Boyer, M.; Sowa, M.; Di Meo, I.; Eftekharian, S.; Steenari, M.; Tiranti, V.; Abdenur, J. Response to medical and a novel dietary treatment in newborn screen identified patients with ethylmalonic encephalopathy. Mol. Genet. Metab. 2018, 124, 57–63. [Google Scholar] [CrossRef]
- Bustamante, J. α-Lipoic Acid in Liver Metabolism and Disease. Free Radic. Biol. Med. 1998, 24, 1023–1039. [Google Scholar] [CrossRef]
- Smith, A.R.; Shenvi, S.V.; Widlansky, M.; Suh, J.H.; Hagen, T.M. Lipoic Acid as a Potential Therapy for Chronic Diseases Associated with Oxidative Stress. Curr. Med. Chem. 2004, 11, 1135–1146. [Google Scholar] [CrossRef]
- Kozlov, A.V.; Gille, L.; Staniek, K.; Nohl, H. Dihydrolipoic Acid Maintains Ubiquinone in the Antioxidant Active Form by Two-Electron Reduction of Ubiquinone and One-Electron Reduction of Ubisemiquinone. Arch. Biochem. Biophys. 1999, 363, 148–154. [Google Scholar] [CrossRef]
- Teichert, J.; Hermann, R.; Ruus, P.; Preiss, R. Plasma Kinetics, Metabolism, and Urinary Excretion of Alpha-Lipoic Acid following Oral Administration in Healthy Volunteers. J. Clin. Pharmacol. 2003, 43, 1257–1267. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.C.; Macdonald, J.R.; Mahoney, D.J.; Parise, G.; Beal, M.F.; Tarnopolsky, M.A. Beneficial effects of creatine, CoQ10, and lipoic acid in mitochondrial disorders. Muscle Nerve 2007, 35, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Beltran, R.S.; Coker, S.B. Familial Spastic Paraparesis: A Case of a Mitochondrial Disorder. Pediatr. Neurosurg. 1990, 16, 40–42. [Google Scholar] [CrossRef] [PubMed]
- Eleff, S.; Kennaway, N.G.; Buist, N.R.; Darley-Usmar, V.M.; Capaldi, R.A.; Bank, W.J.; Chance, B. 31P NMR study of improvement in oxidative phosphorylation by vitamins K3 and C in a patient with a defect in electron transport at complex III in skeletal muscle. Proc. Natl. Acad. Sci. USA 1984, 81, 3529–3533. [Google Scholar] [CrossRef]
- Mowat, D.; Kirby, D.M.; Kamath, K.R.; Kan, A.; Thorburn, D.R.; Christodoulou, J. Respiratory chain complex III deficiency with pruritus: A novel vitamin responsive clinical feature. J. Pediatr. 1999, 134, 352–354. [Google Scholar] [CrossRef]
- Andreu, A.L.; Hanna, M.G.; Reichmann, H.; Bruno, C.; Penn, A.S.; Tanji, K.; Pallotti, F.; Iwata, S.; Bonilla, E.; Lach, B.; et al. Exercise Intolerance Due to Mutations in the CytochromebGene of Mitochondrial DNA. N. Engl. J. Med. 1999, 341, 1037–1044. [Google Scholar] [CrossRef]
- Koopman, W.J.H.; Verkaart, S.; Vries, S.E.V.E.-D.; Grefte, S.; Smeitink, J.A.; Nijtmans, L.G.; Willems, P.H. Mitigation of NADH: Ubiquinone oxidoreductase deficiency by chronic Trolox treatment. Biochim. Biophys. Acta (BBA) Bioenerg. 2008, 1777, 853–859. [Google Scholar] [CrossRef]
- Distelmaier, F.; Visch, H.-J.; Smeitink, J.A.M.; Mayatepek, E.; Koopman, W.J.H.; Willems, P.H.G.M. The antioxidant Trolox restores mitochondrial membrane potential and Ca2+-stimulated ATP production in human complex I deficiency. J. Mol. Med. 2009, 87, 515–522. [Google Scholar] [CrossRef]
- Bentinger, M.; Brismar, K.; Dallner, G. The antioxidant role of coenzyme Q. Mitochondrion 2007, 7, S41–S50. [Google Scholar] [CrossRef]
- Musumeci, O.; Naini, A.; Slonim, A.E.; Skavin, N.; Hadjigeorgiou, G.L.; Krawiecki, N.; Weissman, B.M.; Tsao, C.-Y.; Mendell, J.R.; Shanske, S.; et al. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology 2001, 56, 849–855. [Google Scholar] [CrossRef]
- Lamperti, C.; Naini, A.; Hirano, M.; De Vivo, D.; Bertini, E.; Servidei, S.; Valeriani, M.; Lynch, D.; Banwell, B.; Berg, M.; et al. Cerebellar ataxia and coenzyme Q10 deficiency. Neurology 2003, 60, 1206–1208. [Google Scholar] [CrossRef] [PubMed]
- Argov, Z.; Bank, W.J.; Maris, J.; Eleff, S.; Kennaway, N.G.; Olson, R.E.; Chance, B. Treatment of mitochondrial myopathy due to complex III deficiency with vitamins K3 and C: A31P-NMR follow-up study. Ann. Neurol. 1986, 19, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta (BBA) Biomembr. 2004, 1660, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Geromel, V.; Darin, N.; Chrétien, D.; Bénit, P.; Delonlay, P.; Rötig, A.; Munnich, A.; Rustin, P. Coenzyme Q10 and idebenone in the therapy of respiratory chain diseases: Rationale and comparative benefits. Mol. Genet. Metab. 2002, 77, 21–30. [Google Scholar] [CrossRef]
- Neergheen, V.; Chalasani, A.; Wainwright, L.; Yubero, D.; Montero, R.; Artuch, R.; Hargreaves, I. Coenzyme Q10 in the Treatment of Mitochondrial Disease. J. Inborn Errors Metab. Screen. 2017, 5, 232640981770777. [Google Scholar] [CrossRef]
- Villanueva-Paz, M.; Povea-Cabello, S.; Villalón-García, I.; Álvarez-Córdoba, M.; Suárez-Rivero, J.M.; Talaverón-Rey, M.; Jackson, S.; Falcón-Moya, R.; Rodríguez-Moreno, A.; Sánchez-Alcázar, J.A. Parkin-mediated mitophagy and autophagy flux disruption in cellular models of MERRF syndrome. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165726. [Google Scholar] [CrossRef]
- Ogasahara, S.; Nishikawa, Y.; Yorifuji, S.; Soga, F.; Nakamura, Y.; Takahashi, M.; Hashimoto, S.; Kono, N.; Tarui, S. Treatment of Kearns-Sayre syndrome with coenzyme Q10. Neurology 1986, 36, 45. [Google Scholar] [CrossRef]
- Chan, A.; Reichmann, H.; Kögel, A.; Beck, A.; Gold, R. Metabolic changes in patients with mitochondrial myopathies and effects of coenzyme Q10 therapy. J. Neurol. 1998, 245, 681–685. [Google Scholar] [CrossRef]
- Bendahan, D.; Desnuelle, C.; Vanuxem, D.; Confort-Gouny, S.; Figarella-Branger, D.; Pellissier, J.-F.; Kozak-Ribbens, G.; Pouget, J.; Serratrice, G.; Cozzone, P.J. 31P NMR spectroscopy and ergometer exercise test as evidence for muscle oxidative performance improvement with coenzyme Q in mitochondrial myopathies. Neurology 1992, 42, 1203. [Google Scholar] [CrossRef]
- Goda, S.; Hamada, T.; Ishimoto, S.; Kobayashi, T.; Goto, I.; Kuroiwa, Y. Clinical improvement after administration of coenzyme Q10 in a patient with mitochondrial encephalomyopathy. J. Neurol. 1987, 234, 62–63. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Takahashi, M.; Yorifuji, S.; Nakamura, Y.; Ueno, S.; Tarui, S.; Kozuka, T.; Nishimura, T. Long-term coenzyme Q10 therapy for a mitochondrial encephalomyopathy with cytochrome c oxidase deficiency: A 31P NMR study. Neurology 1989, 39, 399. [Google Scholar] [CrossRef] [PubMed]
- Zierz, S.; Jahns, G.; Jerusalem, F. Coenzyme Q in serum and muscle of 5 patients with Kearns-Sayre syndrome and 12 patients with ophthalmoplegia plus. J. Neurol. 1989, 236, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Barbiroli, B.; Iotti, S.; Lodi, R. Improved brain and muscle mitochondrial respiration with CoQ. Anin vivostudy by31P-MR spectroscopy in patients with mitochondrial cytopathies. BioFactors 1999, 9, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Lott, M.T.; Voljavec, A.S.; Soueidan, S.A.; Costigan, D.A.; Wallace, D.C. Spontaneous Kearns-Sayre/chronic external ophthalmoplegia plus syndrome associated with a mitochondrial DNA deletion: A slip-replication model and metabolic therapy. Proc. Natl. Acad. Sci. USA 1989, 86, 7952–7956. [Google Scholar] [CrossRef]
