Liquid and Solid Self-Emulsifying Drug Delivery Systems (SEDDs) as Carriers for the Oral Delivery of Azithromycin: Optimization, In Vitro Characterization and Stability Assessment

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. HPLC Analysis
2.2.2. Preparation and Characterization of Blank Liquid SEDDs
- Excipients selection and screening
- Solubility study
- Emulsification studies
- Pseudo-ternary diagram construction
2.2.3. Preparation and Characterization of AZM-Loaded L-SEDDs
2.2.4. Preparation and Characterization of AZM-Loaded S-SEDDs
2.2.5. Transmission Electron Microscope (TEM)
2.2.6. Cell Culture Studies
- Cell stock preparation
- Cytotoxicity assay
- Transepithelial electrical resistance evaluation
2.2.7. The In Vitro AZM Release Studies
2.2.8. Stability Studies
2.2.9. Statistical Analysis
3. Results and Discussion
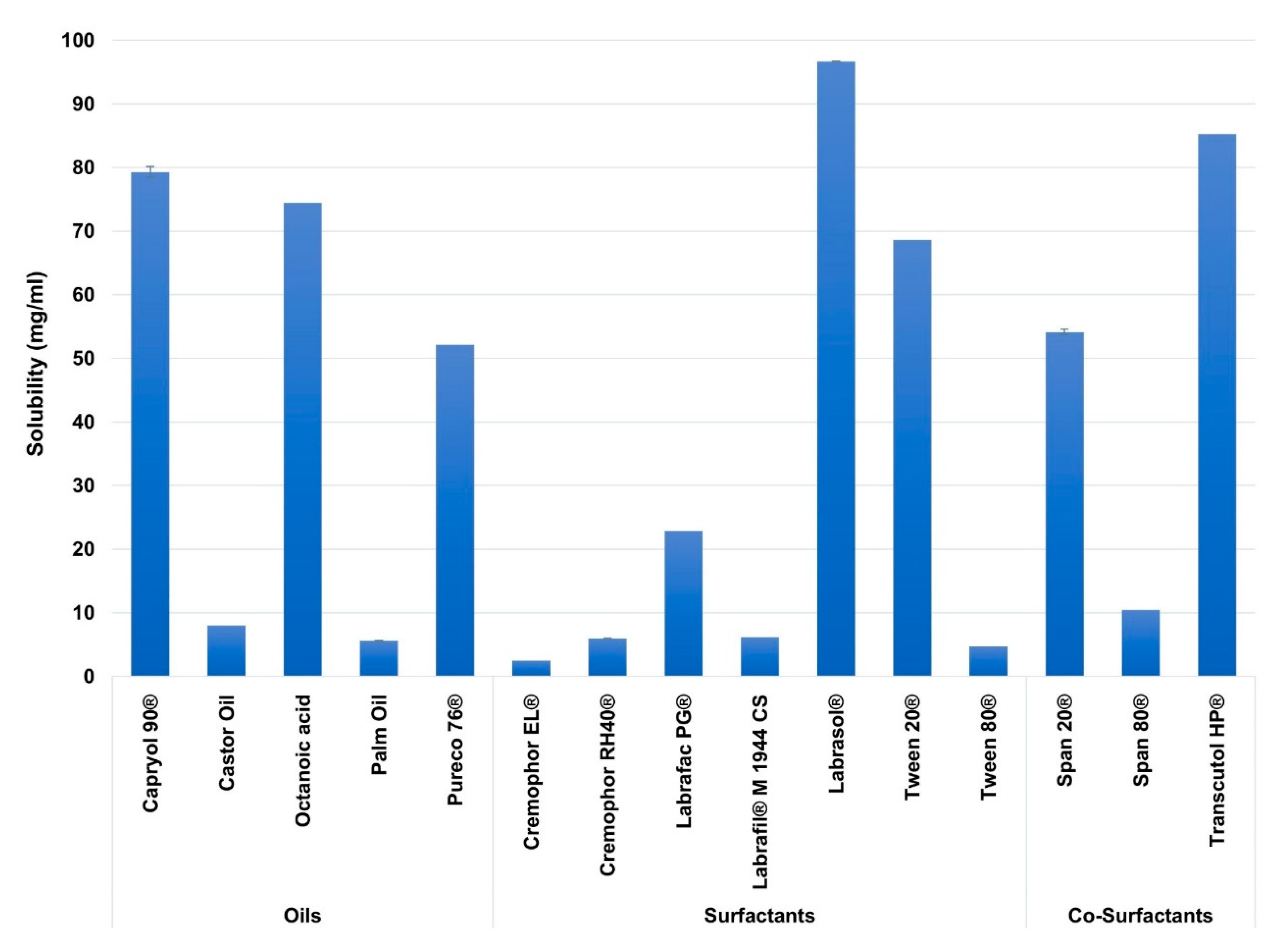
3.1. Excipients Selection and Solubility Studies
3.2. Emulsification Efficiency Studies
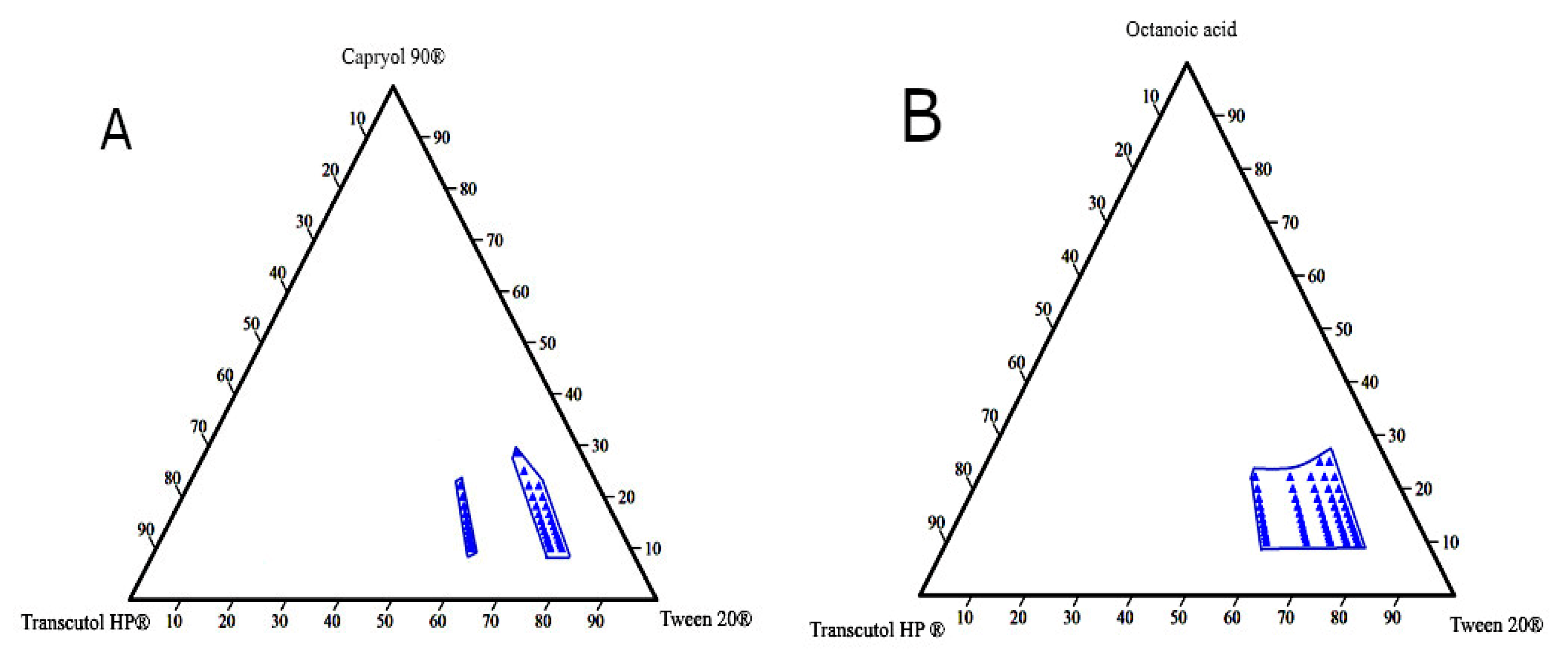
3.3. Construction of Pseudo-Ternary Phase Diagram
3.4. Preparation of AZM Loaded L-SEDDs
AZM Incorporation and Its Impact on L-SEDDs Properties
3.5. Preparation of Solid Self-Emulsifying Drug Delivery System (S-SEDDs)
3.6. Transmission Electron Microscope (TEM)
3.7. Cell Culture
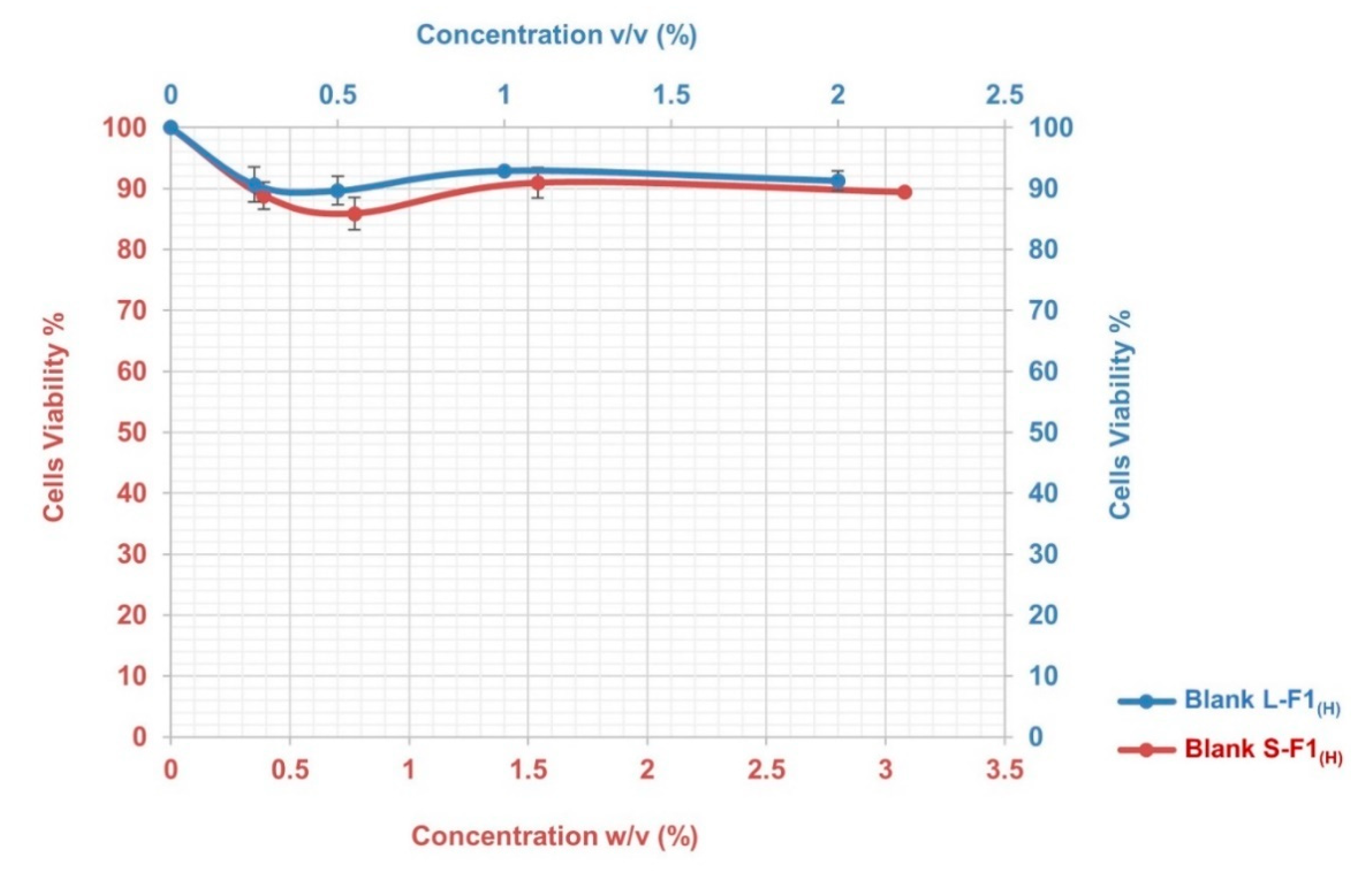
3.7.1. Cytotoxicity Assay
3.7.2. Transepithelial Electrical Resistance Evaluation
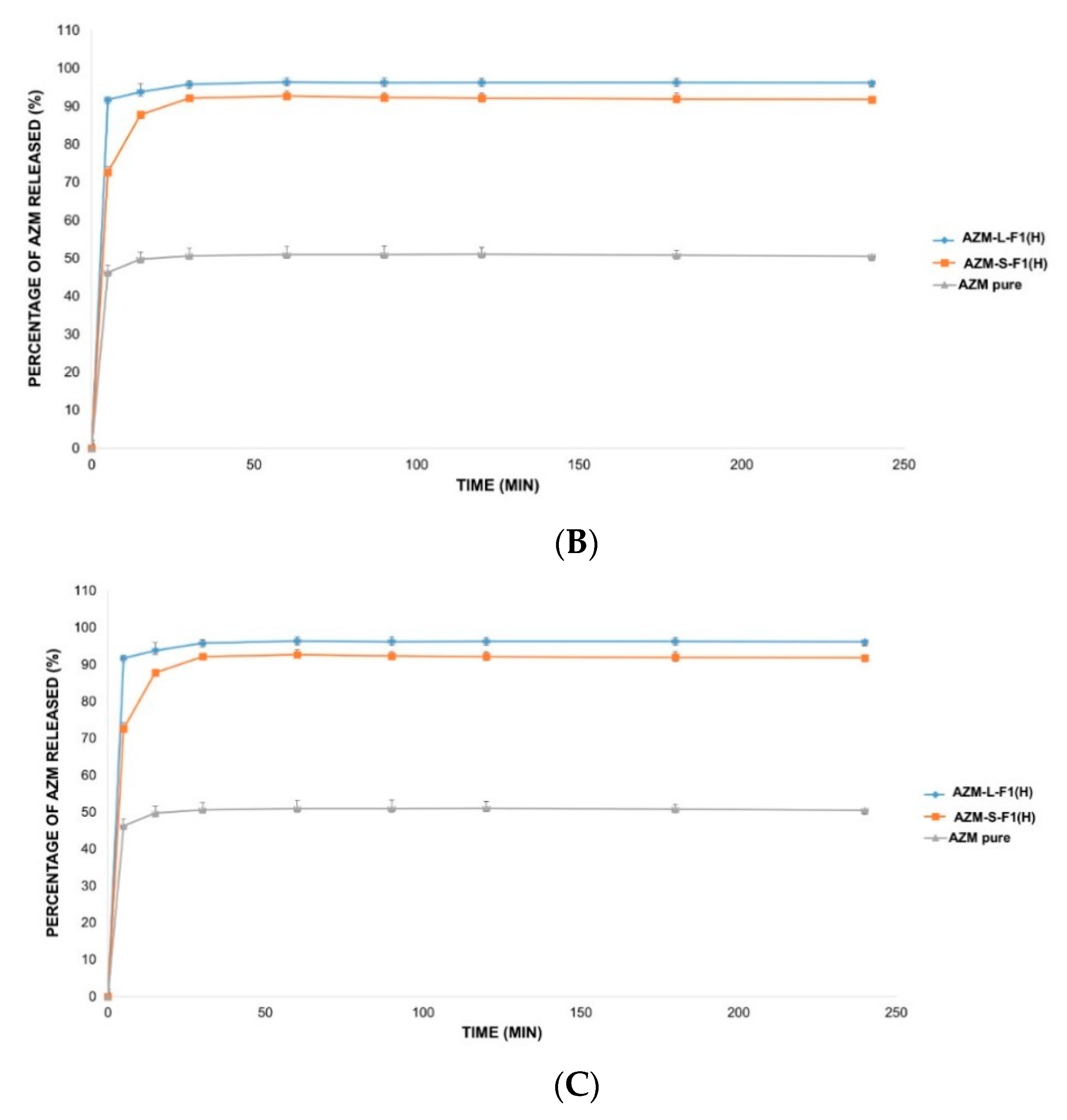
3.8. The In Vitro Release Studies
3.9. Stability Studies
