Timeline of Translational Formulation Technologies for Cancer Therapy: Successes, Failures, and Lessons Learned Therefrom

 and
and
Abstract
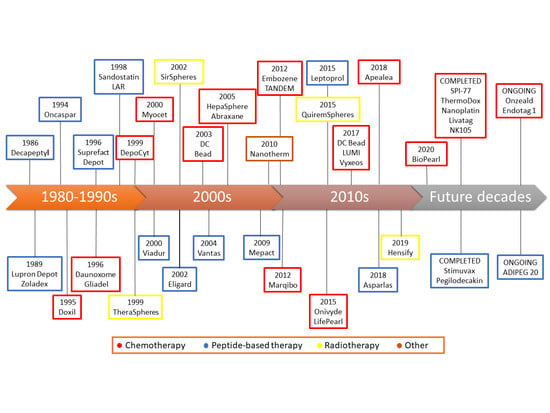
1. Introduction
2. 1990s: The First Steps
2.1. Authorized Formulations for Peptide-Based Therapy
2.2. Authorized Formulations for Chemotherapy
2.3. Authorized Formulations for Radiotherapy
3. 2000s: The Field Matures
3.1. Authorized Formulations for Peptide-Based Therapy
3.2. Authorized Formulations for Chemotherapy
3.3. Authorized Formulations for Radiotherapy
4. 2010s: The End of the Beginning
4.1. Authorized Formulations for Peptide-Based Therapy
4.2. Authorized Formulations for Chemotherapy
4.3. Authorized Formulations for Radiotherapy
4.4. Other Authorized Products
5. The Decades to Come
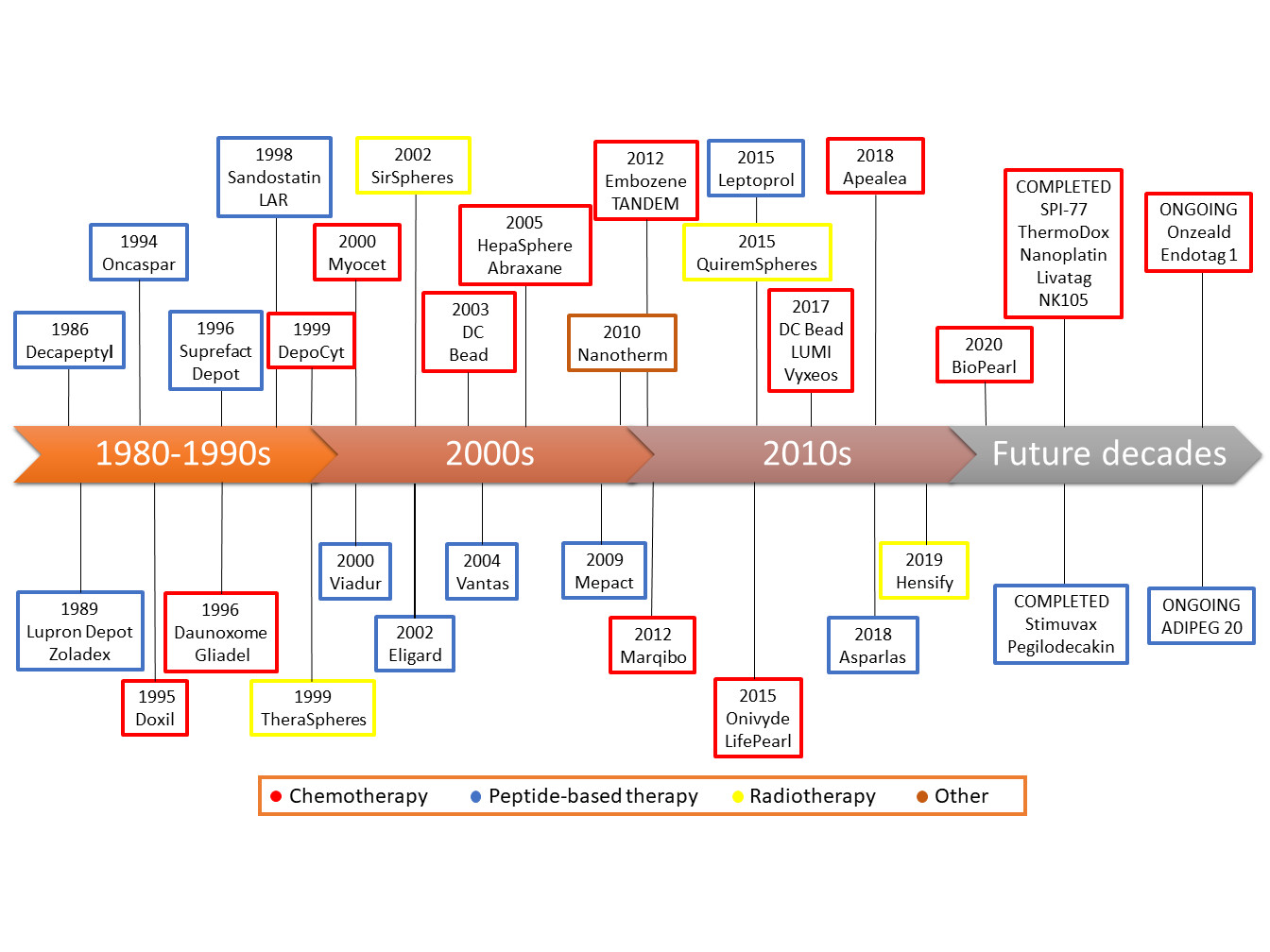
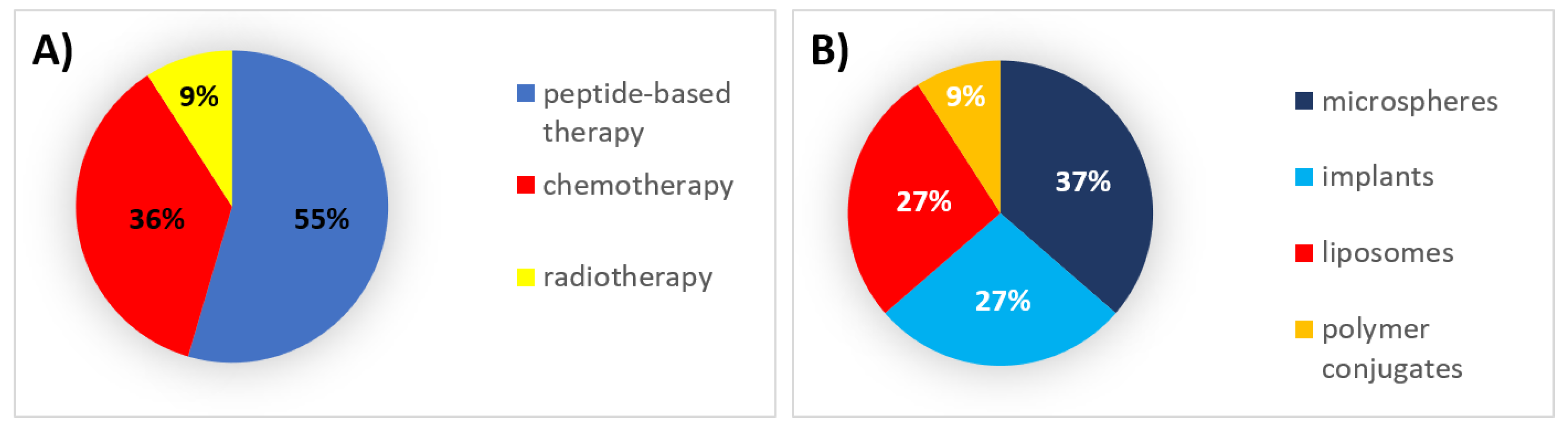
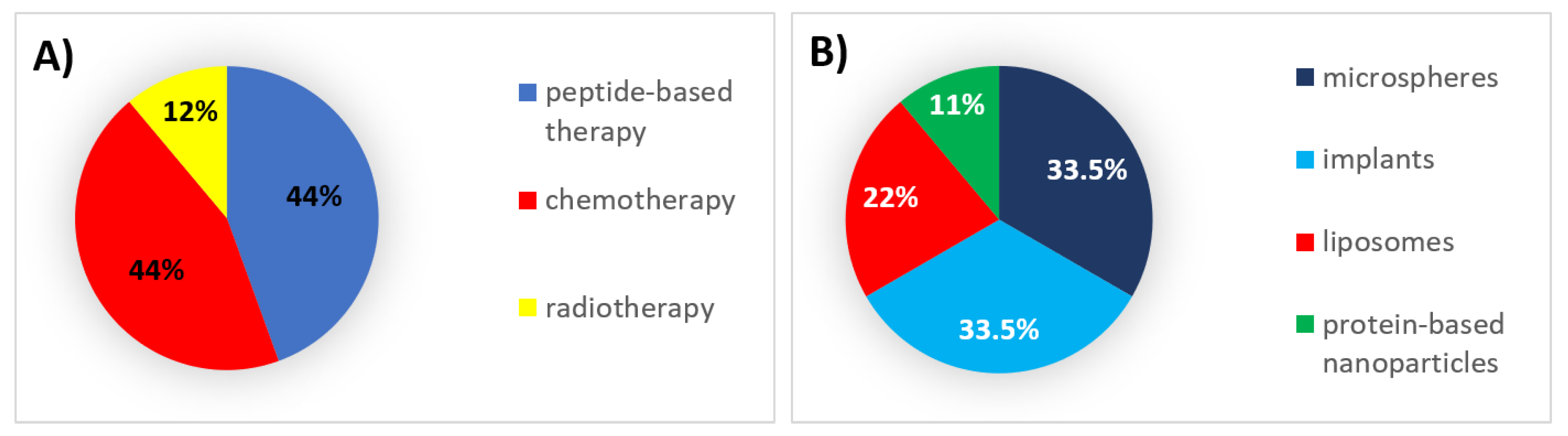
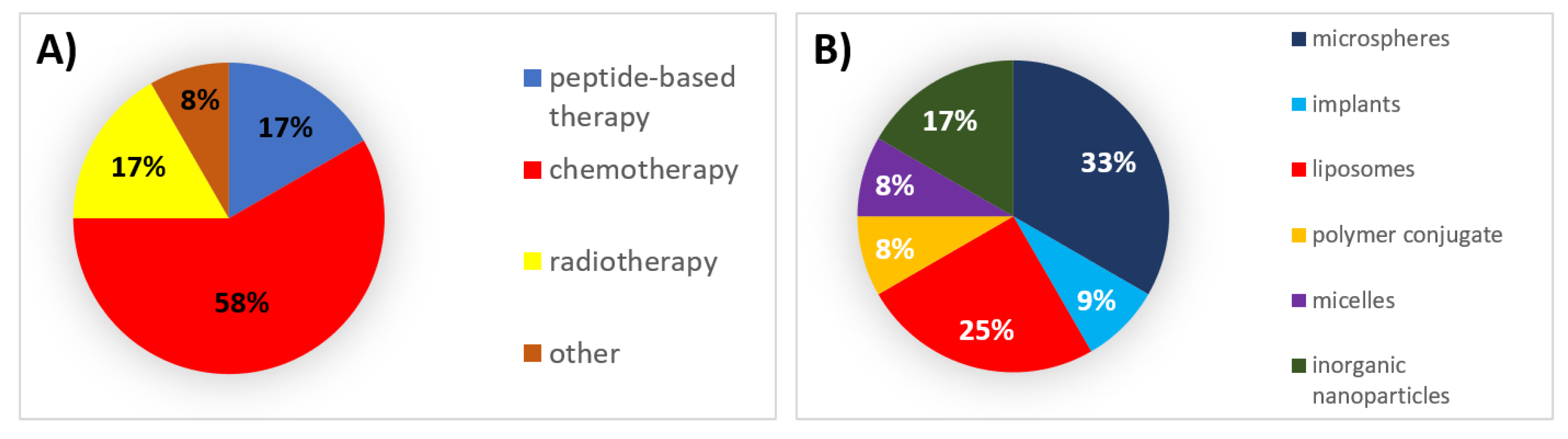
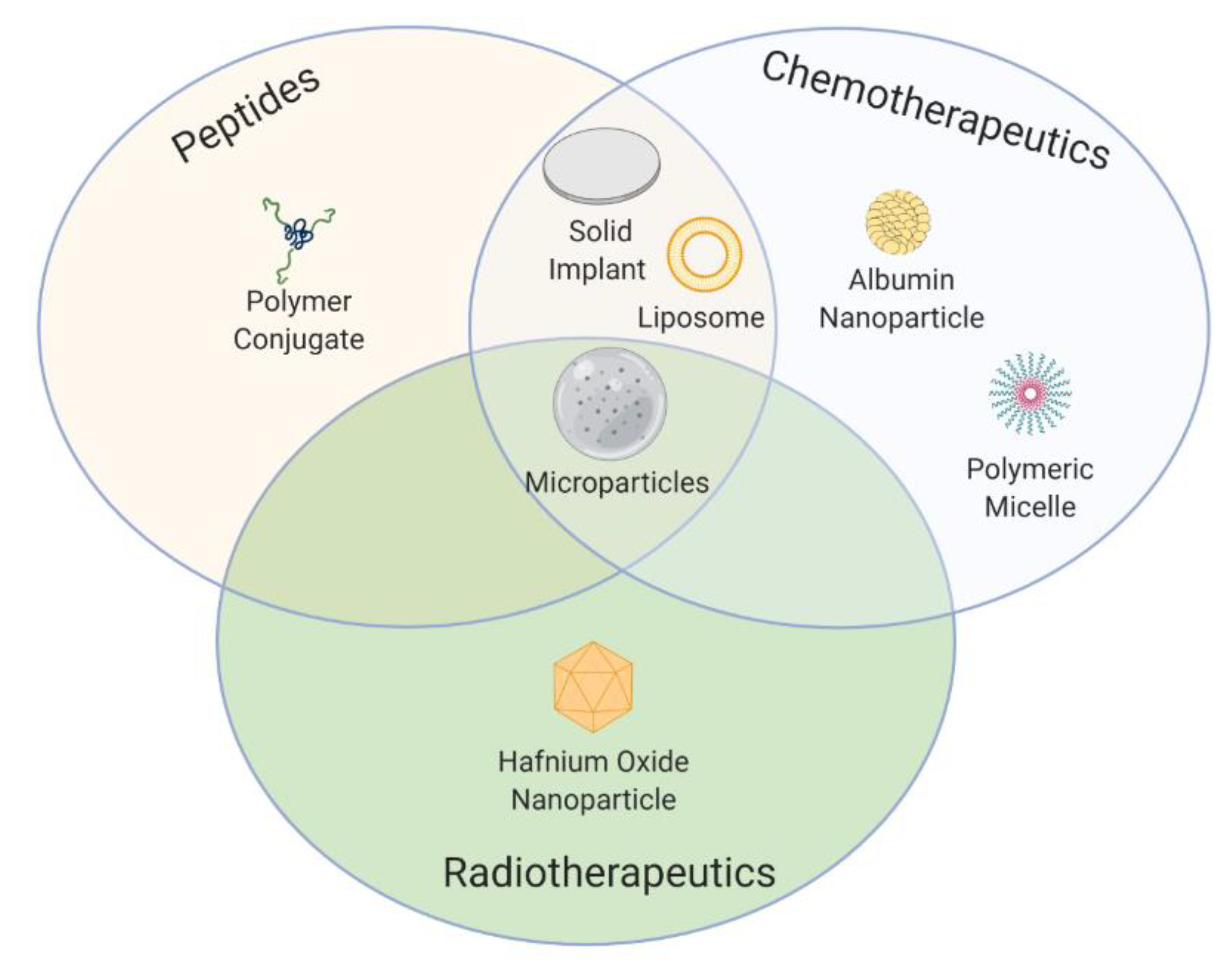
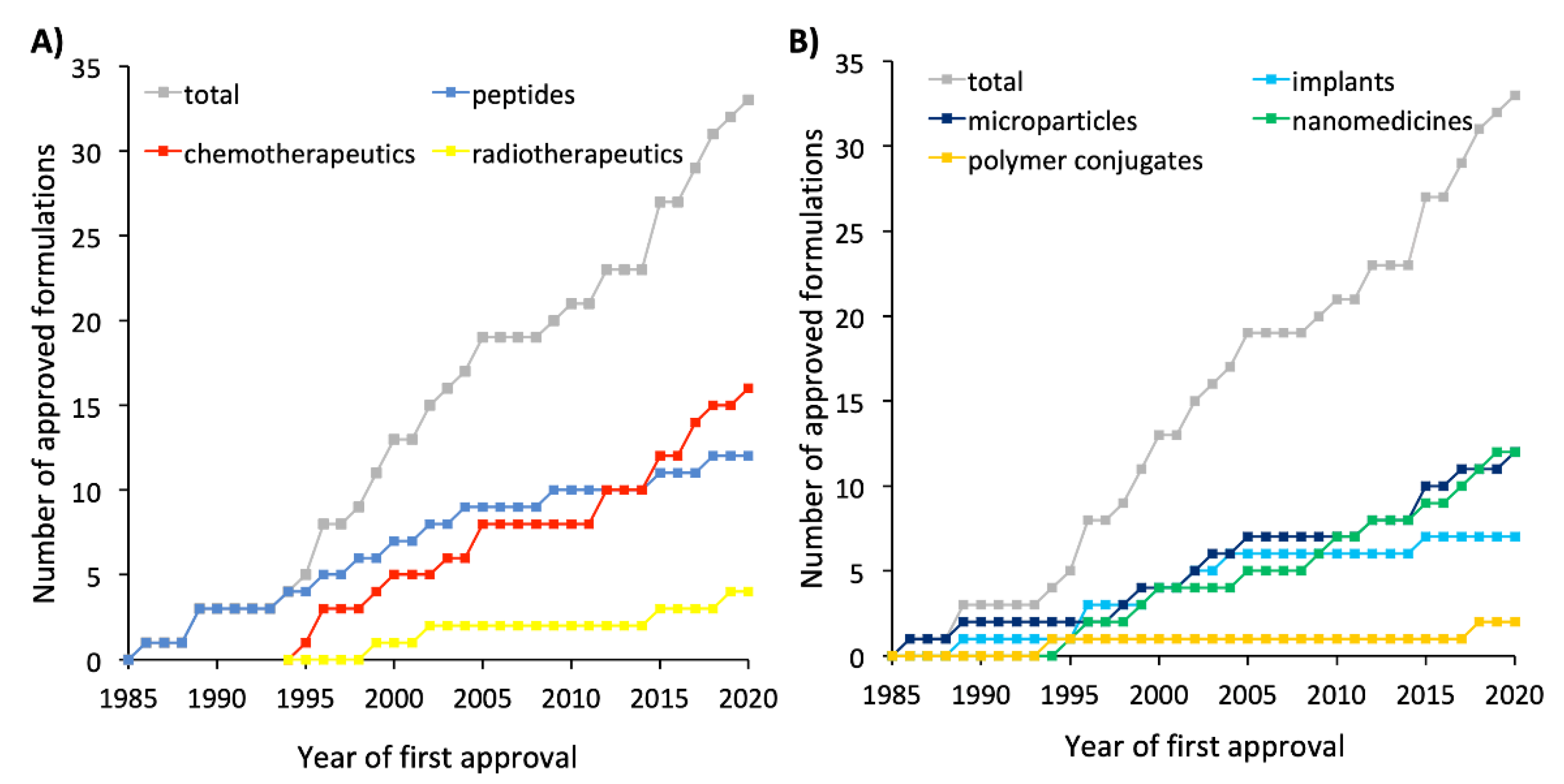
6. Successes, Failures, and Lessons Learned
Funding
Conflicts of Interest
Abbreviations
| ALL | acute lymphoblastic leukemia |
| AML | acute myeloid leukemia |
| CE | Conformité Européenne |
| DEM | drug-eluting microspheres |
| DEM-TACE | drug-eluting beads in transarterial chemoembolization |
| EPR | enhance permeability and retention effect |
| GnRH | gonadotropin-releasing hormone |
| HCC | hepatocellular carcinoma |
| HER-2 | human epidermal growth factor receptor 2 |
| HfO2 | hafnium oxide |
| IA | intra-arterial |
| IC | intracranial |
| IL-10 | interleukin 10 |
| IM | intramuscular |
| IT | intratumoral |
| ITh | intrathecal |
| IV | intravenous |
| MNP | magnetic nanoparticles |
| mPEG | monomethoxy-polyethylene glycol |
| MRI | magnetic resonance imaging |
| Nab | nanoparticle albumin-bound |
| PDT | photodynamic therapy |
| PEG | polyethylene glycol |
| PLA | poly(lactic acid) |
| PLGA | lactic-co-glycolic acid |
| PTT | photothermal therapy |
| RES | reticuloendothelial system |
| RFA | radio-frequency ablation |
| SC | subcutaneous |
| SIRT | selective internal radiation therapy |
| SPECT | single-photon emission computed tomography |
| SPECT/CT | single-photon emission computed tomography/computed tomography |
| SPIONs | superparamagnetic iron oxide nanoparticles |
| TACE | transarterial chemoembolization |
| TTP | time to progression |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T.; Chu, E. A History of Cancer Chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed]
- Arruebo, M.; Vilaboa, N.; Sáez-Gutierrez, B.; Lambea, J.; Tres, A.; Valladares, M.; González-Fernández, Á. Assessment of the evolution of cancer treatment therapies. Cancers 2011, 3, 3279–3330. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.F.; Jie, M.M.; Li, B.S.; Hu, C.J.; Xie, R.; Tang, B.; Yang, S.M. Peptide-Based Treatment: A Promising Cancer Therapy. J. Immunol. Res. 2015, 2015, 761820. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.; Layton, J.; Lewis, B.; Sartor, O. Next Generation of Androgen Deprivation Therapy Combined With Radiotherapy for N0 M0 Prostate Cancer. Cancer J. 2020, 26, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Thundimadathil, J. Cancer Treatment Using Peptides: Current Therapies and Future Prospects. J. Amino Acids 2012, 2012, 1–13. [Google Scholar] [CrossRef]
- Zafar, S.; Beg, S.; Panda, S.K.; Rahman, M.; Alharbi, K.S.; Jain, G.K.; Ahmad, F.J. Novel therapeutic interventions in cancer treatment using protein and peptide-based targeted smart systems. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Marqus, S.; Pirogova, E.; Piva, T.J. Evaluation of the use of therapeutic peptides for cancer treatment. J. Biomed. Sci. 2017, 24, 21. [Google Scholar] [CrossRef]
- Sagnella, S.M.; McCarroll, J.A.; Kavallaris, M. Drug delivery: Beyond active tumour targeting. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1131–1137. [Google Scholar] [CrossRef]
- Kishan, A.U.; Cook, R.R.; Ciezki, J.P.; Ross, A.E.; Pomerantz, M.M.; Nguyen, P.L.; Shaikh, T.; Tran, P.T.; Sandler, K.A.; Stock, R.G.; et al. Radical prostatectomy, external beam radiotherapy, or external beam radiotherapy with brachytherapy boost and disease progression and mortality in patients with gleason score 9-10 prostate cancer. JAMA J. Am. Med. Assoc. 2018, 319, 896–905. [Google Scholar] [CrossRef] [PubMed]
- De Ruysscher, D.; Niedermann, G.; Burnet, N.G.; Siva, S.; Lee, A.W.M.; Hegi-Johnson, F. Radiotherapy toxicity. Nat. Rev. Dis. Prim. 2019, 5, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Faisant, N.; Akiki, J.; Siepmann, F.; Benoit, J.P.; Siepmann, J. Effects of the type of release medium on drug release from PLGA-based microparticles: Experiment and theory. Int. J. Pharm. 2006, 314, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Kempe, S.; Mäder, K. In situ forming implants—An attractive formulation principle for parenteral depot formulations. J. Control. Release 2012, 161, 668–679. [Google Scholar] [CrossRef]
- Hrkach, J.; Langer, R. From micro to nano: Evolution and impact of drug delivery in treating disease. Drug Deliv. Transl. Res. 2020, 10. [Google Scholar] [CrossRef]
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C 2019, 98, 1252–1276. [Google Scholar] [CrossRef]
- Van der Meel, R.; Vehmeijer, L.J.C.; Kok, R.J.; Storm, G.; van Gaal, E.V.B. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: Current status. Adv. Drug Deliv. Rev. 2013, 65, 1284–1298. [Google Scholar] [CrossRef]
- Aparicio-Blanco, J.; Sanz-Arriazu, L.; Lorenzoni, R.; Blanco-Prieto, M.J. Glioblastoma chemotherapeutic agents used in the clinical setting and in clinical trials: Nanomedicine approaches to improve their efficacy. Int. J. Pharm. 2020, 581, 119283. [Google Scholar] [CrossRef]
- Chew, S.A.; Danti, S. Biomaterial-Based Implantable Devices for Cancer Therapy. Adv. Healthc. Mater. 2017, 6, 1600766. [Google Scholar] [CrossRef]