- Matthews, P.M.; Ford, B.; Dandurand, R.J.; Eidelman, D.H.; O’Connor, D.; Sherwin, A.; Karpati, G.; Andermann, F.; Arnold, D.L. Coenzyme Q10 with multiple vitamins is generally ineffective in treatment of mitochondrial disease. Neurology 1993, 43, 884. [Google Scholar] [CrossRef]
- Bresolin, N.; Bet, L.; Binda, A.; Moggio, M.; Comi, G.; Nador, F.; Ferrante, C.; Carenzi, A.; Scarlato, G. Clinical and biochemical correlations in mitochondrial myopathies treated with coenzyme Q10. Neurology 1988, 38, 892. [Google Scholar] [CrossRef]
- Marriage, B.; Clandinin, M.; Macdonald, I.M.; Glerum, D. Cofactor treatment improves ATP synthetic capacity in patients with oxidative phosphorylation disorders. Mol. Genet. Metab. 2004, 81, 263–272. [Google Scholar] [CrossRef]
- Glover, E.I.; Martin, J.; Maher, A.; Thornhill, R.E.; Moran, G.R.; Tarnopolsky, M.A. A randomized trial of coenzyme Q10in mitochondrial disorders. Muscle Nerve 2010, 42, 739–748. [Google Scholar] [CrossRef]
- Sacconi, S.; Trevisson, E.; Salviati, L.; Aymé, S.; Rigal, O.; Redondo, A.G.; Mancuso, M.; Siciliano, G.; Tonin, P.; Angelini, C.; et al. Coenzyme Q10 is frequently reduced in muscle of patients with mitochondrial myopathy. Neuromuscul. Disord. 2010, 20, 44–48. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Degrauw, T.J.; Feigenbaum, A.S.; Hoppel, C.; Kerr, D.S.; McCandless, S.E.; Miles, M.V.; Robinson, B.H.; Tang, P.H. Design and implementation of the first randomized controlled trial of coenzyme Q10 in children with primary mitochondrial diseases. Mitochondrion 2012, 12, 623–629. [Google Scholar] [CrossRef]
- Zs.-Nagy, I. Chemistry, toxicology, pharmacology and pharmacokinetics of idebenone: A review. Arch. Gerontol. Geriatr. 1990, 11, 177–186. [Google Scholar] [CrossRef]
- Di Prospero, N.A.; Sumner, C.J.; Penzak, S.R.; Ravina, B.; Fischbeck, K.H.; Taylor, J.P. Safety, Tolerability, and Pharmacokinetics of High-Dose Idebenone in Patients With Friedreich Ataxia. Arch. Neurol. 2007, 64, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Angebault, C.; Gueguen, N.; Desquiret-Dumas, V.; Chevrollier, A.; Guillet, V.; Verny, C.; Cassereau, J.; Ferré, M.; Milea, D.; Amati-Bonneau, P.; et al. Idebenone increases mitochondrial complex I activity in fibroblasts from LHON patients while producing contradictory effects on respiration. BMC Res. Notes 2011, 4, 557. [Google Scholar] [CrossRef] [PubMed]
- Morvan, D.; Demidem, A. NMR metabolomics of fibroblasts with inherited mitochondrial Complex I mutation reveals treatment-reversible lipid and amino acid metabolism alterations. Metabolomics 2018, 14, 1–10. [Google Scholar] [CrossRef]
- Heitz, F.D.; Erb, M.; Anklin, C.; Robay, D.; Pernet, V.; Gueven, N. Idebenone Protects against Retinal Damage and Loss of Vision in a Mouse Model of Leber’s Hereditary Optic Neuropathy. PLoS ONE 2012, 7, e45182. [Google Scholar] [CrossRef]
- Klopstock, T.; Yu-Wai-Man, P.; Dimitriadis, K.; Rouleau, J.; Heck, S.; Bailie, M.; Atawan, A.; Chattopadhyay, S.; Schubert, M.; Garip, A.; et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain 2011, 134, 2677–2686. [Google Scholar] [CrossRef]
- Rudolph, G.; Dimitriadis, K.; Büchner, B.; Heck, S.; Al-Tamami, J.; Seidensticker, F.; Rummey, C.; Leinonen, M.; Meier, T.; Klopstock, T. Effects of Idebenone on Color Vision in Patients With Leber Hereditary Optic Neuropathy. J. Neuro Ophthalmol. 2013, 33, 30–36. [Google Scholar] [CrossRef]
- Klopstock, T.; Metz, G.; Yu-Wai-Man, P.; Büchner, B.; Gallenmüller, C.; Bailie, M.; Nwali, N.; Griffiths, P.G.; Von Livonius, B.; Reznicek, L.; et al. Persistence of the treatment effect of idebenone in Leber’s hereditary optic neuropathy. Brain 2013, 136, e230. [Google Scholar] [CrossRef]
- Holzerova, E.; Danhauser, K.; Haack, T.B.; Kremer, L.S.; Melcher, M.; Ingold, I.; Kobayashi, S.; Terrile, C.; Wolf, P.; Schaper, J.; et al. Human thioredoxin 2 deficiency impairs mitochondrial redox homeostasis and causes early-onset neurodegeneration. Brain 2015, 139, 346–354. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Smith, T.; Seto, S.; Ganne, P.; Votruba, M. A randomized, placebo-controlled trial of the benzoquinone idebenone in a mouse model of OPA1-related dominant optic atrophy reveals a limited therapeutic effect on retinal ganglion cell dendropathy and visual function. Neuroscience 2016, 319, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Barboni, P.; Valentino, M.L.; La Morgia, C.; Carbonelli, M.; Savini, G.; De Negri, A.; Simonelli, F.; Sadun, F.; Caporali, L.; Maresca, A.; et al. Idebenone treatment in patients with OPA1-mutant dominant optic atrophy. Brain 2013, 136, e231. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, M.; La Morgia, C.; Carbonelli, M.; Di Vito, L.; Amore, G.; Zenesini, C.; Cascavilla, M.L.; Barboni, P.; Carelli, V. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Ann. Clin. Transl. Neurol. 2020, 7, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. USA 2010, 1201, 96–103. [Google Scholar] [CrossRef]
- Asin-Cayuela, J.; Manas, A.-R.B.; James, A.M.; Smith, R.A.J.; Murphy, M.P. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 2004, 571, 9–16. [Google Scholar] [CrossRef]
- James, A.M.; Sharpley, M.S.; Manas, A.-R.B.; Frerman, F.E.; Hirst, J.; Smith, R.A.J.; Murphy, M.P. Interaction of the Mitochondria-targeted Antioxidant MitoQ with Phospholipid Bilayers and Ubiquinone Oxidoreductases. J. Biol. Chem. 2007, 282, 14708–14718. [Google Scholar] [CrossRef]
- Jauslin, M.L.; Meier, T.; Smith, R.A.J.; Murphy, P.M. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003, 17, 1–10. [Google Scholar] [CrossRef]
- Rodriguez-Cuenca, S.; Cochemé, H.M.; Logan, A.; Abakumova, I.; Prime, T.A.; Rose, C.; Vidal-Puig, A.; Smith, A.C.; Rubinsztein, D.C.; Fearnley, I.M.; et al. Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radic. Biol. Med. 2010, 48, 161–172. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef]
- Snow, B.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, R.M.; Salt, I.P.; Miller, W.H.; Logan, A.; Ibrahim, H.A.; Degasperi, A.; Dymott, J.A.; Hamilton, C.A.; Murphy, M.P.; Delles, C.; et al. Mitochondrial reactive oxygen species enhance AMP-activated protein kinase activation in the endothelium of patients with coronary artery disease and diabetes. Clin. Sci. 2012, 124, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Gioscia-Ryan, R.A.; Battson, M.L.; Cuevas, L.M.; Eng, J.S.; Murphy, M.P.; Seals, D.R. Mitochondria-targeted antioxidant therapy with MitoQ ameliorates aortic stiffening in old mice. J. Appl. Physiol. 2018, 124, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Junior, R.F.R.; Dabkowski, E.R.; Shekar, K.C.; O’connell, K.A.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef]
- Enns, G.M.; Kinsman, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Amagata, A.; Barnes, A.; Kheifets, V.; Shrader, W.D.; et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol. Genet. Metab. 2012, 105, 91–102. [Google Scholar] [CrossRef]