3.9.1. Physical Stability
3.9.2. Droplet Size, Đ, and Zeta Potential
3.9.3. Chemical Stability
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Al-Achi, A.; Gupta, M.R.; Stagner, W.C. Integrated Pharmaceutics: Applied Preformulation, Product Design, and Regulatory Science; Wiley: Hoboken, NJ, USA, 2013; pp. 163–164. [Google Scholar]
- Torne, S.R.; Sheela, A.; Sarada, N.C. A Review on Oral Liquid as an Emerging Technology in Controlled Drug Delivery System. Curr. Pharm. Des. 2018, 24, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Ghasemiyeh, P.; Mohammadi-Samani, S. Solid lipid nanoparticles and nanostructured lipid carriers as novel drug delivery systems: Applications, advantages and disadvantages. Res. Pharm. Sci. 2018, 13, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Laksitorini, M.; Prasasty, V.D.; Kiptoo, P.K.; Siahaan, T.J. Pathways and progress in improving drug delivery through the intestinal mucosa and blood-brain barriers. Ther. Deliv. 2014, 5, 1143–1163. [Google Scholar] [CrossRef] [PubMed]
- The United States Pharmacopoeia and National Formulary, 38th ed.; The United States Pharmacopeial Convention: Rockville, MD, USA, 2015; Volume 39, p. 2339.
- Imperi, F.; Leoni, L.; Visca, P. Antivirulence activity of azithromycin in Pseudomonas aeruginosa. Front. Microbiol. 2014, 5, 178. [Google Scholar] [CrossRef]
- World Health Organization. WHO Model List of Essential Medicines. 2013. Available online: https://www.who.int/medicines/publications/essentialmedicines/en/ (accessed on 15 May 2020).