- Bazak, R.; Houri, M.; El Achy, S.; Hussein, W.; Refaat, T. Passive targeting of nanoparticles to cancer: A comprehensive review of the literature. Mol. Clin. Oncol. 2014, 2, 904–908. [Google Scholar] [CrossRef]
- Taha, M.S.; Padmakumar, S.; Singh, A.; Amiji, M.M. Critical quality attributes in the development of therapeutic nanomedicines toward clinical translation. Drug Deliv. Transl. Res. 2020, 10, 766–790. [Google Scholar] [CrossRef]
- Dhaliwal, A.; Zheng, G. Improving accessibility of EPR-insensitive tumor phenotypes using EPR-adaptive strategies: Designing a new perspective in nanomedicine delivery. Theranostics 2019, 9, 8091–8108. [Google Scholar] [CrossRef]
- Jasim, A.; Abdelghany, S.; Greish, K. Current Update on the Role of Enhanced Permeability and Retention Effect in Cancer Nanomedicine. In Nanotechnology-Based Approaches for Targeting and Delivery of Drugs and Genes; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 62–109. ISBN 9780128097182. [Google Scholar]
- Zhang, C.; Wu, L.; Tao, A.; Bera, H.; Tang, X.; Cun, D.; Yang, M. Formulation and in vitro characterization of long-acting PLGA injectable microspheres encapsulating a peptide analog of LHRH. J. Mater. Sci. Technol. 2020. [Google Scholar] [CrossRef]
- Shi, N.-Q.; Zhou, J.; Walker, J.; Li, L.; Hong, J.K.Y.; Olsen, K.F.; Tang, J.; Ackermann, R.; Wang, Y.; Qin, B.; et al. Microencapsulation of luteinizing hormone-releasing hormone agonist in poly (lactic-co-glycolic acid) microspheres by spray-drying. J. Control. Release 2020, 321, 756–772. [Google Scholar] [CrossRef]
- Jain, A.; Kunduru, K.R.; Basu, A.; Mizrahi, B.; Domb, A.J.; Khan, W. Injectable formulations of poly(lactic acid) and its copolymers in clinical use. Adv. Drug Deliv. Rev. 2016, 107, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Kim, S.; Park, K. Issues in long-term protein delivery using biodegradable microparticles. J. Control. Release 2010, 146, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Pandita, D.; Kumar, S.; Lather, V. Hybrid poly(lactic-co-glycolic acid) nanoparticles: Design and delivery prospectives. Drug Discov. Today 2015, 20, 95–104. [Google Scholar] [CrossRef]
- Park, K.; Jung, G.Y.; Kim, M.K.; Park, M.S.; Shin, Y.K.; Hwang, J.K.; Yuk, S.H. Triptorelin acetate-loaded poly(lactide-co-glycolide) (PLGA) microspheres for controlled drug delivery. Macromol. Res. 2012, 20, 847–851. [Google Scholar] [CrossRef]
- Chen, L.; Ahmed, A.M.Q.; Deng, Y.; Cao, D.; Du, H.; Cui, J.; Lee, B.-J.; Cao, Q. Novel triptorelin acetate-loaded microspheres prepared by a liquid/oil/oil method with high encapsulation efficiency and low initial burst release. J. Drug Deliv. Sci. Technol. 2019, 54, 101390. [Google Scholar] [CrossRef]
- Skidmore, S.; Hadar, J.; Garner, J.; Park, H.; Park, K.; Wang, Y.; Jiang, X. (Jeff) Complex sameness: Separation of mixed poly(lactide-co-glycolide)s based on the lactide:glycolide ratio. J. Control. Release 2019, 300, 174–184. [Google Scholar] [CrossRef]
- Wechsel, H.W.; Zerbib, M.; Pagano, F.; Coptcoat, M.J. Randomized Open Labelled Comparative Study of the Efficacy, Safety and Tolerability of Leuprorelin Acetate 1M and 3M Depot in Patients with Advanced Prostatic Cancer. Eur. Urol. 1996, 30, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Skidmore, S.; Hadar, J.; Garner, J.; Park, H.; Otte, A.; Soh, B.K.; Yoon, G.; Yu, D.; Yun, Y.; et al. Injectable, long-acting PLGA formulations: Analyzing PLGA and understanding microparticle formation. J. Control. Release 2019, 304, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.-A.; Syed, Y.Y.; Keam, S.J. Pegaspargase: A Review in Acute Lymphoblastic Leukaemia. Drugs 2019, 79, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Meneguetti, G.P.; Santos, J.H.P.M.; Obreque, K.M.T.; Barbosa, C.M.V.; Monteiro, G.; Farsky, S.H.P.; Marim de Oliveira, A.; Angeli, C.B.; Palmisano, G.; Ventura, S.P.M.; et al. Novel site-specific PEGylated L-asparaginase. PLoS ONE 2019, 14, e0211951. [Google Scholar] [CrossRef]
- van der Meel, R.; Sulheim, E.; Shi, Y.; Kiessling, F.; Mulder, W.J.M.; Lammers, T. Smart cancer nanomedicine. Nat. Nanotechnol. 2019, 14, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Parrish, K.; Sarkaria, J.; Elmquist, W. Improving drug delivery to primary and metastatic brain tumors: Strategies to overcome the blood-brain barrier. Clin. Pharmacol. Ther. 2015, 97, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Shapira-Furman, T.; Serra, R.; Gorelick, N.; Doglioli, M.; Tagliaferri, V.; Cecia, A.; Peters, M.; Kumar, A.; Rottenberg, Y.; Langer, R.; et al. Biodegradable wafers releasing Temozolomide and Carmustine for the treatment of brain cancer. J. Control. Release 2019, 295, 93–101. [Google Scholar] [CrossRef]
- Brem, H.; Piantadosi, S.; Burger, P.; Walker, M.; Selker, R.; Vick, N.; Black, K.; Sisti, M.; Brem, S.; Mohr, G.; et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. Lancet 1995, 345, 1008–1012. [Google Scholar] [CrossRef]
- Xiao, W.; Gao, H. The impact of protein corona on the behavior and targeting capability of nanoparticle-based delivery system. Int. J. Pharm. 2018, 552, 328–339. [Google Scholar] [CrossRef]
- Hadjidemetriou, M.; Kostarelos, K. Evolution of the nanoparticle corona. Nat. Nanotechnol. 2017, 12, 288–290. [Google Scholar] [CrossRef]
- Caracciolo, G.; Farokhzad, O.C.; Mahmoudi, M. Biological Identity of Nanoparticles In Vivo: Clinical Implications of the Protein Corona. Trends Biotechnol. 2017, 35, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, M.; Mignani, S.; Rodrigues, J.; Tomás, H. A glance over doxorubicin based-nanotherapeutics: From proof-of-concept studies to solutions in the market. J. Control. Release 2020, 317, 347–374. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Shmeeda, H.; Barenholz, Y. Pharmacokinetics of Pegylated Liposomal Doxorubicin. Clin. Pharmacokinet. 2003, 42, 419–436. [Google Scholar] [CrossRef]
- Blank, N.; Laskov, I.; Kessous, R.; Kogan, L.; Lau, S.; Sebag, I.A.; Gotlieb, W.H.; Rudski, L. Absence of cardiotoxicity with prolonged treatment and large accumulating doses of pegylated liposomal doxorubicin. Cancer Chemother. Pharmacol. 2017, 80, 737–743. [Google Scholar] [CrossRef]