- Martinelli, D.; Catteruccia, M.; Piemonte, F.; Pastore, A.; Tozzi, G.; Dionisi-Vici, C.; Pontrelli, G.; Corsetti, T.; Livadiotti, S.; Kheifets, V.; et al. EPI-743 reverses the progression of the pediatric mitochondrial disease—Genetically defined Leigh Syndrome. Mol. Genet. Metab. 2012, 107, 383–388. [Google Scholar] [CrossRef]
- Pastore, A.; Petrillo, S.; Tozzi, G.; Carrozzo, R.; Martinelli, D.; Dionisi-Vici, C.; Di Giovamberardino, G.; Ceravolo, F.; Klein, M.B.; Miller, G.; et al. Glutathione: A redox signature in monitoring EPI-743 therapy in children with mitochondrial encephalomyopathies. Mol. Genet. Metab. 2013, 109, 208–214. [Google Scholar] [CrossRef]
- Kouga, T.; Takagi, M.; Miyauchi, A.; Shimbo, H.; Iai, M.; Yamashita, S.; Murayama, K.; Klein, M.B.; Miller, G.; Goto, T.; et al. Japanese Leigh syndrome case treated with EPI-743. Brain Dev. 2018, 40, 145–149. [Google Scholar] [CrossRef]
- Sadun, A.A.; Chicani, C.F.; Ross-Cisneros, F.N.; Barboni, P.; Thoolen, M.; Shrader, W.D.; Kubis, K.; Carelli, V.; Miller, G. Effect of EPI-743 on the Clinical Course of the Mitochondrial Disease Leber Hereditary Optic Neuropathy. Arch. Neurol. 2012, 69, 331–338. [Google Scholar] [CrossRef]
- Lynch, D.R.; Willi, S.M.; Wilson, R.B.; Cotticelli, M.G.; Brigatti, K.W.; Deutsch, E.C.; Kucheruk, O.; Shrader, W.; Rioux, P.; Miller, G.; et al. A0001 in Friedreich ataxia: Biochemical characterization and effects in a clinical trial. Mov. Disord. 2012, 27, 1026–1033. [Google Scholar] [CrossRef]
- Frantz, M.-C.; Skoda, E.M.; Sacher, J.R.; Epperly, M.W.; Goff, J.P.; Greenberger, J.S.; Wipf, P. Synthesis of analogs of the radiation mitigator JP4-039 and visualization of BODIPY derivatives in mitochondria. Org. Biomol. Chem. 2013, 11, 4147–4153. [Google Scholar] [CrossRef] [PubMed]
- Leipnitz, G.; Mohsen, A.-W.; Karunanidhi, A.; Seminotti, B.; Roginskaya, V.Y.; Markantone, D.M.; Grings, M.; Mihalik, S.J.; Wipf, P.; Van Houten, B.; et al. Evaluation of mitochondrial bioenergetics, dynamics, endoplasmic reticulum-mitochondria crosstalk, and reactive oxygen species in fibroblasts from patients with complex I deficiency. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Seminotti, B.; Leipnitz, G.; Karunanidhi, A.; Kochersperger, C.; Roginskaya, V.Y.; Basu, S.; Wang, Y.; Wipf, P.; Van Houten, B.; Mohsen, A.-W.; et al. Mitochondrial energetics is impaired in very long-chain acyl-CoA dehydrogenase deficiency and can be rescued by treatment with mitochondria-targeted electron scavengers. Hum. Mol. Genet. 2019, 28, 928–941. [Google Scholar] [CrossRef] [PubMed]
- Beyrath, J.; Pellegrini, M.; Renkema, H.; Houben, L.; Pecheritsyna, S.; Van Zandvoort, P.; Broek, P.V.D.; Bekel, A.; Eftekhari, P.; Smeitink, J.A.M. KH176 Safeguards Mitochondrial Diseased Cells from Redox Stress-Induced Cell Death by Interacting with the Thioredoxin System/Peroxiredoxin Enzyme Machinery. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- De Haas, R.; Das, D.; Garanto, A.; Renkema, H.G.; Greupink, R.; Broek, P.V.D.; Pertijs, J.; Collin, R.W.J.; Willems, P.; Beyrath, J.; et al. Therapeutic effects of the mitochondrial ROS-redox modulator KH176 in a mammalian model of Leigh Disease. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Frambach, S.J.; Van De Wal, M.A.; Broek, P.H.V.D.; Smeitink, J.A.; Russel, F.G.; De Haas, R.; Schirris, T.J.J. Effects of clofibrate and KH176 on life span and motor function in mitochondrial complex I-deficient mice. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165727. [Google Scholar] [CrossRef]
- Koene, S.; Spaans, E.; Van Bortel, L.M.; Van Lancker, G.; Delafontaine, B.; Badilini, F.; Beyrath, J.; Smeitink, J. KH176 under development for rare mitochondrial disease: A first in man randomized controlled clinical trial in healthy male volunteers. Orphanet J. Rare Dis. 2017, 12, 163. [Google Scholar] [CrossRef]
- Janssen, M.C.; Koene, S.; De Laat, P.; Hemelaar, P.; Pickkers, P.; Spaans, E.; Beukema, R.; Beyrath, J.; Groothuis, J.; Verhaak, C.; et al. The KHENERGY Study: Safety and Efficacy of KH 176 in Mitochondrial m.3243A>G Spectrum Disorders. Clin. Pharmacol. Ther. 2019, 105, 101–111. [Google Scholar] [CrossRef]
- Khondrion Receives Rare Pediatric Disease Designation for Sonlicromanol from US FDA. GlobeNewswire News Room. 28 September 2020. Available online: http://www.globenewswire.com/news-release/2020/09/28/2099659/0/en/Khondrion-Receives-Rare-Pediatric-Disease-Designation-for-Sonlicromanol-from-US-FDA.html (accessed on 6 October 2020).
- Antonenko, Y.N.; Avetisyan, A.V.; Bakeeva, L.E.; Chernyak, B.V.; Chertkov, V.A.; Domnina, L.V.; Ivanova, O.Y.; Izyumov, D.S.; Khailova, L.S.; Klishin, S.S.; et al. Mitochondria-targeted plastoquinone derivatives as tools to interrupt execution of the aging program. 1. Cationic plastoquinone derivatives: Synthesis and in vitro studies. Biochemistry 2008, 73, 1273–1287. [Google Scholar] [CrossRef]
- Severin, F.F.; Severina, I.I.; Antonenko, Y.N.; Rokitskaya, T.I.; Cherepanov, D.A.; Mokhova, E.N.; Vyssokikh, M.Y.; Pustovidko, A.V.; Markova, O.V.; Yaguzhinsky, L.S.; et al. Penetrating cation/fatty acid anion pair as a mitochondria-targeted protonophore. Proc. Natl. Acad. Sci. USA 2010, 107, 663–668. [Google Scholar] [CrossRef]
- Lyamzaev, K.G.; Panteleeva, A.A.; Karpukhina, A.A.; Galkin, I.I.; Popova, E.N.; Pletjushkina, O.Y.; Rieger, B.; Busch, K.B.; Mulkidjanian, A.Y.; Chernyak, B.V. Novel Fluorescent Mitochondria-Targeted Probe MitoCLox Reports Lipid Peroxidation in Response to Oxidative Stress In Vivo. Oxidative Med. Cell. Longev. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.A.; Ershov, N.I.; Kolosova, N.G. Suppression of Alzheimer’s Disease-Like Pathology Progression by Mitochondria-Targeted Antioxidant SkQ1: A Transcriptome Profiling Study. Oxidative Med. Cell. Longev. 2019, 2019, 3984906-17. [Google Scholar] [CrossRef] [PubMed]
- Kolosova, N.G.; Tyumentsev, M.A.; Muraleva, N.A.; Kiseleva, E.; Vitovtov, A.O.; Stefanova, N.A. Antioxidant SkQ1 Alleviates Signs of Alzheimer’s Disease-like Pathology in Old OXYS Rats by Reversing Mitochondrial Deterioration. Curr. Alzheimer Res. 2017, 14, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Fetisova, E.K.; Muntyan, M.S.; Lyamzaev, K.G.; Chernyak, B.V. Therapeutic Effect of the Mitochondria-Targeted Antioxidant SkQ1 on the Culture Model of Multiple Sclerosis. Oxidative Med. Cell. Longev. 2019, 2019, 1–10. [Google Scholar] [CrossRef]
- Veretinskaya, A.G.; Podshivalova, L.S.; Frolova, O.Y.; Belopolskaya, M.V.; Averina, O.A.; Kushnir, E.A.; Marmiy, N.V.; Lovat, M.L. Effects of mitochondrial antioxidant SkQ1 on biochemical and behavioral parameters in a Parkinsonism model in mice. Biochemistry 2017, 82, 1513–1520. [Google Scholar] [CrossRef]
- Shabalina, I.G.; Vyssokikh, M.Y.; Gibanova, N.; Csikasz, R.I.; Edgar, D.; Hallden-Waldemarson, A.; Rozhdestvenskaya, Z.; Bakeeva, L.E.; Vays, V.B.; Pustovidko, A.V.; et al. Improved health-span and lifespan in mtDNA mutator mice treated with the mitochondrially targeted antioxidant SkQ1. Aging 2017, 9, 315–339. [Google Scholar] [CrossRef]