- Peters, D.H.; Friedel, H.A.; McTavish, D. Azithromycin. A review of its antimicrobial activity, pharmacokinetic properties and clinical efficacy. Drugs 1992, 44, 750–799. [Google Scholar] [CrossRef]
- Williams, J.D. Spectrum of Activity of Azithromycin. Eur. J. Clin. Microbiol. Infect. Dis. 1991, 10, 813–820. [Google Scholar] [CrossRef]
- Gautret, P.; Lagier, J.-C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020. [Google Scholar] [CrossRef]
- Adeli, E.; Mortazavi, S.A. Design, formulation and evaluation of Azithromycin binary solid dispersions using Kolliphor series for the solubility and in vitro dissolution rate enhancement. Int. J. Pharm. Investig. 2014, 44, 119–131. [Google Scholar] [CrossRef]
- Aucamp, M.; Odendaal, R.; Liebenberg, W.; Hamman, J. Amorphous azithromycin with improved aqueous solubility and intestinal membrane permeability. Drug Dev. Ind. Pharm. 2015, 41, 1100–1108. [Google Scholar] [CrossRef]
- Adrjanowicz, K.; Zakowiecki, D.; Kaminski, K.; Hawelek, L.; Grzybowska, K.; Tarnacka, M.; Paluch, M.; Cal, K. Molecular dynamics in supercooled liquid and glassy states of antibiotics: Azithromycin, clarithromycin and roxithromycin studied by dielectric spectroscopy. Advantages given by the amorphous state. Mol. Pharm. 2012, 9, 1748–1763. [Google Scholar] [CrossRef]
- Montejo-Bernardo, J.; García-Granda, S.; Bayod, M.; Llorente, I.; Llavona, L. An easy and general method for quantifying Azithromycin dihydrate in a matrix of amorphous Azithromycin. Arkivoc 2005, 2005, 321–331. [Google Scholar] [CrossRef]
- Arora, S.C.; Sharma, P.K.; Irchhaiya, R.; Khatkar, A.; Singh, N.; Gagoria, J. Development, characterization and solubility study of solid dispersions of azithromycin dihydrate by solvent evaporation method. J. Adv. Pharm. Technol. 2010, 1, 221–228. [Google Scholar]
- Tung, N.-T.; Tran, C.-S.; Nguyen, T.-L.; Hoang, T.; Trinh, T.-D.; Nguyen, T.-N. Formulation and biopharmaceutical evaluation of bitter taste masking microparticles containing azithromycin loaded in dispersible tablets. Eur. J. Pharm. Biopharm. 2018, 126, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Luke, D.R.; Foulds, G. Disposition of oral azithromycin in humans. Clin. Pharmacol. Ther. 1997, 61, 641–648. [Google Scholar] [CrossRef]
- Garver, E.; Hugger, E.D.; Shearn, S.P.; Rao, A.; Dawson, P.A.; Davis, C.B.; Han, C. Involvement of intestinal uptake transporters in the absorption of azithromycin and clarithromycin in the rat. Drug Metab. Dispos. 2008, 36, 2492–2498. [Google Scholar] [CrossRef]
- Nožinić, D.; Milić, A.; Mikac, L.; Ralić, J.; Padovan, J.; Antolović, R. Assessment of Macrolide Transport Using Pampa, Caco-2 and Mdckii-Hmdr1 assays. Croat. Chem. Acta 2010, 83, 323–331. [Google Scholar]
- Fohner, A.E.; Sparreboom, A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Macrolide antibiotic pathway, pharmacokinetics/pharmacodynamics. Pharm. Genom. 2017, 27, 164–167. [Google Scholar] [CrossRef]
- Asgrimsson, V.; Gudjonsson, T.; Gudmundsson, G.H.; Baldursson, O. Novel Effects of Azithromycin on Tight Junction Proteins in Human Airway Epithelia. Antimicrob. Agents Chemother. 2006, 50, 1805–1812. [Google Scholar] [CrossRef]
- Arason, A.J.; Joelsson, J.P.; Valdimarsdottir, B.; Sigurdsson, S.; Gudjonsson, A.; Halldorsson, S.; Johannsson, F.; Rolfsson, O.; Lehmann, F.; Ingthorsson, S.; et al. Azithromycin induces epidermal differentiation and multivesicular bodies in airway epithelia. Respir. Res. 2019, 20, 129. [Google Scholar] [CrossRef]
- Salama, N.N.; Eddington, N.D.; Fasano, A. Tight junction modulation and its relationship to drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 15–28. [Google Scholar] [CrossRef]
- U.S. National Center for Biotechnology Information Database. Azithromycin. 2016. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Azithromycin (accessed on 15 May 2020).
- Adeli, E. Preparation and evaluation of azithromycin binary solid dispersions using various polyethylene glycols for the improvement of the drug solubility and dissolution rate. Braz. J. Pharm. 2016, 52, 1–13. [Google Scholar] [CrossRef]
- Hou, C.-D.; Wang, J.-X.; Le, Y.; Zou, H.-K.; Zhao, H. Preparation of azithromycin nanosuspensions by reactive precipitation method. Drug Dev. Ind. Pharm. 2012, 38, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Feng, Y.; Liao, H.; Hu, X.; Wan, S.; Zhu, B.; Zhang, M.; Xiong, H.; Zhou, Y.; Zhang, J. Azithromycin Cationic Non-Lecithoid Nano/Microparticles Improve Bioavailability and Targeting Efficiency. Pharm. Res. 2014, 31, 2857–2867. [Google Scholar] [CrossRef]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000, 11 (Suppl. 2), S93–S98. [Google Scholar] [CrossRef]
- Pouton, C.W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29. [Google Scholar] [CrossRef] [PubMed]
- Yakushiji, K.; Sato, H.; Ogino, M.; Suzuki, H.; Seto, Y.; Onoue, S. Self-Emulsifying Drug Delivery System of Celecoxib for Avoiding Delayed Oral Absorption in Rats with Impaired Gastric Motility. AAPS PharmSciTech 2020, 21, 135. [Google Scholar] [CrossRef] [PubMed]
- Potharaju, S.; Mutyam, S.K.; Liu, M.; Green, C.; Frueh, L.; Nilsen, A.; Pou, S.; Winter, R.; Riscoe, M.K.; Shankar, G. Improving solubility and oral bioavailability of a novel antimalarial prodrug: Comparing spray-dried dispersions with self-emulsifying drug delivery systems. Pharm. Dev. Technol. 2020, 25, 625–639. [Google Scholar] [CrossRef]
- Mazzeti, A.L.; Oliveira, L.T.; Gonçalves, K.R.; Schaun, G.C.; Mosqueira, V.C.F.; Bahia, M.T. Benznidazole self-emulsifying delivery system: A novel alternative dosage form for Chagas disease treatment. Eur. J. Pharm. Sci. 2020, 145, 105234. [Google Scholar] [CrossRef]
- Ukai, H.; Iwasa, K.; Deguchi, T.; Morishita, M.; Katsumi, H.; Yamamoto, A. Enhanced Intestinal Absorption of Insulin by Capryol 90, a Novel Absorption Enhancer in Rats: Implications in Oral Insulin Delivery. Pharmaceutics 2020, 12, 462. [Google Scholar] [CrossRef]
- Kamal, M.M.; Salawi, A.; Lam, M.; Nokhodchi, A.; Abu-Fayyad, A.; El Sayed, K.A.; Nazzal, S. Development and characterization of curcumin-loaded solid self-emulsifying drug delivery system (SEDDS) by spray drying using Soluplus® as solid carrier. Powder Technol. 2020, 369, 137–145. [Google Scholar] [CrossRef]
- Tengshe, S.D.; Karande, K.M. A Review on Self Micro-Emulsifying Drug Delivery System: A Tool for Solubility Enhancement. Int. J. Res. Anal. Rev. 2020, 7, 101–114. [Google Scholar]
- Aloisio, C.; Bueno, M.S.; Ponte, M.P.; Paredes, A.; Palma, S.D.; Longhi, M. Development of solid self-emulsifying drug delivery systems (SEDDS) to improve the solubility of resveratrol. Ther. Deliv. 2019, 10, 626–641. [Google Scholar] [CrossRef] [PubMed]
- Assi, R.A.; Darwis, Y.; Abdulbaqi, I.M.; Asif, S.M. Development and validation of a stability-indicating RP-HPLC method for the detection and quantification of azithromycin in bulk, and selfemulsifying drug delivery system (SEDDs) formulation. J. Appl. Pharm. 2017, 7, 20–29. [Google Scholar]
- Gurram, A.K.; Deshpande, P.B.; Kar, S.S.; Nayak, U.Y.; Udupa, N.; Reddy, M.S. Role of Components in the Formation of Self-microemulsifying Drug Delivery Systems. Indian J. Pharm. Sci. 2015, 77, 249–257. [Google Scholar]
- Ye, J.; Wu, H.; Huang, C.; Lin, W.; Zhang, C.; Huang, B.; Lu, B.; Xu, H.; Li, X.; Long, X. Comparisons of in vitro Fick’s first law, lipolysis, and in vivo rat models for oral absorption on BCS II drugs in SNEDDS. Int. J. Nanomed. 2019, 14, 5623–5636. [Google Scholar] [CrossRef]
- Sobhani, H.; Tarighi, P.; Ostad, S.N.; Shafaati, A.; Nafissi-Varcheh, N.; Aboofazeli, R. Rapamycin-Loaded, Capryol(TM) 90 and Oleic Acid Mediated Nanoemulsions: Formulation Development, Characterization and Toxicity Assessment. Iran. J. Pharm. Res. 2018, 17, 830–850. [Google Scholar]
- Nasr, A.M.; Qushawy, M.K.; Elkhoudary, M.M.; Gawish, A.Y.; Elhady, S.S.; Swidan, S.A. Quality by Design for the Development and Analysis of Enhanced In-Situ Forming Vesicles for the Improvement of the Bioavailability of Fexofenadine HCl in Vitro and in Vivo. Pharmaceutics 2020, 12, 409. [Google Scholar] [CrossRef]
- Lindmark, T.; Kimura, Y.; Artursson, P. Absorption enhancement through intracellular regulation of tight junction permeability by medium chain fatty acids in Caco-2 cells. J. Pharmacol. Exp. Ther. 1998, 284, 362–369. [Google Scholar]
- Suzuki, T. Regulation of the intestinal barrier by nutrients: The role of tight junctions. Anim. Sci. J. 2020, 91, e13357. [Google Scholar] [CrossRef] [PubMed]