- Gabizon, A.; Shmeeda, H.; Grenader, T. Pharmacological basis of pegylated liposomal doxorubicin: Impact on cancer therapy. Eur. J. Pharm. Sci. 2012, 45, 388–398. [Google Scholar] [CrossRef]
- Bellott, R.; Auvrignon, A.; Leblanc, T.; Pérel, Y.; Gandemer, V.; Bertrand, Y.; Méchinaud, F.; Bellenger, P.; Vernois, J.; Leverger, G.; et al. Pharmacokinetics of liposomal daunorubicin (DaunoXome) during a phase I-II study in children with relapsed acute lymphoblastic leukaemia. Cancer Chemother. Pharmacol. 2001, 47, 15–21. [Google Scholar] [CrossRef]
- Yeh, M.-K.; Chang, H.-I.; Cheng, M.-Y. Clinical development of liposome based drugs: Formulation, characterization, and therapeutic efficacy. Int. J. Nanomed. 2011, 7, 49. [Google Scholar] [CrossRef]
- Wolfram, J.; Ferrari, M. Clinical cancer nanomedicine. Nano Today 2019, 25, 85–98. [Google Scholar] [CrossRef]
- Mantripragada, S. A lipid based depot (DepoFoam® technology) for sustained release drug delivery. Prog. Lipid Res. 2002, 41, 392–406. [Google Scholar] [CrossRef]
- Gallio, E.; Richetta, E.; Finessi, M.; Stasi, M.; Pellerito, R.E.; Bisi, G.; Ropolo, R. Calculation of tumour and normal tissue biological effective dose in 90 Y liver radioembolization with different dosimetric methods. Phys. Med. 2016, 32, 1738–1744. [Google Scholar] [CrossRef] [PubMed]
- Bastiaannet, R.; van Roekel, C.; Smits, M.L.J.; Elias, S.G.; van Amsterdam, W.A.C.; Doan, D.; Prince, J.F.; Bruijnen, R.C.G.; de Jong, H.W.A.M.; Lam, M.G.E.H. First Evidence for a Dose-Response Relationship in Patients Treated with 166Ho Radioembolization: A Prospective Study. J. Nucl. Med. 2020, 61, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.W.; Alanis, L.; Cho, S.-K.; Saab, S. Yttrium-90 Selective Internal Radiation Therapy with Glass Microspheres for Hepatocellular Carcinoma: Current and Updated Literature Review. Korean J. Radiol. 2016, 17, 472–488. [Google Scholar] [CrossRef] [PubMed]
- Mantry, P.; Thompson, M.; Khanna, P.; Acharya, P.; Shahin, I. Prolonged Survival With Radioembolization Using Theraspheres in Unresectable HCC. Am. J. Gastroenterol. 2019, 114, S568. [Google Scholar] [CrossRef]
- Memon, K.; Lewandowski, R.J.; Kulik, L.; Riaz, A.; Mulcahy, M.F.; Salem, R. Radioembolization for Primary and Metastatic Liver Cancer. Semin. Radiat. Oncol. 2011, 21, 294–302. [Google Scholar] [CrossRef]
- Desai, N. Nanoparticle Albumin-Bound Paclitaxel (Abraxane®). In Albumin in Medicine; Springer: Singapore, 2016; pp. 101–119. ISBN 9789811021169. [Google Scholar]
- Bernabeu, E.; Cagel, M.; Lagomarsino, E.; Moretton, M.; Chiappetta, D.A. Paclitaxel: What has been done and the challenges remain ahead. Int. J. Pharm. 2017, 526, 474–495. [Google Scholar] [CrossRef]
- Revel-Mouroz, P.; Otal, P.; Jaffro, M.; Petermann, A.; Meyrignac, O.; Rabinel, P.; Mokrane, F.-Z. Other non-surgical treatments for liver cancer. Rep. Pract. Oncol. Radiother. 2017, 22, 181–192. [Google Scholar] [CrossRef][Green Version]
- Chew, S.A.; Moscato, S.; George, S.; Azimi, B.; Danti, S. Liver Cancer: Current and Future Trends Using Biomaterials. Cancers 2019, 11, 2026. [Google Scholar] [CrossRef]
- Liu, Y.-S.; Ou, M.-C.; Tsai, Y.-S.; Lin, X.-Z.; Wang, C.-K.; Tsai, H.-M.; Chuang, M.-T. Transarterial Chemoembolization Using Gelatin Sponges or Microspheres Plus Lipiodol-Doxorubicin versus Doxorubicin-Loaded Beads for the Treatment of Hepatocellular Carcinoma. Korean J. Radiol. 2015, 16, 125–132. [Google Scholar] [CrossRef][Green Version]
- Wright, J.C.; Tao Leonard, S.; Stevenson, C.L.; Beck, J.C.; Chen, G.; Jao, R.M.; Johnson, P.A.; Leonard, J.; Skowronski, R.J. An in vivo/in vitro comparison with a leuprolide osmotic implant for the treatment of prostate cancer. J. Control. Release 2001, 75, 1–10. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef]
- Shore, N. Introducing Vantas: The First Once-Yearly Luteinising Hormone-Releasing Hormone Agonist. Eur. Urol. Suppl. 2010, 9, 701–705. [Google Scholar] [CrossRef]
- Wex, J.; Sidhu, M.; Odeyemi, I.; Abou-Setta, A.M.; Retsa, P.; Tombal, B. Leuprolide acetate 1-, 3- and 6-monthly depot formulations in androgen deprivation therapy for prostate cancer in nine European countries: Evidence review and economic evaluation. Clin. Outcomes Res. 2013, 5, 257. [Google Scholar] [CrossRef] [PubMed]
- Saltzstein, D.; Shore, N.D.; Moul, J.W.; Chu, F.; Concepcion, R.; de la Motte, S.; McLane, J.A.; Atkinson, S.; Yang, A.; Crawford, E.D. Pharmacokinetic and pharmacodynamic comparison of subcutaneous versus intramuscular leuprolide acetate formulations in male subjects. Ther. Adv. Urol. 2018, 10, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.J.; Kleinerman, E.S.; Krailo, M.D.; Chen, Z.; Betcher, D.L.; Healey, J.H.; Conrad, E.U.; Nieder, M.L.; Weiner, M.A.; Wells, R.J.; et al. Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma. Cancer 2009, 115, 5339–5348. [Google Scholar] [CrossRef]
- Bun, S.; Yunokawa, M.; Tamaki, Y.; Shimomura, A.; Shimoi, T.; Kodaira, M.; Shimizu, C.; Yonemori, K.; Fujiwara, Y.; Makino, Y.; et al. Symptom management: The utility of regional cooling for hand-foot syndrome induced by pegylated liposomal doxorubicin in ovarian cancer. Support. Care Cancer 2018, 26, 2161–2166. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, U.; Irfan Bukhari, N.; Ovais, M.; Abass, N.; Hussain, K.; Raza, A. Advances in nano-delivery systems for doxorubicin: An updated insight. J. Drug Target. 2018, 26, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Li, Y.; He, M.; Zhang, H.; Yuan, H.; Johnson, M.; Palmisano, M.; Zhou, S.; Sun, D. Distinct biodistribution of doxorubicin and the altered dispositions mediated by different liposomal formulations. Int. J. Pharm. 2017, 519, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Batist, G.; Harris, L.; Azarnia, N.; Lee, L.W.; Daza-Ramirez, P. Improved anti-tumor response rate with decreased cardiotoxicity of non-pegylated liposomal doxorubicin compared with conventional doxorubicin in first-line treatment of metastatic breast cancer in patients who had received prior adjuvant doxorubicin: Resu. Anticancer. Drugs 2006, 17, 587–595. [Google Scholar] [CrossRef]