- Edgar, D.; Trifunovic, A. The mtDNA mutator mouse: Dissecting mitochondrial involvement in aging. Aging 2009, 1, 1028–1032. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nat. Cell Biol. 2014, 505, 335–343. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; De Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, Á.; et al. Supercomplex Assembly Determines Electron Flux in the Mitochondrial Electron Transport Chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Pelloquin, L.; Belenguer, P.; Hamel, C.P. OPA1 (Kjer type) dominant optic atrophy: A novel mitochondrial disease. Mol. Genet. Metab. 2002, 75, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Delettre, C.; Lenaers, G.; Griffoin, J.-M.; Gigarel, N.; Lorenzo, C.; Belenguer, P.; Pelloquin, L.; Grosgeorge, J.; Turc-Carel, C.; Perret, E.; et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 2000, 26, 207–210. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.M.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.P.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Waterham, H.R.; Koster, J.; Van Roermund, C.W.T.; Mooyer, P.A.W.; Wanders, R.J.A.; Leonard, J.V. A Lethal Defect of Mitochondrial and Peroxisomal Fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef]
- Shamseldin, H.E.; Alshammari, M.; Al-Sheddi, T.; Salih, M.A.; Alkhalidi, H.; Kentab, A.; Repetto, G.M.; Hashem, M.; Alkuraya, F.S. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 2012, 49, 234–241. [Google Scholar] [CrossRef]
- Civiletto, G.; Varanita, T.; Cerutti, R.; Gorletta, T.; Barbaro, S.; Marchet, S.; Lamperti, C.; Viscomi, C.; Scorrano, L.; Zeviani, M. Opa1 Overexpression Ameliorates the Phenotype of Two Mitochondrial Disease Mouse Models. Cell Metab. 2015, 21, 845–854. [Google Scholar] [CrossRef]
- Zeviani, M.; Luna-Sanchez, M.; Scorrano, L.; Viscomi, C.; Calvo, G.B. Opa1 overexpression protects from early onset Mpv17-/--related mouse kidney disease. Mol. Ther. 2020. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Bonamy, G.M.C.; Meeusen, S.; Brusch, R.G.; Turk, C.; Yang, P.-Y.; Schultz, P.G. A Small Molecule Promotes Mitochondrial Fusion in Mammalian Cells. Angew. Chem. Int. Ed. 2012, 51, 9302–9305. [Google Scholar] [CrossRef]
- Qi, X.; Qvit, N.; Su, Y.-C.; Mochly-Rosen, D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Bartolák-Suki, E.; Imsirovic, J.; Nishibori, Y.; Krishnan, R.; Suki, B. Regulation of Mitochondrial Structure and Dynamics by the Cytoskeleton and Mechanical Factors. Int. J. Mol. Sci. 2017, 18, 1812. [Google Scholar] [CrossRef] [PubMed]
- Flatau, G.; Lemichez, E.; Gauthier, M.J.; Chardin, P.; Paris, S.; Fiorentini, C.; Boquet, P. Toxin-induced activation of the G protein p21 Rho by deamidation of glutamine. Nat. Cell Biol. 1997, 387, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, A.; Travaglione, S.; Maroccia, Z.; Guidotti, M.; Pierri, C.; Primiano, G.; Servidei, S.; Loizzo, S.; Fiorentini, C. The Bacterial Protein CNF1 as a Potential Therapeutic Strategy against Mitochondrial Diseases: A Pilot Study. Int. J. Mol. Sci. 2018, 19, 1825. [Google Scholar] [CrossRef] [PubMed]
- Karaa, A.; Haas, R.; Goldstein, A.; Vockley, J.; Weaver, W.D.; Cohen, B.H. Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology 2018, 90, e1212–e1221. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Ren, M. The role of cardiolipin in the structural organization of mitochondrial membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 2080–2083. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mileykovskaya, E.; Dowhan, W. Cardiolipin is essential for organization of complexes III and IV into a supercomplex in intact yeast mitochondria. J. Biol. Chem. 2005, 280, 29403–29408. [Google Scholar] [CrossRef]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-Targeted Antioxidants Protect Against Amyloid-β Toxicity in Alzheimer’s Disease Neurons. J. Alzheimers Dis. 2010, 20, S609–S631. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The Mitochondrial-Targeted Compound SS-31 Re-Energizes Ischemic Mitochondria by Interacting with Cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Dai, D.-F.; Hsieh, E.J.; Chen, T.; Menendez, L.G.; Basisty, N.B.; Tsai, L.; Beyer, R.P.; Crispin, D.A.; Shulman, N.J.; Szeto, H.H.; et al. Global Proteomics and Pathway Analysis of Pressure-Overload–Induced Heart Failure and Its Attenuation by Mitochondrial-Targeted Peptides. Circ. Heart Fail. 2013, 6, 1067–1076. [Google Scholar] [CrossRef]
- Neil, E.E.; Bisaccia, E.K. Nusinersen: A Novel Antisense Oligonucleotide for the Treatment of Spinal Muscular Atrophy. J. Pediatr. Pharmacol. Ther. 2019, 24, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Siegel, M.P.; Kruse, S.E.; Percival, J.M.; Goh, J.; White, C.C.; Hopkins, H.C.; Kavanagh, T.J.; Szeto, H.H.; Rabinovitch, P.S.; Marcinek, D.J. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 2013, 12, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Kloner, R.A.; Shi, J.; Dai, W. New therapies for reducing post-myocardial left ventricular remodeling. Ann. Transl. Med. 2015, 3. [Google Scholar] [CrossRef]
- Karaa, A.; Haas, R.; Goldstein, A.; Vockley, J.; Cohen, B.H. A randomized crossover trial of elamipretide in adults with primary mitochondrial myopathy. J. Cachex Sarcopenia Muscle 2020, 11, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.F.; Renna, M.; Ghislat, G.; Puri, C.; Ashkenazi, A.; Vicinanza, M.; Menzies, F.M.; Rubinsztein, D.C. Mammalian Autophagy: How Does It Work? Annu. Rev. Biochem. 2016, 85, 685–713. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.-B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR Inhibition Alleviates Mitochondrial Disease in a Mouse Model of Leigh Syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef]
- Civiletto, G.; Dogan, S.A.; Cerutti, R.; Fagiolari, G.; Moggio, M.; Lamperti, C.; Benincá, C.; Viscomi, C.; Zeviani, M. Rapamycin rescues mitochondrial myopathy via coordinated activation of autophagy and lysosomal biogenesis. EMBO Mol. Med. 2018, 10, e8799. [Google Scholar] [CrossRef]
- Wang, A.; Mouser, J.; Pitt, J.; Promislow, D.; Kaeberlein, M. Rapamycin enhances survival in a Drosophila model of mitochondrial disease. Oncotarget 2016, 7, 80131–80139. [Google Scholar] [CrossRef]
- Zheng, X.; Boyer, L.; Jin, M.; Kim, Y.; Fan, W.; Bardy, C.; Berggren, T.; Evans, R.M.; Gage, F.H.; Hunter, T. Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria-related neurodegeneration. eLife 2016, 5. [Google Scholar] [CrossRef]
- Peng, M.; Ostrovsky, J.; Kwon, Y.J.; Polyak, E.; Licata, J.; Tsukikawa, M.; Marty, E.; Thomas, J.; Felix, C.A.; Xiao, R.; et al. Inhibiting cytosolic translation and autophagy improves health in mitochondrial disease. Hum. Mol. Genet. 2015, 24, 4829–4847. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Martinez, F.; Bitto, A.; Gonzalez, B.; Tazaerslan, C.; Cohen, C.; Delaval, L.; Timsit, J.; Knebelmann, B.; Terzi, F.; et al. mTOR inhibitors may benefit kidney transplant recipients with mitochondrial diseases. Kidney Int. 2019, 95, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Sage-Schwaede, A.; Engelstad, K.; Salazar, R.; Curcio, A.; Khandji, A.; Jr, J.H.G.; De Vivo, D.C. Exploring mTOR inhibition as treatment for mitochondrial disease. Ann. Clin. Transl. Neurol. 2019, 6, 1877–1881. [Google Scholar] [CrossRef] [PubMed]