- Veeravalli, V.; Cheruvu, H.S.; Srivastava, P.; Vamsi Madgula, L.M. Three-dimensional aspects of formulation excipients in drug discovery: A critical assessment on orphan excipients, matrix effects and drug interactions. J. Pharm. Anal. 2020. [Google Scholar] [CrossRef]
- Zhao, G.; Huang, J.; Xue, K.; Si, L.; Li, G. Enhanced intestinal absorption of etoposide by self-microemulsifying drug delivery systems: Roles of P-glycoprotein and cytochrome P450 3A inhibition. Eur. J. Pharm. Sci. 2013, 50, 429–439. [Google Scholar] [CrossRef]
- Shen, Y.; Lu, Y.; Jv, M.; Hu, J.; Li, Q.; Tu, J. Enhancing effect of Labrasol on the intestinal absorption of ganciclovir in rats. Drug Dev. Ind. Pharm. 2011, 37, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Gurjar, R.; Chan, C.Y.S.; Curley, P.; Sharp, J.; Chiong, J.; Rannard, S.; Siccardi, M.; Owen, A. Inhibitory Effects of Commonly Used Excipients on P-Glycoprotein in Vitro. Mol. Pharm. 2018, 15, 4835–4842. [Google Scholar] [CrossRef]
- Yang, S.; Liu, J.; Chen, Y.; Jiang, J. Reversal effect of Tween-20 on multidrug resistance in tumor cells in vitro. Biomed. Pharmacother. 2012, 66, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Li, G.; Wang, X.; Bi, Y.; Zhang, Y. Inhibitory effect of sixteen pharmaceutical excipients on six major organic cation and anion uptake transporters. Xenobiotica 2020. [Google Scholar] [CrossRef]
- Lin, H.; Gebhardt, M.; Bian, S.; Kwon, K.A.; Shim, C.K.; Chung, S.J.; Kim, D.D. Enhancing Effect of Surfactants on Fexofenadine Hcl Transport across the Human Nasal Epithelial Cell Monolayer. Int. J. Pharm. 2007, 330, 23–31. [Google Scholar] [CrossRef]
- Zhang, H.; Yao, M.; Morrison, R.A.; Chong, S. Commonly used surfactant, Tween 80, improves absorption of P-glycoprotein substrate, digoxin, in rats. Arch. Pharm. Res. 2003, 26, 768–772. [Google Scholar] [CrossRef]
- Prabhakar, K.; Afzal, S.M.; Surender, G.; Kishan, V. Tween 80 containing lipid nanoemulsions for delivery of indinavir to brain. Acta Pharm. Sin. B 2013, 3, 345–353. [Google Scholar] [CrossRef]
- Yamagata, T.; Kusuhara, H.; Morishita, M.; Takayama, K.; Benameur, H.; Sugiyama, Y. Effect of excipients on breast cancer resistance protein substrate uptake activity. J. Control. Release 2007, 124, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Kim, J.K.; Song, Y.K.; Park, J.S.; Kim, C.K. A new self-emulsifying formulation of itraconazole with improved dissolution and oral absorption. J. Control. Release 2006, 110, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Khames, A. Formulation and Characterization of Eplerenone Nanoemulsion Liquisolids, An Oral Delivery System with Higher Release Rate and Improved Bioavailability. Pharmaceutics 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hirschberg, C.; Fanø, M.; Mu, H.; Müllertz, A. Evaluation of self-emulsifying drug delivery systems for oral insulin delivery using an in vitro model simulating the intestinal proteolysis. Eur. J. Pharm. Sci. 2020, 147, 105272. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Mittapelly, N.; Husain, A.; Kushwaha, S.; Chattopadhyay, S.; Kumar, P.; Ramakrishna, E.; Kumar, S.; Maurya, R.; Sanyal, S.; et al. A butanolic fraction from the standardized stem extract of Cassia occidentalis L delivered by a self-emulsifying drug delivery system protects rats from glucocorticoid-induced osteopenia and muscle atrophy. Sci. Rep. 2020, 10, 195. [Google Scholar] [CrossRef] [PubMed]
- Alghananim, A.; Özalp, Y.; Mesut, B.; Serakinci, N.; Özsoy, Y.; Güngör, S. A Solid Ultra Fine Self-Nanoemulsifying Drug Delivery System (S-SNEDDS) of Deferasirox for Improved Solubility: Optimization, Characterization, and In Vitro Cytotoxicity Studies. Pharmaceuticals 2020, 13, 162. [Google Scholar] [CrossRef]
- Jianxian, C.; Saleem, K.; Ijaz, M.; Ur-Rehman, M.; Murtaza, G.; Asim, M.H. Development and in vitro Evaluation of Gastro-protective Aceclofenac-loaded Self-emulsifying Drug Delivery System. Int. J. Nanomed. 2020, 15, 5217–5226. [Google Scholar] [CrossRef]
- Yeom, D.W.; Son, H.Y.; Kim, J.H.; Kim, S.R.; Lee, S.G.; Song, S.H.; Chae, B.R.; Choi, Y.W. Development of a solidified self-microemulsifying drug delivery system (S-SMEDDS) for atorvastatin calcium with improved dissolution and bioavailability. Int. J. Pharm. 2016, 506, 302–311. [Google Scholar] [CrossRef]
- Janković, J.; Djekic, L.; Dobričić, V.; Primorac, M. Evaluation of critical formulation parameters in design and differentiation of self-microemulsifying drug delivery systems (SMEDDSs) for oral delivery of aciclovir. Int. J. Pharm. 2016, 497, 301–311. [Google Scholar] [CrossRef]
- Singh, A.K.; Chaurasiya, A.; Singh, M.; Upadhyay, S.C.; Mukherjee, R.; Khar, R.K. Exemestane Loaded Self-Microemulsifying Drug Delivery System (SMEDDS): Development and Optimization. AAPS PharmSciTech 2008, 9, 628–634. [Google Scholar] [CrossRef]
- Ostolska, I.; Wiśniewska, M. Application of the zeta potential measurements to explanation of colloidal Cr(2)O(3) stability mechanism in the presence of the ionic polyamino acids. Colloid Polym. Sci. 2014, 292, 2453–2464. [Google Scholar] [CrossRef]
- Hwang, C.J.; Na, Y.G.; Huh, H.W.; Kim, M.; Lee, H.K.; Cho, C.W. The Effect of Pharmaceutical Excipients for Applying to Spray-Dried Omega-3 Powder. Appl. Sci. 2019, 9, 1–11. [Google Scholar]
- Tan, A.; Rao, S.; Prestidge, C.A. Transforming Lipid-Based Oral Drug Delivery Systems into Solid Dosage Forms: An Overview of Solid Carriers, Physicochemical Properties, and Biopharmaceutical Performance. Pharm. Res. 2013, 30, 2993–3017. [Google Scholar] [CrossRef] [PubMed]
- Katteboina, S.; Chandrasekhar, P.; Balaji, S. Approaches for the development of solid self-emulsifying drug delivery systems and dosage forms. Asian J. Pharm. Sci. 2009, 4, 240–253. [Google Scholar]
- Beg, S.; Katare, O.P.; Saini, S.; Garg, B.; Khurana, R.K.; Singh, B. Solid self-nanoemulsifying systems of olmesartan medoxomil: Formulation development, micromeritic characterization, in vitro and in vivo evaluation. Powder Technol. 2016, 294, 93–104. [Google Scholar] [CrossRef]
- Kasturi, M.; Agrawal, S.; Janga, K.Y. Development and Characterization of Ramipril Loaded Solid self Nanoemulsifying Drug Delivery System (SNEDDS) for Improved Oral Delivery of Lipophilic Drugs. Int. J. Pharm. Biol. Sci. Arch. 2015, 6, 10–17. [Google Scholar]
- Bhagwat, D.A.; Souza, J.I.D. Formulation and evaluation of solid self micro emulsifying drug delivery system using aerosil 200 as solid carrier. Int. Curr. Pharm. 2012, 1, 414–419. [Google Scholar] [CrossRef]
- Hauptstein Sabine, P.F.; Andreas, B.S. Self-nanoemulsifying drug delivery systems as novel approach for pDNA drug delivery. Int. J. Pharm. 2015, 487, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Watari, A.; Hashegawa, M.; Yagi, K.; Kondoh, M. Homoharringtonine increases intestinal epithelial permeability by modulating specific claudin isoforms in Caco-2 cell monolayers. Eur. J. Pharm. Biopharm. 2015, 89, 232–238. [Google Scholar] [CrossRef]
- Bandivadekar, M.; Pancholi, S.; Kaul-Ghanekar, R.; Choudhari, A.; Koppikar, S. Single non-ionic surfactant based self-nanoemulsifying drug delivery systems: Formulation, characterization, cytotoxicity and permeability enhancement study. Drug Dev. Ind. Pharm. 2013, 39, 696–703. [Google Scholar] [CrossRef]
- Ujhelyi, Z.; Kalantari, A.; Vecsernyés, M.; Róka, E.; Fenyvesi, F.; Póka, R.; Kozma, B.; Bácskay, I. The enhanced inhibitory effect of different antitumor agents in self-microemulsifying drug delivery systems on human cervical cancer HeLa cells. Molecules 2015, 20, 13226–13239. [Google Scholar] [CrossRef]
- Zhang, Q.; He, N.; Zhang, L.; Zhu, F.; Chen, Q.; Qin, Y.; Zhang, Z.; Zhang, Q.; Wang, S.; He, Q. The in vitro and in vivo study on self-nanoemulsifying drug delivery system (SNEDDS) based on insulin-phospholipid complex. J. Biomed. Nanotechnol. 2012, 8, 90–97. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. TEER measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef] [PubMed]
- Mine, Y.; Zhang, J.W. Surfactants enhance the tight-junction permeability of food allergens in human intestinal epithelial Caco-2 cells. Int. Arch. Allergy Immunol. 2003, 130, 135–142. [Google Scholar] [CrossRef]
- Lea, T. Epithelial Cell Models; General Introduction. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Verhoeckx, K., Cotter, P., López-Expósito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 95–102. [Google Scholar] [CrossRef]
- USFDA. Guidance for Industry: Dissolution Methods of Azithromycin Pharmaceutical Forms. 2006. Available online: https://www.accessdata.fda.gov/scripts/cder/dissolution/ (accessed on 18 May 2020).