- Yardley, D.A. nab-Paclitaxel mechanisms of action and delivery. J. Control. Release 2013, 170, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J. Albumin-bound paclitaxel: A next-generation taxane. Expert Opin. Pharmacother. 2006, 7, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.R.; Dahut, W.L.; Scripture, C.D.; Jones, J.; Aragon-Ching, J.B.; Desai, N.; Hawkins, M.J.; Sparreboom, A.; Figg, W.D. Randomized Crossover Pharmacokinetic Study of Solvent-Based Paclitaxel and nab-Paclitaxel. Clin. Cancer Res. 2008, 14, 4200–4205. [Google Scholar] [CrossRef]
- Barkat, M.A.; Beg, S.; Pottoo, F.H.; Ahmad, F.J. Nanopaclitaxel therapy: An evidence based review on the battle for next-generation formulation challenges. Nanomedicine 2019, 14, 1323–1341. [Google Scholar] [CrossRef] [PubMed]
- Nouri, Y.M.; Kim, J.H.; Yoon, H.-K.; Ko, H.-K.; Shin, J.H.; Gwon, D. Il Update on Transarterial Chemoembolization with Drug-Eluting Microspheres for Hepatocellular Carcinoma. Korean J. Radiol. 2019, 20, 34–49. [Google Scholar] [CrossRef]
- Chen, Y.-P.; Zhang, J.-L.; Zou, Y.; Wu, Y.-L. Recent Advances on Polymeric Beads or Hydrogels as Embolization Agents for Improved Transcatheter Arterial Chemoembolization (TACE). Front. Chem. 2019, 7, 408. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Irurzun, J.; Munchart, J.; Trofimov, I.; Scupchenko, A.; Tatum, C.; Narayanan, G. Optimal technique and response of doxorubicin beads in hepatocellular cancer: Bead size and dose. Korean J. Hepatol. 2011, 17, 51. [Google Scholar] [CrossRef] [PubMed]
- Malagari, K.; Emmanouil, E.; Pomoni, M.; Kelekis, D. Chemoembolization with DC BeadTM for the treatment of hepatocellular carcinoma: An update. Hepatic Oncol. 2014, 1, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Martinetti, L.; Crespi, S.; Maggioni, M.; Sangiovanni, A. Transarterial Chemoembolization with Epirubicin-eluting Beads versus Transarterial Embolization before Liver Transplantation for Hepatocellular Carcinoma. J. Vasc. Interv. Radiol. 2010, 21, 327–332. [Google Scholar] [CrossRef]
- Song, M.J.; Park, C.-H.; Kim, J.D.; Kim, H.Y.; Bae, S.H.; Choi, J.Y.; Yoon, S.K.; Chun, H.J.; Choi, B.G.; Lee, H.G. Drug-eluting bead loaded with doxorubicin versus conventional Lipiodol-based transarterial chemoembolization in the treatment of hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2011, 23, 521–527. [Google Scholar] [CrossRef]
- Poon, R.T.P.; Tso, W.K.; Pang, R.W.C.; Ng, K.K.C.; Woo, R.; Tai, K.S.; Fan, S.T. A Phase I/II Trial of Chemoembolization for Hepatocellular Carcinoma Using a Novel Intra-Arterial Drug-Eluting Bead. Clin. Gastroenterol. Hepatol. 2007, 5, 1100–1108. [Google Scholar] [CrossRef]
- Malagari, K.; Pomoni, M.; Moschouris, H.; Kelekis, A.; Charokopakis, A.; Bouma, E.; Spyridopoulos, T.; Chatziioannou, A.; Sotirchos, V.; Karampelas, T.; et al. Chemoembolization of Hepatocellular Carcinoma with Hepasphere 30–60 μm. Safety and Efficacy Study. Cardiovasc. Intervent. Radiol. 2014, 37, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Bouvry, C.; Palard, X.; Edeline, J.; Ardisson, V.; Loyer, P.; Garin, E.; Lepareur, N. Transarterial Radioembolization (TARE) Agents beyond 90 Y-Microspheres. Biomed. Res. Int. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Li, R.-J.; Jin, R.; Liu, C.; Cao, X.; Manning, M.L.; Di, X.M.; Przepiorka, D.; Namuswe, F.; Deisseroth, A.; Goldberg, K.B.; et al. FDA Approval Summary: Calaspargase Pegol-mknl For Treatment of Acute Lymphoblastic Leukemia in Children and Young Adults. Clin. Cancer Res. 2020, 26, 328–331. [Google Scholar] [CrossRef]
- Lew, G. Space for Calaspargase? A New Asparaginase for Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2020, 26, 325–327. [Google Scholar] [CrossRef]
- Vrooman, L.M.; Blonquist, T.M.; Supko, J.G.; Hunt, S.K.; O’Brien, J.E.; Kay-Green, S.; Athale, U.H.; Clavell, L.A.; Cole, P.D.; Harris, M.H.; et al. Efficacy and toxicity of pegaspargase and calaspargase pegol in childhood acute lymphoblastic leukemia/lymphoma: Results of DFCI 11-001. J. Clin. Oncol. 2019, 37, 10006. [Google Scholar] [CrossRef]
- Douer, D. Efficacy and Safety of Vincristine Sulfate Liposome Injection in the Treatment of Adult Acute Lymphocytic Leukemia. Oncologist 2016, 21, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.; Soosay Raj, T.; Smith, A. Vincristine sulfate liposomal injection for acute lymphoblastic leukemia. Int. J. Nanomed. 2013, 8, 4361. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.A.; Deitcher, S.R. Marqibo® (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother. Pharmacol. 2013, 71, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Passero, F.C.; Grapsa, D.; Syrigos, K.N.; Saif, M.W. The safety and efficacy of Onivyde (irinotecan liposome injection) for the treatment of metastatic pancreatic cancer following gemcitabine-based therapy. Expert Rev. Anticancer. Ther. 2016, 16, 697–703. [Google Scholar] [CrossRef]
- Zhang, H. Onivyde for the therapy of multiple solid tumors. Onco Targets Ther. 2016, 9, 3001. [Google Scholar] [CrossRef] [PubMed]
- Alfayez, M.; Kantarjian, H.; Kadia, T.; Ravandi-Kashani, F.; Daver, N. CPX-351 (vyxeos) in AML. Leuk. Lymphoma 2020, 61, 288–297. [Google Scholar] [CrossRef]
- Blair, H.A. Daunorubicin/Cytarabine Liposome: A Review in Acute Myeloid Leukaemia. Drugs 2018, 78, 1903–1910. [Google Scholar] [CrossRef]
- Chen, E.C.; Fathi, A.T.; Brunner, A.M. Reformulating acute myeloid leukemia: Liposomal cytarabine and daunorubicin (CPX-351) as an emerging therapy for secondary AML. OncoTargets Ther. 2018, 11, 3425–3434. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.T.J.; Gilabert-Oriol, R.; Bally, M.B.; Leung, A.W.Y. Recent Treatment Advances and the Role of Nanotechnology, Combination Products, and Immunotherapy in Changing the Therapeutic Landscape of Acute Myeloid Leukemia. Pharm. Res. 2019, 36, 125. [Google Scholar] [CrossRef] [PubMed]