- Barriocanal-Casado, E.; Hidalgo-Gutiérrez, A.; Raimundo, N.; González-García, P.; Acuña-Castroviejo, D.; Escames, G.; López, L.C. Rapamycin administration is not a valid therapeutic strategy for every case of mitochondrial disease. EBioMedicine 2019, 42, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Opelz, G.; Unterrainer, C.; Süsal, C.; Döhler, B. Immunosuppression with mammalian target of rapamycin inhibitor and incidence of post-transplant cancer in kidney transplant recipients. Nephrol. Dial. Transplant. 2016, 31, 1360–1367. [Google Scholar] [CrossRef]
- Seo, B.B.; Matsuno-Yagi, A.; Yagi, T. Modulation of oxidative phosphorylation of human kidney 293 cells by transfection with the internal rotenone-insensitive NADH-quinone oxidoreductase (NDI1) gene of Saccharomyces cerevisiae. Biochim. Biophys. Acta (BBA) Bioenerg. 1999, 1412, 56–65. [Google Scholar] [CrossRef]
- Rasmusson, A.G.; Soole, K.L.; Elthon, T.E. ALTERNATIVE NAD(P)H DEHYDROGENASES OF PLANT MITOCHONDRIA. Annu. Rev. Plant Biol. 2004, 55, 23–39. [Google Scholar] [CrossRef]
- Sanz, A.; Soikkeli, M.; Portero-Otín, M.; Wilson, A.; Kemppainen, E.; McIlroy, G.; Ellilä, S.; Kemppainen, K.K.; Tuomela, T.; Lakanmaa, M.; et al. Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc. Natl. Acad. Sci. USA 2010, 107, 9105–9110. [Google Scholar] [CrossRef]
- Perales-Clemente, E.; Bayona-Bafaluy, M.P.; Pérez-Martos, A.; Barrientos, A.; Fernández-Silva, P.; Enriquez, J.A. Restoration of electron transport without proton pumping in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 18735–18739. [Google Scholar] [CrossRef]
- Dassa, E.P.; Dufour, E.; Goncalves, S.; Paupe, V.; Hakkaart, G.A.J.; Jacobs, H.T.; Rustin, P. Expression of the alternative oxidase complements cytochrome c oxidase deficiency in human cells. EMBO Mol. Med. 2009, 1, 30–36. [Google Scholar] [CrossRef]
- Fernandez-Ayala, D.J.; Sanz, A.; Vartiainen, S.; Kemppainen, K.K.; Babusiak, M.; Mustalahti, E.; Costa, R.; Tuomela, T.; Zeviani, M.; Chung, J.; et al. Expression of the Ciona intestinalis Alternative Oxidase (AOX) in Drosophila Complements Defects in Mitochondrial Oxidative Phosphorylation. Cell Metab. 2009, 9, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Chadderton, N.; Palfi, A.; Millington-Ward, S.; Gobbo, O.; Overlack, N.; Carrigan, M.; O’Reilly, M.; Campbell, M.T.; Ehrhardt, C.; Wolfrum, U.; et al. Intravitreal delivery of AAV-NDI1 provides functional benefit in a murine model of Leber hereditary optic neuropathy. Eur. J. Hum. Genet. 2013, 21, 62–68. [Google Scholar] [CrossRef] [PubMed]
- McElroy, G.S.; Reczek, C.R.; Reyfman, P.A.; Mithal, D.S.; Horbinski, C.M.; Chandel, N.S. NAD+ Regeneration Rescues Lifespan, but Not Ataxia, in a Mouse Model of Brain Mitochondrial Complex I Dysfunction. Cell Metab. 2020, 32, 301–308.e6. [Google Scholar] [CrossRef] [PubMed]
- Rapola, J.; Heikkilä, P.; Fellman, V. Pathology of lethal fetal growth retardation syndrome with aminoaciduria, iron overload, and lactic acidosis (GRACILE). Pediatr. Pathol. Mol. Med. 2002, 21, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Fellman, V.; Rapola, J.; Pihko, H.; Varilo, T.; Raivio, K.O. Iron-overload disease in infants involving fetal growth retardation, lactic acidosis, liver haemosiderosis, and aminoaciduria. Lancet 1998, 351, 490–493. [Google Scholar] [CrossRef]
- Dogan, S.A.; Cerutti, R.; Benincá, C.; Brea-Calvo, G.; Jacobs, H.T.; Zeviani, M.; Szibor, M.; Viscomi, C. Perturbed Redox Signaling Exacerbates a Mitochondrial Myopathy. Cell Metab. 2018, 28, 764–775.e5. [Google Scholar] [CrossRef]
- Catania, A.; Iuso, A.; Bouchereau, J.; Kremer, L.S.; Paviolo, M.; Terrile, C.; Bénit, P.; Rasmusson, A.G.; Schwarzmayr, T.; Tiranti, V.; et al. Arabidopsis thaliana alternative dehydrogenases: A potential therapy for mitochondrial complex I deficiency? Perspectives and pitfalls. Orphanet J. Rare Dis. 2019, 14, 236. [Google Scholar] [CrossRef]
- Houshmand, M.; Holme, E.; Oldfors, A.; Holmberg, E. De novo mutation in the mitochondrial ATP synthase subunit 6 gene (T8993G) with rapid segregation resulting in Leigh syndrome in the offspring. Qual. Life Res. 1995, 96, 290–294. [Google Scholar] [CrossRef]
- Carling, P.J.; Cree, L.M.; Chinnery, P.F. The implications of mitochondrial DNA copy number regulation during embryogenesis. Mitochondrion 2011, 11, 686–692. [Google Scholar] [CrossRef]
- Wei, W.; Chinnery, P.F. Inheritance of mitochondrial DNA in humans: Implications for rare and common diseases. J. Intern. Med. 2020, 287, 634–644. [Google Scholar] [CrossRef]
- Rai, P.K.; Craven, L.; Hoogewijs, K.; Russell, O.M.; Chrzanowska-Lightowlers, Z. Advances in methods for reducing mitochondrial DNA disease by replacing or manipulating the mitochondrial genome. Essays Biochem. 2018, 62, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Sallevelt, S.C.E.H.; Dreesen, J.C.F.M.; Coonen, E.; Paulussen, A.D.C.; EI Hellebrekers, D.M.; Die-Smulders, C.E.M.D.; Smeets, H.J.M.; Lindsey, P. Preimplantation genetic diagnosis for mitochondrial DNA mutations: Analysis of one blastomere suffices. J. Med. Genet. 2017, 54, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Treff, N.R.; Campos, J.; Tao, X.; Levy, B.; Ferry, K.M.; Scott, R.T. Blastocyst preimplantation genetic diagnosis (PGD) of a mitochondrial DNA disorder. Fertil. Steril. 2012, 98, 1236–1240. [Google Scholar] [CrossRef]
- Craven, L.; Tuppen, H.A.; Greggains, G.D.; Harbottle, S.J.; Murphy, J.L.; Cree, L.M.; Murdoch, A.P.; Chinnery, P.F.; Taylor, R.W.; Lightowlers, R.N.; et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nat. Cell Biol. 2010, 465, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.; Turnbull, D. Progress in mitochondrial replacement therapies. Nat. Rev. Mol. Cell Biol. 2018, 19, 71–72. [Google Scholar] [CrossRef]
- Craven, L.; Tang, M.-X.; Gorman, G.S.; De Sutter, P.; Heindryckx, B. Novel reproductive technologies to prevent mitochondrial disease. Hum. Reprod. Update 2017, 23, 501–519. [Google Scholar] [CrossRef]
- Ishii, T.; Hibino, Y. Mitochondrial manipulation in fertility clinics: Regulation and responsibility. Reprod. Biomed. Soc. Online 2018, 5, 93–109. [Google Scholar] [CrossRef]
- Tachibana, M.; Sparman, M.; Sritanaudomchai, H.; Ma, H.; Clepper, L.; Woodward, J.; Li, Y.; Ramsey, C.; Kolotushkina, O.; Mitalipov, S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nat. Cell Biol. 2009, 461, 367–372. [Google Scholar] [CrossRef]