- Yeom, D.W.; Song, Y.S.; Kim, S.R.; Lee, S.G.; Kang, M.H.; Lee, S.; Choi, Y.W. Development and optimization of a self-microemulsifying drug delivery system for ator vastatin calcium by using d-optimal mixture design. Int. J. Nanomed. 2015, 10, 3865–3878. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, W.; Liu, X.; Sun, J. Preparation and evaluation of sustained-release azithromycin tablets in vitro and in vivo. Asian J. Pharm. Sci. 2014, 9, 155–161. [Google Scholar] [CrossRef]
- Izgelov, D.; Shmoeli, E.; Domb, A.J.; Hoffman, A. The effect of medium chain and long chain triglycerides incorporated in self-nano emulsifying drug delivery systems on oral absorption of cannabinoids in rats. Int. J. Pharm. 2020, 580, 119201. [Google Scholar] [CrossRef]
- Grove, M.; Pedersen, G.P.; Nielsen, J.L.; Müllertz, A. Bioavailability of seocalcitol I: Relating solubility in biorelevant media with oral bioavailability in rats—Effect of medium and long chain triglycerides. J. Pharm. Sci. 2005, 94, 1830–1838. [Google Scholar] [CrossRef] [PubMed]
- Grove, M.; Müllertz, A.; Nielsen, J.L.; Pedersen, G.P. Bioavailability of seocalcitol: II: Development and characterisation of self-microemulsifying drug delivery systems (SMEDDS) for oral administration containing medium and long chain triglycerides. Eur. J. Pharm. Sci. 2006, 28, 233–242. [Google Scholar] [CrossRef]
- Patel, A.R.; Vavia, P.R. Preparation and in vivo evaluation of SMEDDS (Self-microemulsifying drug delivery system) containing fenofibrate. AAPS J. 2007, 9, E344–E352. [Google Scholar] [CrossRef]
- Bhandari, V.; Avachat, A. Formulation and characterization of self emulsifing pellets of carvedilol. Braz. J. Pharm. 2015, 51, 663–671. [Google Scholar] [CrossRef]
- Behrens, D.; Fricker, R.; Bodoky, A.; Drewe, J.; Harder, F.; Heberer, M. Comparison of Cyclosporin A Absorption from LCT and MCT Solutions following Intrajejunal Administration in Conscious Dogs. J. Pharm. Sci. 1996, 85, 666–668. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Kaukonen, A.M.; Boyd, B.J.; Edwards, G.A.; Charman, W.N. Susceptibility to Lipase-Mediated Digestion Reduces the Oral Bioavailability of Danazol After Administration as a Medium-Chain Lipid-Based Microemulsion Formulation. Pharm. Res. 2004, 21, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Persson, L.C.; Porter, C.J.H.; Charman, W.N.; Bergström, C.A.S. Computational Prediction of Drug Solubility in Lipid Based Formulation Excipients. Pharm. Res. 2013, 30, 3225–3237. [Google Scholar] [CrossRef] [PubMed]
- Kaukonen, A.M.; Boyd, B.J.; Porter, C.J.; Charman, W.N. Drug solubilization behavior during in vitro digestion of simple triglyceride lipid solution formulations. Pharm. Res. 2004, 21, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Kollipara, S.; Gandhi, R.K. Pharmacokinetic aspects and in vitro–in vivo correlation potential for lipid-based formulations. Acta Pharm. Sin. B 2014, 4, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wei, Y.; Cao, Z.; Xu, Y.; Lu, C.; Zhao, M.; Gou, J.; Yin, T.; Zhang, Y.; He, H.; et al. Aprepitant Intravenous Emulsion Based on Ion Pairing/Phospholipid Complex for Improving Physical and Chemical Stability During Thermal Sterilization. AAPS PharmSciTech 2020, 21, 75. [Google Scholar] [CrossRef] [PubMed]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral lipid-based drug delivery systems—An overview. Acta Pharm. Sin. B 2013, 3, 361–372. [Google Scholar] [CrossRef]
- Pouton, C.W. Formulation of self-emulsifying drug delivery systems. Adv. Drug Deliv. Rev. 1997, 25, 47–58. [Google Scholar] [CrossRef]
- Gupta, S.; Kesarla, R.; Omri, A. Formulation Strategies to Improve the Bioavailability of Poorly Absorbed Drugs with Special Emphasis on Self-Emulsifying Systems. ISRN Pharm. 2013, 2013, 848043. [Google Scholar] [CrossRef]
- Agrawal, S.; Giri, T.K.; Tripathi, D.K.; Alexander, A. A Review on Novel Therapeutic Strategies for the Enhancement of Solubility for Hydrophobic Drugs through Lipid and Surfactant Based Self Micro Emulsifying Drug Delivery System: A Novel Approach. Am. J. Drug Discov. Devel. 2012, 2, 143–183. [Google Scholar] [CrossRef]
- Yan, B.; Ma, Y.; Guo, J.; Wang, Y. Self-microemulsifying delivery system for improving bioavailability of water insoluble drugs. J. Nanopart. Res. 2020, 22, 18. [Google Scholar] [CrossRef]
- Chatterjee, B.; Hamed Almurisi, S.; Ahmed Mahdi Dukhan, A.; Mandal, U.K.; Sengupta, P. Controversies with self-emulsifying drug delivery system from pharmacokinetic point of view. Drug Deliv. 2016, 23, 3639–3652. [Google Scholar] [CrossRef]
- Akula, S.; Gurram, A.K.; Devireddy, S.R. Self-Microemulsifying Drug Delivery Systems: An Attractive Strategy for Enhanced Therapeutic Profile. Int. Sch. Res. Not. 2014, 2014, 11. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Z.; Mao, J.; Wang, S.; Zheng, Y. Quantitative evaluation of inclusion homogeneity in composites and the applications. J. Mater. Res. Technol. 2020, 9, 6790–6807. [Google Scholar] [CrossRef]
- Pouton, C.W. Self-emulsifying drug delivery systems: Assessment of the efficiency of emulsification. Int. J. Pharm. 1985, 27, 335–348. [Google Scholar] [CrossRef]
- Madan, J.R.; Patil, K.; Awasthi, R.; Dua, K. Formulation and evaluation of solid self-microemulsifying drug delivery system for azilsartan medoxomil. Int. J. Polym. Mater. 2019, 1–17. [Google Scholar] [CrossRef]
- Gupta, S.; Chavhan, S.; Sawant, K.K. Self-nanoemulsifying drug delivery system for adefovir dipivoxil: Design, characterization, in vitro and ex vivo evaluation. Colloids Surf. A Physicochem. Eng. Asp. 2011, 392, 145–155. [Google Scholar] [CrossRef]
- Kohli, K.; Chopra, S.; Dhar, D.; Arora, S.; Khar, R.K. Self-emulsifying drug delivery systems: An approach to enhance oral bioavailability. Drug Discov. Today 2010, 15, 958–965. [Google Scholar] [CrossRef]
- Buya, A.B.; Ucakar, B.; Beloqui, A.; Memvanga, P.B.; Préat, V. Design and evaluation of self-nanoemulsifying drug delivery systems (SNEDDSs) for senicapoc. Int. J. Pharm. 2020, 580, 119180. [Google Scholar] [CrossRef] [PubMed]
- Constantinides, P.P. Lipid Microemulsions for Improving Drug Dissolution and Oral Absorption: Physical and Biopharmaceutical Aspects. Pharm. Res. 1995, 12, 1561–1572. [Google Scholar] [CrossRef]
- Azman, N.A.Z.; Raman, I.A.; Jantan, I.; Derawi, D. Formulation Screening of Palm-based Nanoemulsion for an OralDrug Vehicle of Phyllanthin. Malays. J. Chem. 2019, 21, 43–49. [Google Scholar]
- Singh, K.; Sharma, M.; Gandhi, K. Recent approaches in self emulsifying drug delivery system. Int. J. Pharm. Sci. Res. 2012, 3, 4192–4201. [Google Scholar]
- Zaichik, S.; Steinbring, C.; Caliskan, C.; Bernkop-Schnürch, A. Development and in vitro evaluation of a self-emulsifying drug delivery system (SEDDS) for oral vancomycin administration. Int. J. Pharm. 2019, 554, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, Y.; Feng, N.; Xu, J. Preparation and evaluation of self-microemulsifying drug delivery system of oridonin. Int. J. Pharm. 2008, 355, 269–276. [Google Scholar] [CrossRef]