- Germain, M.; Caputo, F.; Metcalfe, S.; Tosi, G.; Spring, K.; Åslund, A.K.O.; Pottier, A.; Schiffelers, R.; Ceccaldi, A.; Schmid, R. Delivering the power of nanomedicine to patients today. J. Control. Release 2020, 326, 164–171. [Google Scholar] [CrossRef]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef]
- Tzogani, K.; Penttilä, K.; Lapveteläinen, T.; Hemmings, R.; Koenig, J.; Freire, J.; Márcia, S.; Cole, S.; Coppola, P.; Flores, B.; et al. EMA Review of Daunorubicin and Cytarabine Encapsulated in Liposomes (Vyxeos, CPX-351) for the Treatment of Adults with Newly Diagnosed, Therapy-Related Acute Myeloid Leukemia or Acute Myeloid Leukemia with Myelodysplasia-Related Changes. Oncologist 2020. [Google Scholar] [CrossRef]
- Borgå, O.; Lilienberg, E.; Bjermo, H.; Hansson, F.; Heldring, N.; Dediu, R. Pharmacokinetics of Total and Unbound Paclitaxel After Administration of Paclitaxel Micellar or Nab-Paclitaxel: An Open, Randomized, Cross-Over, Explorative Study in Breast Cancer Patients. Adv. Ther. 2019, 36, 2825–2837. [Google Scholar] [CrossRef]
- Guiu, B.; Schmitt, A.; Reinhardt, S.; Fohlen, A.; Pohl, T.; Wendremaire, M.; Denys, A.; Blümmel, J.; Boulin, M. Idarubicin-Loaded ONCOZENE Drug-Eluting Embolic Agents for Chemoembolization of Hepatocellular Carcinoma: In Vitro Loading and Release and In Vivo Pharmacokinetics. J. Vasc. Interv. Radiol. 2015, 26, 262–270. [Google Scholar] [CrossRef]
- Delicque, J.; Guiu, B.; Boulin, M.; Schwanz, H.; Piron, L.; Cassinotto, C. Liver chemoembolization of hepatocellular carcinoma using TANDEM® microspheres. Futur. Oncol. 2018, 14, 2761–2772. [Google Scholar] [CrossRef] [PubMed]
- Richter, G.; Radeleff, B.; Stroszczynski, C.; Pereira, P.; Helmberger, T.; Barakat, M.; Huppert, P. Safety and Feasibility of Chemoembolization with Doxorubicin-Loaded Small Calibrated Microspheres in Patients with Hepatocellular Carcinoma: Results of the MIRACLE I Prospective Multicenter Study. Cardiovasc. Interv. Radiol. 2018, 41, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Aliberti, C.; Carandina, R.; Sarti, D.; Mulazzani, L.; Pizzirani, E.; Guadagni, S.; Fiorentini, G. Chemoembolization Adopting Polyethylene Glycol Drug-Eluting Embolics Loaded With Doxorubicin for the Treatment of Hepatocellular Carcinoma. Am. J. Roentgenol. 2017, 209, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Aliberti, C.; Carandina, R.; Sarti, D.; Mulazzani, L.; Catalano, V.; Felicioli, A.; Coschiera, P.; Fiorentini, G. Hepatic arterial infusion of polyethylene glycol drug-eluting beads for primary and metastatic liver cancer therapy. Anticancer. Res. 2016, 36, 3515–3521. [Google Scholar] [PubMed]
- Pottier, A.; Borghi, E.; Levy, L. Metals as radio-enhancers in oncology: The industry perspective. Biochem. Biophys. Res. Commun. 2015, 468, 471–475. [Google Scholar] [CrossRef]
- Reinders, M.T.M.; Smits, M.L.J.; van Roekel, C.; Braat, A.J.A.T. Holmium-166 Microsphere Radioembolization of Hepatic Malignancies. Semin. Nucl. Med. 2019, 49, 237–243. [Google Scholar] [CrossRef]
- Bonvalot, S.; Rutkowski, P.L.; Thariat, J.; Carrère, S.; Ducassou, A.; Sunyach, M.-P.; Agoston, P.; Hong, A.; Mervoyer, A.; Rastrelli, M.; et al. NBTXR3, a first-in-class radioenhancer hafnium oxide nanoparticle, plus radiotherapy versus radiotherapy alone in patients with locally advanced soft-tissue sarcoma (Act.In.Sarc): A multicentre, phase 2–3, randomised, controlled trial. Lancet Oncol. 2019, 20, 1148–1159. [Google Scholar] [CrossRef]
- Bisso, S.; Leroux, J.-C. Nanopharmaceuticals: A focus on their clinical translatability. Int. J. Pharm. 2020, 578, 119098. [Google Scholar] [CrossRef]
- Chajon, E.; Pracht, M.; De Baere, T.; Nguyen, F.; Bronowicki, J.-P.; Vendrely, V.; Baumann, A.-S.; Croisé-Laurent, V.; Deutsch, E. A phase I/II trial of NBTXR3 nanoparticles activated by SBRT in the treatment of liver cancers. J. Clin. Oncol. 2018, 36, TPS551. [Google Scholar] [CrossRef]
- Bonvalot, S.; Le Pechoux, C.; De Baere, T.; Buy, X.; Italiano, A.; Stockle, E.; Terrier, P.; Lassau, N.; Le Cesne, A.; Sargos, P.; et al. Phase I study of NBTXR3 nanoparticles, in patients with advanced soft tissue sarcoma (STS). J. Clin. Oncol. 2014, 32, 10563. [Google Scholar] [CrossRef]
- Bonvalot, S.; Le Pechoux, C.; De Baere, T.; Kantor, G.; Buy, X.; Stoeckle, E.; Terrier, P.; Sargos, P.; Coindre, J.M.; Lassau, N.; et al. First-in-Human Study Testing a New Radioenhancer Using Nanoparticles (NBTXR3) Activated by Radiation Therapy in Patients with Locally Advanced Soft Tissue Sarcomas. Clin. Cancer Res. 2017, 23, 908–917. [Google Scholar] [CrossRef]
- Grauer, O.; Jaber, M.; Hess, K.; Weckesser, M.; Schwindt, W.; Maring, S.; Wölfer, J.; Stummer, W. Combined intracavitary thermotherapy with iron oxide nanoparticles and radiotherapy as local treatment modality in recurrent glioblastoma patients. J. Neurooncol. 2019, 141, 83–94. [Google Scholar] [CrossRef]
- Mahmoudi, K.; Bouras, A.; Bozec, D.; Ivkov, R.; Hadjipanayis, C. Magnetic hyperthermia therapy for the treatment of glioblastoma: A review of the therapy’s history, efficacy and application in humans. Int. J. Hyperth. 2018, 34, 1316–1328. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, C.; Pucci, C.; Ciofani, G. Nanostructured carriers as innovative tools for cancer diagnosis and therapy. APL Bioeng. 2019, 3, 011502. [Google Scholar] [CrossRef] [PubMed]
- El-Boubbou, K. Magnetic iron oxide nanoparticles as drug carriers: Clinical relevance. Nanomedicine 2018, 13, 953–971. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Qin, S.; Ryoo, B.-Y.; Lu, S.-N.; Yen, C.-J.; Feng, Y.-H.; Lim, H.Y.; Izzo, F.; Colombo, M.; Sarker, D.; et al. Phase III randomized study of second line ADI-PEG 20 plus best supportive care versus placebo plus best supportive care in patients with advanced hepatocellular carcinoma. Ann. Oncol. 2018, 29, 1402–1408. [Google Scholar] [CrossRef]