- Kang, E.; Wu, J.; Gutierrez, N.M.; Koski, A.; Tippner-Hedges, R.; Agaronyan, K.; Platero-Luengo, A.; Martinez-Redondo, P.; Ma, H.; Lee, Y.; et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nat. Cell Biol. 2016, 540, 270–275. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, H.; Luo, S.; Chavez-Badiola, A.; Liu, Z.; Yang, M.; Munne, S.; Konstantinidis, M.; Wells, D.; Huang, T. First live birth using human oocytes reconstituted by spindle nuclear transfer for mitochondrial DNA mutation causing Leigh syndrome. Fertil. Steril. 2016, 106, e375–e376. [Google Scholar] [CrossRef]
- Hudson, G.; Takeda, Y.; Herbert, M. Reversion after replacement of mitochondrial DNA. Nature 2019, 574. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, H.; Luo, S.; Lu, Z.; Chávez-Badiola, A.; Liu, Z.; Yang, M.; Merhi, Z.; Silber, S.J.; Munné, S.; et al. Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod. Biomed. Online 2017, 34, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Kono, T.; Nakada, K.; Ishikawa, K.; Inoue, S.-I.; Yonekawa, H.; Hayashi, J.-I. Gene therapy for progeny of mito-mice carrying pathogenic mtDNA by nuclear transplantation. Proc. Natl. Acad. Sci. USA 2005, 102, 16765–16770. [Google Scholar] [CrossRef] [PubMed]
- Hyslop, L.A.; Blakeley, P.; Craven, L.; Richardson, J.; Fogarty, N.M.E.; Fragouli, E.; Lamb, M.; Wamaitha, S.E.; Prathalingam, N.; Zhang, Q.; et al. Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nat. Cell Biol. 2016, 534, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Yasuzaki, Y.; Yamada, Y.; Harashima, H. Mitochondrial matrix delivery using MITO-Porter, a liposome-based carrier that specifies fusion with mitochondrial membranes. Biochem. Biophys. Res. Commun. 2010, 397, 181–186. [Google Scholar] [CrossRef]
- Yamada, Y.; Harashima, H. Targeting the Mitochondrial Genome via a Dual Function MITO-Porter: Evaluation of mtDNA Levels and Mitochondrial Function. Methods Mol. Biol. 2015, 1265, 123–133. [Google Scholar] [CrossRef]
- Lyrawati, D.; Trounson, A.; Cram, D. Expression of GFP in the Mitochondrial Compartment Using DQAsome-Mediated Delivery of an Artificial Mini-mitochondrial Genome. Pharm. Res. 2011, 28, 2848–2862. [Google Scholar] [CrossRef]
- Mahata, B.; Mukherjee, S.; Mishra, S.; Bandyopadhyay, A.; Adhya, S. Functional Delivery of a Cytosolic tRNA into Mutant Mitochondria of Human Cells. Science 2006, 314, 471–474. [Google Scholar] [CrossRef]
- Kawamura, E.; Maruyama, M.; Abe, J.; Sudo, A.; Takeda, A.; Takada, S.; Yokota, T.; Kinugawa, S.; Harashima, H.; Yamada, Y. Validation of Gene Therapy for Mutant Mitochondria by Delivering Mitochondrial RNA Using a MITO-Porter. Mol. Ther. Nucleic Acids 2020, 20, 687–698. [Google Scholar] [CrossRef]
- Yamada, Y.; Somiya, K.; Miyauchi, A.; Osaka, H.; Harashima, H. Validation of a mitochondrial RNA therapeutic strategy using fibroblasts from a Leigh syndrome patient with a mutation in the mitochondrial ND3 gene. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Weissig, V. DQAsomes as the Prototype of Mitochondria-Targeted Pharmaceutical Nanocarriers: Preparation, Characterization, and Use. Methods Mol. Biol. 2015, 1265, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Adhya, S. Leishmania mitochondrial tRNA importers. Int. J. Biochem. Cell Biol. 2008, 40, 2681–2685. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Borgeld, H.-J.; Zhang, J.; Muramatsu, S.-I.; Gong, J.-S.; Yoneda, M.; Maruyama, W.; Naoi, M.; Ibi, T.; Sahashi, K.; et al. Gene Therapy for Mitochondrial Disease by Delivering Restriction Endonuclease SmaI into Mitochondria. J. Biomed. Sci. 2002, 9, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Comte, C.; Tonin, Y.; Heckel-Mager, A.-M.; Boucheham, A.; Smirnov, A.; Auré, K.; Lombès, A.; Martin, R.P.; Entelis, N.; Tarassov, I. Mitochondrial targeting of recombinant RNAs modulates the level of a heteroplasmic mutation in human mitochondrial DNA associated with Kearns Sayre Syndrome. Nucleic Acids Res. 2013, 41, 418–433. [Google Scholar] [CrossRef]
- Jo, A.; Ham, S.; Lee, G.H.; Lee, Y.-S.; Kim, S.; Shin, J.-H.; Lee, Y. Efficient Mitochondrial Genome Editing by CRISPR/Cas9. BioMed Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Moretton, A.; Morel, F.; Macao, B.; Lachaume, P.; Ishak, L.; Lefebvre, M.; Garreau-Balandier, I.; Vernet, P.; Falkenberg, M.; Farge, G. Selective mitochondrial DNA degradation following double-strand breaks. PLoS ONE 2017, 12, e0176795. [Google Scholar] [CrossRef]
- Hussain, S.-R.A.; Yalvac, M.E.; Khoo, B.; Eckardt, S.; McLaughlin, K.J. Adapting CRISPR/Cas9 System for Targeting Mitochondrial Genome. BioRxiv 2020. [Google Scholar] [CrossRef]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial Genome Engineering: The Revolution May Not Be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef]
- Kauppila, J.H.; Baines, H.L.; Bratic, A.; Simard, M.-L.; Freyer, C.; Mourier, A.; Stamp, C.; Filograna, R.; Larsson, N.-G.; Greaves, L.C.; et al. A Phenotype-Driven Approach to Generate Mouse Models with Pathogenic mtDNA Mutations Causing Mitochondrial Disease. Cell Rep. 2016, 16, 2980–2990. [Google Scholar] [CrossRef]
- Gammage, P.A.; Viscomi, C.; Simard, M.-L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef]
- Bacman, S.R.; Kauppila, J.H.K.; Pereira, C.V.; Nissanka, N.; Miranda, M.; Pinto, M.; Williams, S.L.; Larsson, N.-G.; Stewart, J.B.; Moraes, C.T. MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 2018, 24, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.Y.; De Moraes, M.H.; Zeng, J.; Yeh, M.M.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nat. Cell Biol. 2020, 583, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, G.; Fu, J.; Ojaimi, J.; Sadlock, J.E.; Kwong, J.Q.; Guy, J.; Schon, E.A. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat. Genet. 2002, 30, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Ellouze, S.; Augustin, S.; Bouaita, A.; Bonnet, C.; Simonutti, M.; Forster, V.; Picaud, S.; Sahel, J.-A.; Corral-Debrinski, M. Optimized Allotopic Expression of the Human Mitochondrial ND4 Prevents Blindness in a Rat Model of Mitochondrial Dysfunction. Am. J. Hum. Genet. 2008, 83, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Feuer, W.J.; Davis, J.L.; Porciatti, V.; Gonzalez, P.J.; Koilkonda, R.D.; Yuan, H.; Hauswirth, W.W.; Lam, B.L. Gene Therapy for Leber Hereditary Optic Neuropathy. Ophthalmology 2017, 124, 1621–1634. [Google Scholar] [CrossRef]
- Wan, X.; Pei, H.; Zhao, M.-J.; Yang, S.; Hu, W.-K.; He, H.; Ma, S.-Q.; Zhang, G.; Dong, X.-Y.; Chen, C.; et al. Efficacy and Safety of rAAV2-ND4 Treatment for Leber’s Hereditary Optic Neuropathy. Sci. Rep. 2016, 6, 21587. [Google Scholar] [CrossRef]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; Wani, M.R.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef]
- Yosef, O.B.; Jacoby, E.; Gruber, N.; Varda-Bloom, N.; Azaria, E.; Eisenstein, E.; Barak, S.; Ahonniska-Assa, J.; Anikster, Y.; Toren, A. Promising Results for Kearns-Sayre Syndrome of First in Man Treatment by Mitochondrial Augmentation Therapy (457). Neurology 2020, 94, 457. [Google Scholar]
- Mingozzi, F.; High, K.A. Therapeutic in vivo gene transfer for genetic disease using AAV: Progress and challenges. Nat. Rev. Genet. 2011, 12, 341–355. [Google Scholar] [CrossRef]