- Parmar, B.; Patel, U.; Bhimani, B.; Sanghavi, K.; Patel, G.; Daslaniya, D. SMEDDS: A Dominant Dosage Form Which Improve Bioavailability. Am. J. PharmTech Res. 2012, 2, 54–72. [Google Scholar]
- Almeida, S.R.D.; Tippavajhala, V.K. A Rundown Through Various Methods Used in the Formulation of Solid Self-Emulsifying Drug Delivery Systems (S-SEDDS). AAPS PharmSciTech 2019, 20, 323. [Google Scholar] [CrossRef]
- Mantry, S.; Majumder, D. Development of Liquid and Solid Self-Emulsifying Drug Delivery System of Silymarin. J. Drug Deliv. Ther. 2019, 9, 54–61. [Google Scholar]
- Dash, R.N.; Mohammed, H.; Humaira, T.; Ramesh, D. Design, optimization and evaluation of glipizide solid self-nanoemulsifying drug delivery for enhanced solubility and dissolution. Saudi Pharm. J. 2015, 23, 528–540. [Google Scholar] [CrossRef]
- Kim, D.W.; Kwon, M.S.; Yousaf, A.M.; Balakrishnan, P.; Park, J.H.; Kim, D.S.; Lee, B.-J.; Park, Y.J.; Yong, C.S.; Kim, J.O.; et al. Comparison of a solid SMEDDS and solid dispersion for enhanced stability and bioavailability of clopidogrel napadisilate. Carbohydr. Polym. 2014, 114, 365–374. [Google Scholar] [CrossRef]
- Cho, W.; Kim, M.-S.; Kim, J.-S.; Park, J.; Park, H.J.; Cha, K.-H.; Park, J.-S.; Hwang, S.-J. Optimized formulation of solid self-microemulsifying sirolimus delivery systems. Int. J. Nanomed. 2013, 8, 1673–1682. [Google Scholar] [CrossRef]
- Dixit, R.P.; Nagarsenker, M.S. Self-nanoemulsifying granules of ezetimibe: Design, optimization and evaluation. Eur. J. Pharm. Sci. 2008, 35, 183–192. [Google Scholar] [CrossRef]
- Guan, Q.; Zhang, G.; Sun, S.; Fan, H.; Sun, C.; Zhang, S. Enhanced Oral Bioavailability of Pueraria Flavones by a Novel Solid Self-microemulsifying Drug Delivery System (SMEDDS) Dropping Pills. Biol. Pharm. Bull. 2016, 39, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Man, N.; Wang, Q.; Li, H.; Adu-Frimpong, M.; Sun, C.; Zhang, K.; Yang, Q.; Wei, Q.; Ji, H.; Toreniyazov, E.; et al. Improved oral bioavailability of myricitrin by liquid self-microemulsifying drug delivery systems. J. Drug Deliv. Sci. Technol. 2019, 52, 597–606. [Google Scholar] [CrossRef]
- Sha, X.; Wu, J.; Chen, Y.; Fang, X. Self-microemulsifying drug-delivery system for improved oral bioavailability of probucol: Preparation and evaluation. Int. J. Nanomed. 2012, 7, 705–712. [Google Scholar] [CrossRef]
- Ameeduzzafar; El-Bagory, I.; Alruwaili, N.K.; Elkomy, M.H.; Ahmad, J.; Afzal, M.; Ahmad, N.; Elmowafy, M.; Alharbi, K.S.; Md Shoaib, A. Development of novel dapagliflozin loaded solid self-nanoemulsifying oral delivery system: Physiochemical characterization and in vivo antidiabetic activity. J. Drug Deliv. Sci. Technol. 2019, 54, 101279. [Google Scholar] [CrossRef]
- Wei, Y.; Ye, X.; Shang, X.; Peng, X.; Bao, Q.; Liu, M.; Guo, M.; Li, F. Enhanced oral bioavailability of silybin by a supersaturatable self-emulsifying drug delivery system (S-SEDDS). Colloids Surf. A Physicochem. Eng. Asp. 2012, 396, 22–28. [Google Scholar] [CrossRef]
- Mencucci, R.; Pellegrini-Giampietro, D.E.; Paladini, I.; Favuzza, E.; Menchini, U.; Scartabelli, T. Azithromycin: Assessment of intrinsic cytotoxic effects on corneal epithelial cell cultures. Clin. Ophthalmol. 2013, 7, 965–971. [Google Scholar] [CrossRef]
- Schögler, A.; Kopf, B.S.; Edwards, M.R.; Johnston, S.L.; Casaulta, C.; Kieninger, E.; Jung, A.; Moeller, A.; Geiser, T.; Regamey, N.; et al. Novel antiviral properties of azithromycin in cystic fibrosis airway epithelial cells. Eur. Respir. J. 2015, 45, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.W., Jr.; Gad, S.C.; Julien, M. A review of the nonclinical safety of Transcutol®, a highly purified form of diethylene glycol monoethyl ether (DEGEE) used as a pharmaceutical excipient. Food Chem. Toxicol. 2014, 72, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Cox, S.; Sandall, A.; Smith, L.; Rossi, M.; Whelan, K. Food additive emulsifiers: A review of their role in foods, legislation and classifications, presence in food supply, dietary exposure, and safety assessment. Nutr. Rev. 2020. [Google Scholar] [CrossRef]
- USFDA. Center for Drug Evaluation and Research. Application Number 209394orig1s000. Non-clinical Review(s). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209394Orig1s000PharmR.pdf (accessed on 30 August 2020).
- Larregieu, C.A.; Benet, L.Z. Drug discovery and regulatory considerations for improving in silico and in vitro predictions that use Caco-2 as a surrogate for human intestinal permeability measurements. AAPS J. 2013, 15, 483–497. [Google Scholar] [CrossRef]
- Shah, P.; Jogani, V.; Bagchi, T.; Misra, A. Role of Caco-2 Cell Monolayers in Prediction of Intestinal Drug Absorption. Biotechnol. Prog. 2006, 22, 186–198. [Google Scholar] [CrossRef]
- Li, W.; Zhou, J.; Xu, Y. Study of the in vitro cytotoxicity testing of medical devices (Review). Biomed. Rep. 2015, 3, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar] [CrossRef] [PubMed]
- Tscheik, C.; Blasig, I.E.; Winkler, L. Trends in drug delivery through tissue barriers containing tight junctions. Tissue Barriers 2013, 1, e24565. [Google Scholar] [CrossRef]
- Sha, X.; Yan, G.; Wu, Y.; Li, J.; Fang, X. Effect of self-microemulsifying drug delivery systems containing Labrasol on tight junctions in Caco-2 cells. Eur. J. Pharm. Sci. 2005, 24, 477–486. [Google Scholar] [CrossRef]
- Deli, M.A. Potential use of tight junction modulators to reversibly open membranous barriers and improve drug delivery. BBA-Bioenergetics 2009, 1788, 892–910. [Google Scholar] [CrossRef]
- Dimitrijevic, D.; Shaw, A.J.; Florence, A.T. Effects of some non-ionic surfactants on transepithelial permeability in Caco-2 cells. J. Pharm. Pharmacol. 2000, 52, 157–162. [Google Scholar] [CrossRef]
- Gopalakrishnan, S.; Durai, M.; Kitchens, K.; Tamiz, A.P.; Somerville, R.; Ginski, M.; Paterson, B.M.; Murray, J.A.; Verdu, E.F.; Alkan, S.S.; et al. Larazotide acetate regulates epithelial tight junctions in vitro and in vivo. Peptides 2012, 35, 86–94. [Google Scholar] [CrossRef]
- Mariano, C.; Sasaki, H.; Brites, D.; Brito, M.A. A look at tricellulin and its role in tight junction formation and maintenance. Eur. J. Cell Biol. 2011, 90, 787–796. [Google Scholar] [CrossRef]
- Liu, Y.; Chiu, G.N.C. Dual-Functionalized PAMAM Dendrimers with Improved P-Glycoprotein Inhibition and Tight Junction Modulating Effect. Biomacromolecules 2013, 14, 4226–4235. [Google Scholar] [CrossRef]
- USFDA. Centre for Drug Evaluation and Research Approval Package for: Application Number 50-784. Clinical Pharmacology and Biopharmaceutics Review. 2002. Available online: www.accessdata.fda.gov (accessed on 25 May 2020).
- USFDA. Zithromax® (Azithromycin Tablets and Azithromycin for Oral Suspension). Reference ID: 3141345; 2012; pp. 1–22. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/050710s039,050711s036,050784s023lbl.pdf (accessed on 30 August 2020).