- Doherty, G.J.; Tempero, M.; Corrie, P.G. HALO-109–301: A Phase III trial of PEGPH20 (with gemcitabine and nab-paclitaxel) in hyaluronic acid-high stage IV pancreatic cancer. Futur. Oncol. 2018, 14, 13–22. [Google Scholar] [CrossRef]
- Naing, A.; Papadopoulos, K.P.; Autio, K.A.; Ott, P.A.; Patel, M.R.; Wong, D.J.; Falchook, G.S.; Pant, S.; Whiteside, M.; Rasco, D.R.; et al. Safety, Antitumor Activity, and Immune Activation of Pegylated Recombinant Human Interleukin-10 (AM0010) in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 3562–3569. [Google Scholar] [CrossRef] [PubMed]
- Butts, C.; Socinski, M.A.; Mitchell, P.L.; Thatcher, N.; Havel, L.; Krzakowski, M.; Nawrocki, S.; Ciuleanu, T.-E.; Bosquée, L.; Trigo, J.M.; et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 59–68. [Google Scholar] [CrossRef]
- Zisman, N.; Dos Santos, N.; Johnstone, S.; Tsang, A.; Bermudes, D.; Mayer, L.; Tardi, P. Optimizing Liposomal Cisplatin Efficacy through Membrane Composition Manipulations. Chemother. Res. Pract. 2011, 2011, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chauffert, B.; Mornex, F.; Bonnetain, F.; Rougier, P.; Mariette, C.; Bouché, O.; Bosset, J.F.; Aparicio, T.; Mineur, L.; Azzedine, A.; et al. Phase III trial comparing intensive induction chemoradiotherapy (60 Gy, infusional 5-FU and intermittent cisplatin) followed by maintenance gemcitabine with gemcitabine alone for locally advanced unresectable pancreatic cancer. Definitive results of the 2. Ann. Oncol. 2008, 19, 1592–1599. [Google Scholar] [CrossRef]
- Osada, A. NC-6004, a novel cisplatin nanoparticle, in combination with pembrolizumab for head and neck cancer. Integr. Clin. Med. 2019, 3. [Google Scholar] [CrossRef]
- Dunne, M.; Epp-Ducharme, B.; Sofias, A.M.; Regenold, M.; Dubins, D.N.; Allen, C. Heat-activated drug delivery increases tumor accumulation of synergistic chemotherapies. J. Control. Release 2019, 308, 197–208. [Google Scholar] [CrossRef]
- Wood, B.J.; Poon, R.T.; Locklin, J.K.; Dreher, M.R.; Ng, K.K.; Eugeni, M.; Seidel, G.; Dromi, S.; Neeman, Z.; Kolf, M.; et al. Phase I Study of Heat-Deployed Liposomal Doxorubicin during Radiofrequency Ablation for Hepatic Malignancies. J. Vasc. Interv. Radiol. 2012, 23, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Merle, P.; Blanc, J.-F.; Phelip, J.-M.; Pelletier, G.; Bronowicki, J.-P.; Touchefeu, Y.; Pageaux, G.; Gerolami, R.; Habersetzer, F.; Nguyen-Khac, E.; et al. Doxorubicin-loaded nanoparticles for patients with advanced hepatocellular carcinoma after sorafenib treatment failure (RELIVE): A phase 3 randomised controlled trial. Lancet Gastroenterol. Hepatol. 2019, 4, 454–465. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Zardavas, D.; Lemort, M.; Wilke, C.; Vanderbeeken, M.-C.; D’Hondt, V.; De Azambuja, E.; Gombos, A.; Lebrun, F.; Dal Lago, L.; et al. Feasibility Study of EndoTAG-1, a Tumor Endothelial Targeting Agent, in Combination with Paclitaxel followed by FEC as Induction Therapy in HER2-Negative Breast Cancer. PLoS ONE 2016, 11, e0154009. [Google Scholar] [CrossRef]
- Negishi, T.; Koizumi, F.; Uchino, H.; Kuroda, J.; Kawaguchi, T.; Naito, S.; Matsumura, Y. NK105, a paclitaxel-incorporating micellar nanoparticle, is a more potent radiosensitising agent compared to free paclitaxel. Br. J. Cancer 2006, 95, 601–606. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Mukai, H.; Saeki, T.; Ro, J.; Lin, Y.-C.; Nagai, S.E.; Lee, K.S.; Watanabe, J.; Ohtani, S.; Kim, S.B.; et al. A multi-national, randomised, open-label, parallel, phase III non-inferiority study comparing NK105 and paclitaxel in metastatic or recurrent breast cancer patients. Br. J. Cancer 2019, 120, 475–480. [Google Scholar] [CrossRef]
- Perez, E.A.; Awada, A.; O’Shaughnessy, J.; Rugo, H.S.; Twelves, C.; Im, S.-A.; Gómez-Pardo, P.; Schwartzberg, L.S.; Diéras, V.; Yardley, D.A.; et al. Etirinotecan pegol (NKTR-102) versus treatment of physician’s choice in women with advanced breast cancer previously treated with an anthracycline, a taxane, and capecitabine (BEACON): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 1556–1568. [Google Scholar] [CrossRef]
- Tripathy, D.; Tolaney, S.M.; Seidman, A.D.; Anders, C.K.; Ibrahim, N.; Rugo, H.S.; Twelves, C.; Dieras, V.; Müller, V.; Tagliaferri, M.; et al. ATTAIN: Phase III study of etirinotecan pegol versus treatment of physician’s choice in patients with metastatic breast cancer and brain metastases. Futur. Oncol. 2019, 15, 2211–2225. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Li, T.; Xie, X.; Feng, Y.; Chen, Z.; Yang, H.; Wu, C.; Deng, S.; Liu, Y. PLGA-Based Drug Delivery Systems for Remotely Triggered Cancer Therapeutic and Diagnostic Applications. Front. Bioeng. Biotechnol. 2020, 8, 381. [Google Scholar] [CrossRef]
- Li, C.; Wang, J.; Wang, Y.; Gao, H.; Wei, G.; Huang, Y.; Yu, H.; Gan, Y.; Wang, Y.; Mei, L.; et al. Recent progress in drug delivery. Acta Pharm. Sin. B 2019, 9, 1145–1162. [Google Scholar] [CrossRef]
- Pillai, G. Nanomedicines for Cancer Therapy: An Update of FDA Approved and Those under Various Stages of Development. SOJ Pharm. Pharm. Sci. 2014. [Google Scholar] [CrossRef]
- Baeza, A. Tumor Targeted Nanocarriers for Immunotherapy. Molecules 2020, 25, 1508. [Google Scholar] [CrossRef]
- Pearce, A.K.; O’Reilly, R.K. Insights into Active Targeting of Nanoparticles in Drug Delivery: Advances in Clinical Studies and Design Considerations for Cancer Nanomedicine. Bioconjug. Chem. 2019, 30, 2300–2311. [Google Scholar] [CrossRef]
- You, J.; Li, X.; de Cui, F.; Du, Y.-Z.; Yuan, H.; Hu, F.Q. Folate-conjugated polymer micelles for active targeting to cancer cells: Preparation, in vitro evaluation of targeting ability and cytotoxicity. Nanotechnology 2008, 19, 045102. [Google Scholar] [CrossRef]
- Landeros-Martínez, L.-L.; Glossman-Mitnik, D.; Flores-Holguín, N. Studying the chemical reactivity properties of the target tumor-environment tripeptides NGR (asparagine-glycine-arginine) and RGD (arginine-glycine-aspartic acid) in their interactions with tamoxifen through conceptual density functional theory. J. Mol. Model. 2018, 24, 336. [Google Scholar] [CrossRef]