- Gao, G.-P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef] [PubMed]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.-M.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nat. Cell Biol. 2003, 426, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Gropp, T.; Brustovetsky, N.; Klingenberg, M.; Müller, V.; Fendler, K.; Bamberg, E. Kinetics of electrogenic transport by the ADP/ATP carrier. Biophys. J. 1999, 77, 714–726. [Google Scholar] [CrossRef]
- Walter, M.C.; Czermin, B.; Müller-Ziermann, S.; Bulst, S.; Stewart, J.D.; Hudson, G.; Schneiderat, P.; Abicht, A.; Holinski-Feder, E.; Lochmuller, H.; et al. Late-onset ptosis and myopathy in a patient with a heterozygous insertion in POLG2. J. Neurol. 2010, 257, 1517–1523. [Google Scholar] [CrossRef]
- Flierl, A.; Chen, Y.; Coskun, P.E.; Samulski, R.J.; Wallace, D.C. Adeno-associated virus-mediated gene transfer of the heart/muscle adenine nucleotide translocator (ANT) in mouse. Gene Ther. 2005, 12, 570–578. [Google Scholar] [CrossRef]
- Di Meo, I.; Marchet, S.; Lamperti, C.; Zeviani, M.; Viscomi, C. AAV9-based gene therapy partially ameliorates the clinical phenotype of a mouse model of Leigh syndrome. Gene Ther. 2017, 24, 661–667. [Google Scholar] [CrossRef]
- Parikh, S.; Karaa, A.; Goldstein, A.; Ng, Y.S.; Gorman, G.; Feigenbaum, A.; Christodoulou, J.; Haas, R.; Tarnopolsky, M.; Cohen, B.K.; et al. Solid organ transplantation in primary mitochondrial disease: Proceed with caution. Mol. Genet. Metab. 2016, 118, 178–184. [Google Scholar] [CrossRef]
- Spinazzola, A.; Viscomi, C.; Fernandez-Vizarra, E.; Carrara, F.; D’Adamo, P.; Calvo, S.E.; Marsano, R.M.; Donnini, C.; Weiher, H.; Strisciuglio, P.; et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 2006, 38, 570–575. [Google Scholar] [CrossRef]
- Shimura, M.; Kuranobu, N.; Ogawa-Tominaga, M.; Akiyama, N.; Sugiyama, Y.; Ebihara, T.; Fushimi, T.; Ichimoto, K.; Matsunaga, A.; Tsuruoka, T.; et al. Clinical and molecular basis of hepatocerebral mitochondrial DNA depletion syndrome in Japan: Evaluation of outcomes after liver transplantation. Orphanet J. Rare Dis. 2020, 15, 1–9. [Google Scholar] [CrossRef]
- Hassan, S.; Mahmoud, A.; Mohammed, T.O.; Mohammad, S. Pediatric liver transplantation from a living donor in mitochondrial disease: Good outcomes in DGUOK deficiency? Pediatr. Transplant. 2020, 24, 13714. [Google Scholar] [CrossRef]
- Tiranti, V.; Briem, E.; Lamantea, E.; Mineri, R.; Papaleo, E.; De Gioia, L.; Forlani, F.; Rinaldo, P.; Dickson, P.; Abu-Libdeh, B.; et al. ETHE1 mutations are specific to ethylmalonic encephalopathy. J. Med. Genet. 2005, 43, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Viscomi, C.; Bottani, E.; Zeviani, M. Emerging concepts in the therapy of mitochondrial disease. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1847, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, I.; Auricchio, A.; Lamperti, C.; Burlina, A.; Viscomi, C.; Zeviani, M. Effective AAV-mediated gene therapy in a mouse model of ethylmalonic encephalopathy. EMBO Mol. Med. 2012, 4, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Torres-Torronteras, J.; Viscomi, C.; Cabrera-Pérez, R.; Cámara, Y.; Di Meo, I.; Barquinero, J.; Auricchio, A.; Pizzorno, G.; Hirano, M.; Zeviani, M.; et al. Gene Therapy Using a Liver-targeted AAV Vector Restores Nucleoside and Nucleotide Homeostasis in a Murine Model of MNGIE. Mol. Ther. 2014, 22, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.-L.; Sánchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; Deverman, B.E.; et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Nishino, I. MNGIE (mitochondrial neurogastrointestinal encephalomyopathy). Ryoikibetsu Shokogun Shirizu 2001, 47, 792–800. [Google Scholar]
- Hirano, M.; Garone, C.; Quinzii, C.M. CoQ10 deficiencies and MNGIE: Two treatable mitochondrial disorders. Biochim. Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 625–631. [Google Scholar] [CrossRef]
- Spinazzola, A.; Marti, R.; Nishino, I.; Andreu, A.L.; Naini, A.; Tadesse, S.; Pela, I.; Zammarchi, E.; Donati, M.A.; Oliver, J.A.; et al. Altered Thymidine Metabolism Due to Defects of Thymidine Phosphorylase. J. Biol. Chem. 2001, 277, 4128–4133. [Google Scholar] [CrossRef]
- De Giorgio, R.; Pironi, L.; Rinaldi, R.; Boschetti, E.; Caporali, L.; Capristo, M.; Casali, C.; Cenacchi, G.; Contin, M.; D’Angelo, R.; et al. Liver transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Ann. Neurol. 2016, 80, 448–455. [Google Scholar] [CrossRef]
- Dionisi-Vici, C.; Diodato, D.; Torre, G.; Picca, S.; Pariante, R.; Picardo, S.G.; Di Meo, I.; Rizzo, C.; Tiranti, V.; Zeviani, M.; et al. Liver transplant in ethylmalonic encephalopathy: A new treatment for an otherwise fatal disease. Brain 2016, 139, 1045–1051. [Google Scholar] [CrossRef]
- Tam, A.; Aldhaheri, N.S.; Mysore, K.; Tessier, M.E.; Goss, J.; Fernandez, L.A.; D’Alessandro, A.M.; Schwoerer, J.S.; Rice, G.M.; Elsea, S.H.; et al. Improved clinical outcome following liver transplant in patients with ethylmalonic encephalopathy. Am. J. Med. Genet. Part A 2019, 179, 1015–1019. [Google Scholar] [CrossRef]
- Grabhorn, E.; Tsiakas, K.; Herden, U.; Fischer, L.; Freisinger, P.; Marquardt, T.; Ganschow, R.; Briem-Richter, A.; Santer, R. Long-term outcomes after liver transplantation for deoxyguanosine kinase deficiency: A single-center experience and a review of the literature. Liver Transplant. 2014, 20, 464–472. [Google Scholar] [CrossRef]
- Lindvall, O.; Kokaia, Z.; Martinez-Serrano, A. Stem cell therapy for human neurodegenerative disorders—how to make it work. Nat. Med. 2004, 10, S42–S50. [Google Scholar] [CrossRef]
- Hirano, M.; Martí, R.; Casali, C.; Tadesse, S.; Uldrick, T.; Fine, B.; Escolar, D.M.; Valentino, M.L.; Nishino, I.; Hesdorffer, C.; et al. Allogeneic stem cell transplantation corrects biochemical derangements in MNGIE. Neurology 2006, 67, 1458–1460. [Google Scholar] [CrossRef]
- Filosto, M.; Scarpelli, M.; Tonin, P.; Lucchini, G.; Pavan, F.; Santus, F.; Parini, R.; Donati, M.A.; Cotelli, M.S.; Vielmi, V.; et al. Course and management of allogeneic stem cell transplantation in patients with mitochondrial neurogastrointestinal encephalomyopathy. J. Neurol. 2012, 259, 2699–2706. [Google Scholar] [CrossRef]
- Garone, C.; Viscomi, C. Towards a therapy for mitochondrial disease: An update. Biochem. Soc. Trans. 2018, 46, 1247–1261. [Google Scholar] [CrossRef]
- Wang, L.; Saada, A.; Eriksson, S. Kinetic Properties of Mutant Human Thymidine Kinase 2 Suggest a Mechanism for Mitochondrial DNA Depletion Myopathy. J. Biol. Chem. 2002, 278, 6963–6968. [Google Scholar] [CrossRef]
- Taanman, J.-W.; Muddle, J.R.; Muntau, A.C. Mitochondrial DNA depletion can be prevented by dGMP and dAMP supplementation in a resting culture of deoxyguanosine kinase-deficient fibroblasts. Hum. Mol. Genet. 2003, 12, 1839–1845. [Google Scholar] [CrossRef]