- Czajkowska-Kosnik, A.; Szekalska, M.; Amelian, A.; Szymanska, E.; Winnicka, K. Development and Evaluation of Liquid and Solid Self-Emulsifying Drug Delivery Systems for Atorvastatin. Molecules 2015, 20, 21010–21022. [Google Scholar] [CrossRef]
- Mohd, A.B.; Sanka, K.; Bandi, S.; Diwan, P.V.; Shastri, N. Solid self-nanoemulsifying drug delivery system (S-SNEDDS) for oral delivery of glimepiride: Development and antidiabetic activity in albino rabbits. Drug Deliv. 2015, 22, 499–508. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Role | HLB | Chain Length | Reported Cellular Activity (s) | References |
|---|---|---|---|---|---|
| Capryol 90® | Oil | 5 | Medium-chain (C8) | TJ opening | [33,40,41] |
| Castor Oil | - | Long-chain (C18) | - | - | |
| Octanoic acid (caprylic acid or caprylate) | - | Medium-chain (C8) | TJ opening | [42,43] | |
| Palm Oil | - | Long-chain. Mixture of C16 and C18 | - | - | |
| Pureco 76® | - | Medium-chain (C8–C10) | - | - | |
| Cremophor EL® | Surfactant | 12–14 | Medium-chain C8 | TJ opening and P-gp inhibitor | [44] |
| Cremophor RH40® | 14–16 | Medium-chain C10 | TJ opening and P-gp inhibitor | [45] | |
| Labrafac PG® | 1 | Medium-chain. Mixture of C8 and C10 | - | - | |
| Labrafil® M 1944 CS | 9 | Long-chain C18 | - | - | |
| Labrasol® | 12–14 | Medium-chain (C8–C10) | TJ opening and P-gp inhibitor | [44,46] | |
| Tween 20® | 16.7 | Medium-chain C12 | P-gp inhibitor | [47,48,49] | |
| Tween 80® | 15 | Long-chain C18 | TJ opening and P-gp inhibitor | [50,51,52] | |
| Span 20® | Co-surfactant | 8.6 | Medium-chain C12 | P-gp inhibitor | [53] |
| Span 80® | 4.3 | Long-chain C18 | - | - | |
| Transcutol HP® | - | - | P-gp inhibitor | [54] |
| No. | Formulation Code | Oil | Surfactant | Co-Surfactant |
|---|---|---|---|---|
| 1 | A1 | Capryol 90® | Labrasol® | Transcutol HP® |
| 2 | B1 | Labrasol® | Span 20® | |
| 3 | C1 | Tween 20® | Transcutol HP® | |
| 4 | D1 | Tween 20® | Span 20® | |
| 5 | A2 | Octanoic acid | Labrasol® | Transcutol HP® |
| 6 | B2 | Labrasol® | Span 20® | |
| 7 | C2 | Tween 20® | Transcutol HP® | |
| 8 | D2 | Tween 20® | Span 20® |
| Oil % | Surfactant % | Co-Surfactant % | DS (nm) |
|---|---|---|---|
| Diagram 1: Capryol 90®, Tween 20®, and Transcutol HP® at Smix ratio of 2:1 | |||
| 11.11111 | 59.25926 | 29.62963 | 113.6 ± 1.3 |
| 16.66667 | 55.55556 | 27.77778 | 126.03 ± 1.3 |
| 22.22222 | 51.85185 | 25.92593 | 139.2 ± 0.69 |
| Diagram 2: Capryol 90®, Tween 20®, and Transcutol HP® at a Smix ratio of 5:1 | |||
| 11.11111 | 74.07407 | 14.81481 | 56.17 ± 0.76 |
| 16.66667 | 69.44444 | 13.88889 | 76 ± 1 |
| 22.22222 | 64.81481 | 12.96296 | 94 ± 1 |
| Diagram 3: Capryol 90®, Tween 20®, and Transcutol HP® at a Smix ratio of 6:1 | |||
| 11.11111 | 76.19048 | 12.69841 | 10.1 ± 0.3 |
| 16.66667 | 71.42857 | 11.90476 | 12.13 ± 0.31 |
| 22.22222 | 66.66667 | 11.11111 | 60.27 ± 1.1 |
| Diagram 4: Octanoic acid, Tween 20®, and Transcutol HP® at a Smix ratio of 2:1 | |||
| 11.11111 | 59.25926 | 29.62963 | 125.1 ± 1.1 |
| 16.66667 | 55.55556 | 27.77778 | 140.03 ± 0.95 |
| 22.22222 | 51.85185 | 25.92593 | 145 ± 1 |
| Diagram 5: Octanoic acid, Tween 20®, and Transcutol HP® at a Smix ratio of 3:1 | |||
| 11.11111 | 66.66667 | 22.22222 | 98.13 ± 0.91 |
| 16.66667 | 62.5 | 20.83333 | 112.73 ± 1.14 |
| 22.22222 | 58.33333 | 19.44444 | 144.23 ± 0.93 |
| Diagram 6: Octanoic acid, Tween 20®, and Transcutol HP® at a Smix ratio of 4:1 | |||
| 11.11111 | 71.11111 | 17.77778 | 86.37 ± 1.1 |
| 16.66667 | 66.66667 | 16.66667 | 103.6 ± 0.36 |
| 22.22222 | 62.22222 | 15.55556 | 112.2 ± 0.72 |
| Diagram 7: Octanoic acid, Tween 20®, and Transcutol HP® at a Smix ratio of 5:1. | |||
| 11.11111 | 74.07407 | 14.81481 | 63.67 ± 1.5 |
| 16.66667 | 69.44444 | 13.88889 | 87.1 ± 1.2 |
| 22.22222 | 64.81481 | 12.96296 | 109.07 ± 0.86 |
| Diagram 8: Octanoic acid, Tween 20®, and Transcutol HP® at a Smix ratio of 6:1 | |||
| 11.11111 | 76.19048 | 12.69841 | 10.13 ± 0.15 |
| 16.66667 | 71.42857 | 11.90476 | 13.43 ± 0.35 |
| 22.22222 | 66.66667 | 11.11111 | 97.43 ± 1.09 |
| Blank L-SEDDs | AZM-L-SEDDs | |||||||
|---|---|---|---|---|---|---|---|---|
| DW | ||||||||
| Code | DS (nm) | Đ | ZP (mV) | T% | DS (nm) | Đ | ZP (mV) | T% |
| L-F1(L) | 112.63 ± 1.02 | 0.56 ± 0.05 | −18.5 ± 0.96 | 90.17 ± 0.56 | 112.17 ± 1.26 | 0.41 ± 0.01 | −21.73 ± 0.96 | 91.51 ± 0.19 |
| L-F1(M) | 125.4 ± 1.02 | 0.57 ± 0.002 | −23.3 ± 1.4 | 90.01 ± 0.66 | 124.9 ± 1.65 | 0.47 ± 0.01 | −23.1 ± 0.96 | 90.33 ± 0.31 |
| L-F1(H) | 138.6 ± 0.85 | 0.59 ± 0.009 | −23.03 ± 1.1 | 90.1 ± 0.11 | 141.57 ± 1.1 | 0.52 ± 0.004 | −26.47 ± 0.65 | 90.1 ± 0.1 |
| 0.1 mM HCl (pH = 4) | ||||||||
| L-F1(L) | 116.83 ± 1.3 | 0.56 ± 0.004 | 1.24 ± 1.5 | 94.75 ± 0.11 | 97.6 ± 1.6 | 0.52 ± 0.003 | 0.33 ± 1.7 | 98.2 ± 0.23 |
| L-F1(M) | 112.63 ± 1.4 | 0.54 ± 0.01 | −0.89 ± 0.28 | 94.45 ± 0.11 | 91.23 ± 0.76 | 0.54 ± 0.003 | −0.896 ± 0.35 | 98.04 ± 0.06 |
| L-F1(H) | 93.5 ± 1.3 | 0.62 ± 0.002 | 0.24 ± 0.47 | 94.75 ± 0.05 | 86.6 ± 1.44 | 0.52 ± 0.003 | 0.567 ± 2.2 | 97.99 ± 0.09 |
| SIF (pH = 6.8) | ||||||||
| L-F1(L) | 89.43 ± 0.53 | 0.628 ± 0.005 | −3.23 ± 0.11 | 93.3 ± 0.1 | 166.27 ± 1.96 | 0.824 ± 0.01 | −3.27 ± 0.55 | 97.47 ± 0.08 |
| L-F1(M) | 86.63 ± 1.25 | 0.63 ± 0.01 | −5.86 ± 0.68 | 93.48 ± 0.13 | 154.67 ± 2.1 | 0.83 ± 0.02 | −6.05 ± 1.3 | 95.23 ± 0.12 |
| L-F1(H) | 95.47 ± 1.7 | 0.657 ± 0.04 | −5.28 ± 0.89 | 93.38 ± 0.13 | 148.1 ± 1.6 | 0.68 ± 0.006 | −5.97 ± 1.8 | 94.55 ± 0.48 |
| Blank L-SEDDs | AZM-L-SEDDs | |||||||
|---|---|---|---|---|---|---|---|---|
| DW | ||||||||
| Code | DS (nm) | Đ | ZP (mV) | T% | DS (nm) | Đ | ZP (mV) | T% |
| L-F2(L) | 124.93 ± 1.5 | 0.64 ± 0.003 | −17 ± 0.76 | 89.44 ± 0.26 | 143.1 ± 1.4 | 0.84 ± 0.02 | −16.3 ± 0.87 | 90.47 ± 0.24 |
| L-F2(M) | 139.73 ± 1.31 | 0.59 ± 0.005 | −17.1 ± 0.53 | 88.69 ± 0.08 | 148.43 ± 1.3 | 0.71 ± 0.003 | −17.77 ± 0.96 | 89.03 ± 0.15 |
| L-F2(H) | 143.9 ± 1.64 | 0.68 ± 0.002 | −18.23 ± 0.59 | 88.83 ± 0.32 | 267.8 ± 1.9 | 1 ± 0 | −18.43 ± 0.97 | 77.96 ± 0.48 |
| 0.1 mM HCl (pH = 4) | ||||||||