- Hong, M.; Zhu, S.; Jiang, Y.; Tang, G.; Pei, Y. Efficient tumor targeting of hydroxycamptothecin loaded PEGylated niosomes modified with transferrin. J. Control. Release 2009, 133, 96–102. [Google Scholar] [CrossRef]
- Zhai, J.; Scoble, J.A.; Li, N.; Lovrecz, G.; Waddington, L.J.; Tran, N.; Muir, B.W.; Coia, G.; Kirby, N.; Drummond, C.J.; et al. Epidermal growth factor receptor-targeted lipid nanoparticles retain self-assembled nanostructures and provide high specificity. Nanoscale 2015, 7, 2905–2913. [Google Scholar] [CrossRef]
- Smith, J.E.; Medley, C.D.; Tang, Z.; Shangguan, D.; Lofton, C.; Tan, W. Aptamer-Conjugated Nanoparticles for the Collection and Detection of Multiple Cancer Cells. Anal. Chem. 2007, 79, 3075–3082. [Google Scholar] [CrossRef]
- Arranja, A.G.; Pathak, V.; Lammers, T.; Shi, Y. Tumor-targeted nanomedicines for cancer theranostics. Pharmacol. Res. 2017, 115, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Mi, P. Stimuli-responsive nanocarriers for drug delivery, tumor imaging, therapy and theranostics. Theranostics 2020, 10, 4557–4588. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Li, W.; Liu, R.; Li, X.; Li, H.; Liu, L.; Chen, Y.; Lv, C.; Liu, Y. pH- and enzyme-triggered drug release as an important process in the design of anti-tumor drug delivery systems. Biomed. Pharmacother. 2019, 118, 109340. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, Y.; Kumar, A.; Tan, A.; Jin, S.; Mozhi, A.; Liang, X.-J. pH-Sensitive nano-systems for drug delivery in cancer therapy. Biotechnol. Adv. 2014, 32, 693–710. [Google Scholar] [CrossRef]
- Gao, G.H.; Park, M.J.; Li, Y.; Im, G.H.; Kim, J.-H.; Kim, H.N.; Lee, J.W.; Jeon, P.; Bang, O.Y.; Lee, J.H.; et al. The use of pH-sensitive positively charged polymeric micelles for protein delivery. Biomaterials 2012, 33, 9157–9164. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Hu, J.-J.; Xu, Q.; Chen, S.; Jia, H.-Z.; Sun, Y.-X.; Zhuo, R.-X.; Zhang, X.-Z. A redox-responsive drug delivery system based on RGD containing peptide-capped mesoporous silica nanoparticles. J. Mater. Chem. B 2015, 3, 39–44. [Google Scholar] [CrossRef]
- Xin, X.; Teng, C.; Du, X.; Lv, Y.; Xiao, Q.; Wu, Y.; He, W.; Yin, L. Drug-delivering-drug platform-mediated potent protein therapeutics via a non-endo-lysosomal route. Theranostics 2018, 8, 3474–3489. [Google Scholar] [CrossRef]
- Fu, H.; Shi, K.; Hu, G.; Yang, Y.; Kuang, Q.; Lu, L.; Zhang, L.; Chen, W.; Dong, M.; Chen, Y.; et al. Tumor-Targeted Paclitaxel Delivery and Enhanced Penetration Using TAT-Decorated Liposomes Comprising Redox-Responsive Poly(Ethylene Glycol). J. Pharm. Sci. 2015, 104, 1160–1173. [Google Scholar] [CrossRef]
- Alsehli, M. Polymeric nanocarriers as stimuli-responsive systems for targeted tumor (cancer) therapy: Recent advances in drug delivery. Saudi Pharm. J. 2020, 28, 255–265. [Google Scholar] [CrossRef]
- Chiang, C.-S.; Shen, Y.-S.; Liu, J.-J.; Shyu, W.-C.; Chen, S.-Y. Synergistic Combination of Multistage Magnetic Guidance and Optimized Ligand Density in Targeting a Nanoplatform for Enhanced Cancer Therapy. Adv. Healthc. Mater. 2016, 5, 2131–2141. [Google Scholar] [CrossRef]
- Farzin, A.; Etesami, S.A.; Quint, J.; Memic, A.; Tamayol, A. Magnetic Nanoparticles in Cancer Therapy and Diagnosis. Adv. Healthc. Mater. 2020, 9, 1901058. [Google Scholar] [CrossRef]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Al-Ahmady, Z.S.; Al-Jamal, W.T.; Bossche, J.V.; Bui, T.T.; Drake, A.F.; Mason, A.J.; Kostarelos, K. Lipid–Peptide Vesicle Nanoscale Hybrids for Triggered Drug Release by Mild Hyperthermia in Vitro and in Vivo. ACS Nano 2012, 6, 9335–9346. [Google Scholar] [CrossRef]
- Karimi, M.; Ghasemi, A.; Sahandi Zangabad, P.; Rahighi, R.; Moosavi Basri, S.M.; Mirshekari, H.; Amiri, M.; Shafaei Pishabad, Z.; Aslani, A.; Bozorgomid, M.; et al. Smart micro/nanoparticles in stimulus-responsive drug/gene delivery systems. Chem. Soc. Rev. 2016, 45, 1457–1501. [Google Scholar] [CrossRef]
- Luo, D.; Carter, K.A.; Razi, A.; Geng, J.; Shao, S.; Giraldo, D.; Sunar, U.; Ortega, J.; Lovell, J.F. Doxorubicin encapsulated in stealth liposomes conferred with light-triggered drug release. Biomaterials 2016, 75, 193–202. [Google Scholar] [CrossRef]
- Aryal, M.; Arvanitis, C.D.; Alexander, P.M.; McDannold, N. Ultrasound-mediated blood–brain barrier disruption for targeted drug delivery in the central nervous system. Adv. Drug Deliv. Rev. 2014, 72, 94–109. [Google Scholar] [CrossRef]
- Nam, J.; Son, S.; Park, K.S.; Zou, W.; Shea, L.D.; Moon, J.J. Cancer nanomedicine for combination cancer immunotherapy. Nat. Rev. Mater. 2019, 4, 398–414. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Jin, N.; Mao, C.; Yang, M. Protein-Induced Gold Nanoparticle Assembly for Improving the Photothermal Effect in Cancer Therapy. ACS Appl. Mater. Interfaces 2019, 11, 11136–11143. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, C.; Cheng, L.; He, W.; Cheng, Z.; Liu, Z. Protein modified upconversion nanoparticles for imaging-guided combined photothermal and photodynamic therapy. Biomaterials 2014, 35, 2915–2923. [Google Scholar] [CrossRef]
- Aparicio-Blanco, J.; Torres-Suárez, A.I. Towards tailored management of malignant brain tumors with nanotheranostics. Acta Biomater. 2018, 73, 52–63. [Google Scholar] [CrossRef]
- Sonali; Viswanadh, M.K.; Singh, R.P.; Agrawal, P.; Mehata, A.K.; Pawde, D.M.; Narendra; Sonkar, R.; Muthu, M.S. Nanotheranostics: Emerging Strategies for Early Diagnosis and Therapy of Brain Cancer. Nanotheranostics 2018, 2, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Cao, L.; Zhang, Y.; Yi, P.; Wang, M.; Tan, B.; Deng, Z.; Wu, D.; Wang, Q. Preoperative Detection and Intraoperative Visualization of Brain Tumors for More Precise Surgery: A New Dual-Modality MRI and NIR Nanoprobe. Small 2015, 11, 4517–4525. [Google Scholar] [CrossRef] [PubMed]