- Rampazzo, C.; Miazzi, C.; Franzolin, E.; Pontarin, G.; Ferraro, P.; Frangini, M.; Reichard, P.; Bianchi, V. Regulation by degradation, a cellular defense against deoxyribonucleotide pool imbalances. Mutat. Res. Toxicol. Environ. Mutagen. 2010, 703, 2–10. [Google Scholar] [CrossRef]
- Garone, C.; García-Díaz, B.; Emmanuele, V.; López, L.C.; Tadesse, S.; Akman, H.O.; Tanji, K.; Quinzii, C.M.; Hirano, M. Deoxypyrimidine monophosphate bypass therapy for thymidine kinase 2 deficiency. EMBO Mol. Med. 2014, 6, 1016–1027. [Google Scholar] [CrossRef]
- Lopez-Gomez, C.; Levy, R.J.; Sanchez-Quintero, M.J.; Juanola-Falgarona, M.; Barca, E.; Garcia-Diaz, B.; Tadesse, S.; Garone, C.; Hirano, M. Deoxycytidine and Deoxythymidine Treatment for Thymidine Kinase 2 Deficiency. Ann. Neurol. 2017, 81, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Bulst, S.; Abicht, A.; Holinski-Feder, E.; Müller-Ziermann, S.; Koehler, U.; Thirion, C.; Walter, M.C.; Stewart, J.D.; Chinnery, P.F.; Lochmuller, H.; et al. In vitro supplementation with dAMP/dGMP leads to partial restoration of mtDNA levels in mitochondrial depletion syndromes. Hum. Mol. Genet. 2009, 18, 1590–1599. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Munro, B.; Horvath, R.; Müller, J.S. Nucleoside supplementation modulates mitochondrial DNA copy number in the dguok -/- zebrafish. Hum. Mol. Genet. 2019, 28, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-González, C.; Madruga-Garrido, M.; Mavillard, F.; Garone, C.; Aguirre-Rodríguez, F.J.; Donati, M.A.; Kleinsteuber, K.; Martí, I.; Martín-Hernández, E.; Morealejo-Aycinena, J.P.; et al. Deoxynucleoside Therapy for Thymidine Kinase 2–Deficient Myopathy. Ann. Neurol. 2019, 86, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Carrabba, G.; Macchini, F.; Fabietti, I.; Schisano, L.; Meccariello, G.; Campanella, R.; Bertani, G.; Locatelli, M.; Boito, S.; Porro, G.A.; et al. Minimally invasive fetal surgery for myelomeningocele: Preliminary report from a single center. Neurosurg. Focus 2019, 47, E12. [Google Scholar] [CrossRef] [PubMed]
- Lindenburg, I.T.M.; Van Kamp, I.L.; Oepkes, D. Intrauterine Blood Transfusion: Current Indications and Associated Risks. Fetal Diagn. Ther. 2014, 36, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Rashnonejad, A.; Chermahini, G.A.; Gündüz, C.; Onay, H.; Aykut, A.; Durmaz, B.; Baka, M.; Su, Q.; Gao, G.; Özkınay, F. Fetal Gene Therapy Using a Single Injection of Recombinant AAV9 Rescued SMA Phenotype in Mice. Mol. Ther. 2019, 27, 2123–2133. [Google Scholar] [CrossRef]
- Peranteau, W.H.; Flake, A.W. The Future of In Utero Gene Therapy. Mol. Diagn. Ther. 2020, 24, 135–142. [Google Scholar] [CrossRef]
- Visapää, I.; Fellman, V.; Vesa, J.; Dasvarma, A.; Hutton, J.L.; Kumar, V.; Payne, G.S.; Makarow, M.; Van Coster, R.; Taylor, R.W.; et al. GRACILE Syndrome, a Lethal Metabolic Disorder with Iron Overload, Is Caused by a Point Mutation in BCS1L. Am. J. Hum. Genet. 2002, 71, 863–876. [Google Scholar] [CrossRef]
- Inak, G.; Rybak-Wolf, A.; Lisowski, P.; Juettner, R.; Zink, A.; Mlody, B.; Glazar, P.; Secker, C.; Ciptasari, U.H.; Stenzel, W.; et al. SURF1 mutations causative of Leigh syndrome impair human neurogenesis. bioRxiv 2019, 551390. [Google Scholar] [CrossRef]
- Quadalti, C.; Brunetti, D.; Lagutina, I.; Duchi, R.; Perota, A.; Lazzari, G.; Cerutti, R.; Di Meo, I.; Johnson, M.; Bottani, E.; et al. SURF1 knockout cloned pigs: Early onset of a severe lethal phenotype. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Romero-Morales, A.I.; Rastogi, A.; Temuri, H.; Rasmussen, M.L.; McElroy, G.S.; Hsu, L.; Almonacid, P.M.; Millis, B.A.; Chandel, N.S.; Cartailler, J.P.; et al. Human iPSC-derived cerebral organoids model features of Leigh Syndrome and reveal abnormal corticogenesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Massaro, G.; Mattar, C.N.Z.; Wong, A.M.S.; Sirka, E.; Buckley, S.M.K.; Herbert, B.R.; Karlsson, S.; Perocheau, D.P.; Burke, D.; Heales, S.; et al. Fetal gene therapy for neurodegenerative disease of infants. Nat. Med. 2018, 24, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, T.C. Future AAVenues for In Utero Gene Therapy. Cell Stem Cell 2018, 23, 320–321. [Google Scholar] [CrossRef]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Mandel, G.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife 2016, 5, 13374. [Google Scholar] [CrossRef]
- Bifari, F.; Dolci, S.; Bottani, E.; Pino, A.; Di Chio, M.; Zorzin, S.; Ragni, M.; Zamfir, R.G.; Brunetti, D.; Bardelli, D.; et al. Complete neural stem cell (NSC) neuronal differentiation requires a branched chain amino acids-induced persistent metabolic shift towards energy metabolism. Pharmacol. Res. 2020, 158, 104863. [Google Scholar] [CrossRef]
- Brunetti, D.; Bottani, E.; Segala, A.; Marchet, S.; Rossi, F.; Orlando, F.; Malavolta, M.; Carruba, M.O.; Lamperti, C.; Provinciali, M.; et al. Targeting Multiple Mitochondrial Processes by a Metabolic Modulator Prevents Sarcopenia and Cognitive Decline in SAMP8 Mice. Front. Pharmacol. 2020, 11, 1171. [Google Scholar] [CrossRef]
- Langer, Y.; Aran, A.; Gulsuner, S.; Abu Libdeh, B.; Renbaum, P.; Brunetti, D.; Teixeira, P.-F.; Walsh, T.; Zeligson, S.; Ruotolo, R.; et al. Mitochondrial PITRM1 peptidase loss-of-function in childhood cerebellar atrophy. J. Med. Genet. 2018, 55, 599–606. [Google Scholar] [CrossRef]
- Pérez, M.J.; Ivanyuk, D.; Panagiotakopoulou, V.; Di Napoli, G.; Kalb, S.; Brunetti, D.; Al-Shaana, R.; Kaeser, S.A.; Fraschka, S.A.-K.; Jucker, M.; et al. Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer’s disease-like pathology in human cerebral organoids. Mol. Psychiatry 2020, 1–18. [Google Scholar] [CrossRef]
- Motori, E.; Atanassov, I.; Kochan, S.M.V.; Folz-Donahue, K.; Sakthivelu, V.; Giavalisco, P.; Toni, N.; Puyal, J.; Larsson, N.-G. Neuronal metabolic rewiring promotes resilience to neurodegeneration caused by mitochondrial dysfunction. Sci. Adv. 2020, 6, eaba8271. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bottani, E.; Lamperti, C.; Prigione, A.; Tiranti, V.; Persico, N.; Brunetti, D. Therapeutic Approaches to Treat Mitochondrial Diseases: “One-Size-Fits-All” and “Precision Medicine” Strategies. Pharmaceutics 2020, 12, 1083. https://doi.org/10.3390/pharmaceutics12111083
Bottani E, Lamperti C, Prigione A, Tiranti V, Persico N, Brunetti D. Therapeutic Approaches to Treat Mitochondrial Diseases: “One-Size-Fits-All” and “Precision Medicine” Strategies. Pharmaceutics. 2020; 12(11):1083. https://doi.org/10.3390/pharmaceutics12111083
Chicago/Turabian StyleBottani, Emanuela, Costanza Lamperti, Alessandro Prigione, Valeria Tiranti, Nicola Persico, and Dario Brunetti. 2020. "Therapeutic Approaches to Treat Mitochondrial Diseases: “One-Size-Fits-All” and “Precision Medicine” Strategies" Pharmaceutics 12, no. 11: 1083. https://doi.org/10.3390/pharmaceutics12111083
APA StyleBottani, E., Lamperti, C., Prigione, A., Tiranti, V., Persico, N., & Brunetti, D. (2020). Therapeutic Approaches to Treat Mitochondrial Diseases: “One-Size-Fits-All” and “Precision Medicine” Strategies. Pharmaceutics, 12(11), 1083. https://doi.org/10.3390/pharmaceutics12111083