| L-F2(L) | 104.65 ± 0.5 | 0.45 ± 0.002 | 0.27 ± 1.8 | 94.75 ± 0.1 | 113.7 ± 1.8 | 0.61 ± 0.07 | 0.086 ± 1.1 | 97.17 ± 0.55 |
| L-F2(M) | 113.53 ± 1.8 | 0.47 ± 0.003 | −2.6 ± 0.4 | 93.77 ± 0.04 | 127.6 ± 1.5 | 0.54 ± 0.009 | −0.58 ± 0.83 | 96.56 ± 0.57 |
| L-F2(H) | 130.37 ± 0.6 | 0.55 ± 0.002 | 0.24 ± 0.5 | 90.75 ± 0.07 | 136.7 ± 1.9 | 0.54 ± 0.02 | 0.62 ± 0.8 | 91.76 ± 0.58 |
| SIF (pH = 6.8) | ||||||||
| L-F2(L) | 126.77 ± 1.2 | 1 ± 0 | −3.61 ± 0.5 | 93.03 ± 0.11 | 149.77 ± 1.5 | 0.68 ± 0.01 | −1.53 ± 0.17 | 89.1 ± 0.68 |
| L-F2(M) | 115.6 ± 1.1 | 1 ± 0 | −4.18 ± 0.7 | 93.47 ± 0.12 | 159.47 ± 0.93 | 1 ± 0 | −4.16 ± 1.02 | 86.62 ± 0.29 |
| L-F2(H) | 127.2 ± 1.4 | 1 ± 0 | −4.44 ± 0.7 | 93.35 ± 0.13 | 226.3 ± 2.01 | 0.99 ± 0.006 | −3.88 ± 0.69 | 75.02 ± 0.08 |
| Solidifying Agent | Adsorption Capacity (L-F1(H)): Solidifying Agent | DS (nm) | Đ | ZP (mV) |
|---|---|---|---|---|
| Calcium carbonate | (1:4) | 1863.67 ± 10.3 | 1 ± 0 | −14.47 ± 1.001 |
| Aerosil 200® | (2:1) | 156.67 ± 1.5 | 0.62 ± 0.004 | −21.7 ± 1.4 |
| Lactose | (1:3) | 384.7 ± 4.04 | 1 ± 0 | −40.33 ± 1.07 |
| Mannitol | (1:2) | 845.63 ± 6.2 | 1 ± 0 | −23.4 ± 1.3 |
| Blank S-SEDDs (S-F1(H)) | AZM S-SEDDs (S-F1(H)) | ||||
|---|---|---|---|---|---|
| DW | |||||
| DS (nm) | Đ | ZP (mV) | DS (nm) | Đ | ZP (mV) |
| 157.1 ± 1.85 | 0.61 ± 0.01 | −22.67 ± 2.3 | 155.3 ± 1.91 | 0.62 ± 0.03 | −19.43 ± 0.15 |
| 0.1 mM HCl (pH = 4) | |||||
| 139.03 ± 1.8 | 1 ± 0 | 2.1 ± 0.2 | 136.6 ± 1.9 | 0.73 ± 0.01 | 0.88 ± 0.02 |
| SIF (pH = 6.8) | |||||
| 195.32 ± 1.9 | 1 ± 0 | −5.93 ± 1.1 | 191.5 ± 1.4 | 1 ± 0 | −7.43 ± 0.81 |
| Parameters | Time (Months) | ||||
|---|---|---|---|---|---|
| 0 | 0.5 | 1 | 2 | 3 | |
| Refrigerator (4 ± 2 °C) | |||||
| Physical stability | |||||
| DS (nm) | 141.23 ± 0.38 | 141.6 ± 0.99 | 142 ± 1.15 | 146.87 ± 1.3 | 149.97 ± 1.16 |
| Đ | 0.528 ± 0.006 | 0.522 ± 0.01 | 0.534 ± 0.005 | 0.541 ± 0.009 | 0.56 ± 0.01 |
| ZP (mV) | −26.78 ± 1.056 | −27.43 ± 1.23 | −26.73 ± 1.07 | −25.69 ± 0.56 | −26.3 ± 0.5 |
| Chemical stability | |||||
| DC (%) | 99.843 ± 0.353 | 99.79 ± 0.45 | 99.66 ± 0.162 | 99.37 ± 0.12 | 99.43 ± 0.22 |
| Room condition (25 ± 2 °C/60 ± 5% RH) | |||||
| Physical stability | |||||
| DS (nm) | 141.23 ± 0.38 | 142.03 ± 0.42 | 143.7 ± 1.31 | 148.23 ± 1.23 | 156.83 ± 1.5 |
| Đ | 0.528 ± 0.006 | 0.524 ± 0.04 | 0.53 ± 0.002 | 0.54 ± 0.003 | 0.552 ± 0.01 |
| ZP (mV) | −26.78 ± 1.056 | −26.7 ± 1.1 | −26.37 ± 0.42 | −25.73 ± 51 | −25.47 ± 0.65 |
| Chemical stability | |||||
| DC (%) | 99.843 ± 0.353 | 99.65 ± 0.2 | 99.42 ± 0.14 | 99.241 ± 0.17 | 99.11 ± 0.12 |
| Humidity chamber (40 ± 2 °C/75 ± 5% RH) | |||||
| Physical stability | |||||
| DS (nm) | 141.23 ± 0.38 | 142.03 ± 1.16 | 147.55 ± 1.05 | 149.57 ± 1.37 | 166.63 ± 1.56 |
| Đ | 0.528 ± 0.006 | 0.529 ± 0.007 | 0.53 ± 0.003 | 0.542 ± 0.007 | 0.613 ± 0.05 |
| ZP (mV) | −26.78 ± 1.056 | −26.93 ± 0.15 | −25.97 ± 0.7 | −25.67 ± 0.32 | −23.3 ± 0.56 |
| Chemical stability | |||||
| DC (%) | 99.843 ± 0.353 | 99.74 ± 0.13 | 99.12 ± 0.08 | 98.95 ± 0.1 | 98.58 ± 0.43 |
| Parameters | Time (Months) | ||||
|---|---|---|---|---|---|
| 0 | 0.5 | 1 | 2 | 3 | |
| Refrigerator (4 ± 3 °C) | |||||
| Physical stability | |||||
| DS (nm) | 155.93 ± 1.39 | 155.13 ± 1.35 | 156.27 ± 1.19 | 157.33 ± 0.59 | 159.2 ± 0.78 |
| Đ | 0.64 ± 0.04 | 0.65 ± 0.015 | 0.64 ± 0.015 | 0.66 ± 0.021 | 0.66 ± 0.01 |
| ZP (mV) | −19.28 ± 0.45 | −19.23 ± 0.32 | −19.27 ± 0.45 | −18.97 ± 0.75 | −18.49 ± 0.6 |
| Chemical stability | |||||
| DC (%) | 98.599 ± 0.32 | 98.52 ± 0.38 | 98.41 ± 0.57 | 98.37 ± 0.2 | 98.09 ± 0.5 |
| Room condition (25 ± 2 °C/60 ± 5% RH) | |||||
| Physical stability | |||||
| DS (nm) | 155.93 ± 1.39 | 155.8 ± 1.67 | 155.6 ± 1.25 | 158.3 ± 1.25 | 161.55 ± 1.1 |
| Đ | 0.64 ± 0.04 | 0.627 ± 0.025 | 0.65 ± 0.02 | 0.647 ± 0.03 | 0.68 ± 0.006 |
| ZP (mV) | −19.28 ± 0.45 | −19.15 ± 0.53 | −18.97 ± 0.31 | −18.77 ± 0.31 | −16.27 ± 0.45 |
| Chemical stability | |||||
| DC (%) | 98.599 ± 0.32 | 98.42 ± 0.12 | 98.34 ± 0.36 | 98.06 ± 0.33 | 97.92 ± 0.43 |
| Humidity chamber (40 ± 2 °C/75 ± 5% RH) | |||||
| Physical stability | |||||
| DS (nm) | 155.93 ± 1.39 | 156.4 ± 0.6 | 158 ± 1 | 162.37 ± 1.1 | 164.1 ± 1.02 |
| Đ | 0.64 ± 0.04 | 0.65 ± 0.01 | 0.653 ± 0.015 | 0.657 ± 0.015 | 0.603 ± 0.006 |
| ZP (mV) | −19.28 ± 0.45 | −19.03 ± 0.4 | −18.9 ± 0.3 | −18.63 ± 0.42 | −15.3 ± 0.56 |
| Chemical stability | |||||
| DC (%) | 98.599 ± 0.32 | 98.44 ± 0.28 | 98.014 ± 0.4 | 97.86 ± 0.37 | 97.48 ± 0.58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abou Assi, R.; M. Abdulbaqi, I.; Seok Ming, T.; Siok Yee, C.; A. Wahab, H.; Asif, S.M.; Darwis, Y. Liquid and Solid Self-Emulsifying Drug Delivery Systems (SEDDs) as Carriers for the Oral Delivery of Azithromycin: Optimization, In Vitro Characterization and Stability Assessment. Pharmaceutics 2020, 12, 1052. https://doi.org/10.3390/pharmaceutics12111052
Abou Assi R, M. Abdulbaqi I, Seok Ming T, Siok Yee C, A. Wahab H, Asif SM, Darwis Y. Liquid and Solid Self-Emulsifying Drug Delivery Systems (SEDDs) as Carriers for the Oral Delivery of Azithromycin: Optimization, In Vitro Characterization and Stability Assessment. Pharmaceutics. 2020; 12(11):1052. https://doi.org/10.3390/pharmaceutics12111052
Chicago/Turabian StyleAbou Assi, Reem, Ibrahim M. Abdulbaqi, Toh Seok Ming, Chan Siok Yee, Habibah A. Wahab, Shaik Mohammed Asif, and Yusrida Darwis. 2020. "Liquid and Solid Self-Emulsifying Drug Delivery Systems (SEDDs) as Carriers for the Oral Delivery of Azithromycin: Optimization, In Vitro Characterization and Stability Assessment" Pharmaceutics 12, no. 11: 1052. https://doi.org/10.3390/pharmaceutics12111052
APA StyleAbou Assi, R., M. Abdulbaqi, I., Seok Ming, T., Siok Yee, C., A. Wahab, H., Asif, S. M., & Darwis, Y. (2020). Liquid and Solid Self-Emulsifying Drug Delivery Systems (SEDDs) as Carriers for the Oral Delivery of Azithromycin: Optimization, In Vitro Characterization and Stability Assessment. Pharmaceutics, 12(11), 1052. https://doi.org/10.3390/pharmaceutics12111052