- Lungu, I.I.; Grumezescu, A.M.; Volceanov, A.; Andronescu, E. Nanobiomaterials Used in Cancer Therapy: An Up-To-Date Overview. Molecules 2019, 24, 3547. [Google Scholar] [CrossRef]
- Zhou, M.; Wei, W.; Chen, X.; Xu, X.; Zhang, X.; Zhang, X. pH and redox dual responsive carrier-free anticancer drug nanoparticles for targeted delivery and synergistic therapy. Nanomed. Nanotechnol. Biol. Med. 2019, 20, 102008. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; He, H.; Liu, Y.; Cao, D.; Yan, J.; Duan, S.; Chen, Y.; Yin, L. Cancer-Selective Bioreductive Chemotherapy Mediated by Dual Hypoxia-Responsive Nanomedicine upon Photodynamic Therapy-Induced Hypoxia Aggravation. Biomacromolecules 2019, 20, 2649–2656. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.E. Patient-reported experience with the Viadur 12-month leuprolide implant for prostate cancer. Urology 2001, 58, 430–434. [Google Scholar] [CrossRef]
- Kanwar, N.; Sinha, V.R. In situ forming depot as sustained-release drug delivery systems. Crit. Rev. Ther. Drug Carr. Syst. 2019, 36, 93–136. [Google Scholar] [CrossRef]
- Salvioni, L.; Rizzuto, M.A.; Bertolini, J.A.; Pandolfi, L.; Colombo, M.; Prosperi, D. Thirty years of cancer nanomedicine: Success, frustration, and hope. Cancers 2019, 11, 1855. [Google Scholar] [CrossRef]
- Rafiyath, S.M.; Rasul, M.; Lee, B.; Wei, G.; Lamba, G.; Liu, D. Comparison of safety and toxicity of liposomal doxorubicin vs. conventional anthracyclines: A meta-analysis. Exp. Hematol. Oncol. 2012, 1, 10. [Google Scholar] [CrossRef]
- Hua, S.; de Matos, M.B.C.; Metselaar, J.M.; Storm, G. Current trends and challenges in the clinical translation of nanoparticulate nanomedicines: Pathways for translational development and commercialization. Front. Pharmacol. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Hussain, Z.; Khan, S.; Imran, M.; Sohail, M.; Shah, S.W.A.; de Matas, M. PEGylation: A promising strategy to overcome challenges to cancer-targeted nanomedicines: A review of challenges to clinical transition and promising resolution. Drug Deliv. Transl. Res. 2019, 9, 721–734. [Google Scholar] [CrossRef]
- Fang, Y.; Xue, J.; Gao, S.; Lu, A.; Yang, D.; Jiang, H.; He, Y.; Shi, K. Cleavable PEGylation: A strategy for overcoming the “PEG dilemma” in efficient drug delivery. Drug Deliv. 2017, 24, 22–32. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Liu, L.; Morin, E.E.; Liu, M.; Schwendeman, A. Survey of Clinical Translation of Cancer Nanomedicines—Lessons Learned from Successes and Failures. Acc. Chem. Res. 2019, 52, 2445–2461. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Drug-Eluting Beads|Radiology Key. Available online: https://radiologykey.com/drug-eluting-beads/ (accessed on 27 August 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Therapy | Formulation Type | Mechanism of Action | Drug Substance | Trade Name | Administration Route | Dosing Frequency | Indications | First Approval | Current Status |
|---|---|---|---|---|---|---|---|---|---|
| Peptide-based therapy | Microspheres | Inhibition of gonadotropin secretion | Triptorelin acetate | Decapeptyl | IM | 1–6 months | Prostate cancer | 1986 | Active |
| Microspheres | Inhibition of gonadotropin secretion | Leuprolide acetate | Lupron Depot | IM | 1–6 months | Prostate cancer | 1989 | Active | |
| Solid implant | Inhibition of gonadotropin secretion | Goserelin acetate | Zoladex | SC | 1–3 months | Prostate and breast cancer | 1989 | Active | |
| Polymer conjugate | l-asparagine depletion | Pegaspargase | Oncaspar | IV, IM | 2 weeks | Lymphoblastic leukemia | 1994 | Active | |
| Solid implant | Inhibition of gonadotropin secretion | Buserelin acetate | Suprefact Depot | SC | 2–3 months | Prostate cancer | 1996 | Active | |
| Microspheres | Inhibition of secretion of peptides from the endocrine gastrointestinal system | Octreotide acetate | Sandostatin LAR | IM | 1 month | Neuroendocrine tumors | 1998 | Active | |
| Chemotherapy | Liposomes | Topoisomerase-II inhibition, DNA intercalation | Doxorubicin | Doxil | IV | 3–4 weeks | AIDS-related Kaposi’s sarcoma, ovarian, and breast neoplasms, multiple myeloma | 1995 | Active |
| Liposomes | Topoisomerase-II inhibition, DNA intercalation | Daunorubicin | Daunoxome | IV | 2 weeks | AIDS-related Kaposi’s sarcoma | 1996 | Discontinued | |
| Solid implant | DNA alkylation | Carmustine | Gliadel | IC | 3 weeks * | Malignant glioma | 1996 | Active | |
| Liposomes | DNA polymerase inhibition | Cytarabine | DepoCyt | ITh | 4 weeks | Lymphomatous meningitis | 1999 | Discontinued | |
| Radiotherapy | Microspheres | Beta particle emission | Yttrium-90 | Theraspheres | IA | - | Hepatocellular carcinoma | 1999 | Active |
| Type of Therapy | Formulation Type | Mechanism of Action | Drug Substance | Trade Name | Administration Route | Dosing Frequency | Indications | First Approval | Current Status |
|---|---|---|---|---|---|---|---|---|---|
| Peptide-based therapy | Solid implant | Inhibition of gonadotropin secretion | Leuprolide acetate | Viadur | SC | 12 months | Prostate cancer | 2000 | Discontinued |
| In-situ-forming implants | Inhibition of gonadotropin secretion | Leuprolide acetate | Eligard | SC | 1–6 months | Prostate cancer | 2002 | Active | |
| Solid implant | Inhibition of gonadotropin secretion | Histrelin acetate | Vantas | SC | 12 months | Prostate cancer | 2004 | Active | |
| Liposomes | Immunomodulation | Mifamurtide | Mepact | IV | 1/2–1 week | Osteosarcoma | 2009 | Active | |
| Chemotherapy | Liposomes | Topoisomerase-II inhibition, DNA intercalation | Doxorubicin | Myocet | IV | 3 weeks | Metastatic breast cancer | 2000 | Active |
| Microspheres | - | Various * | DC Bead | IA | - | Hepatocellular carcinoma | 2003 | Active | |
| Protein-based nanoparticles | Microtubule inhibition | Paclitaxel | Abraxane | IV | 1–3 weeks | Breast neoplasms, pancreatic neoplasms, non-small-cell lung cancer | 2005 | Active | |
| Microspheres | - | Various * | Hepasphere | IA | - | Hepatocellular carcinoma | 2005 | Active | |
| Radiotherapy | Microspheres | Beta particle emission | Yttrium-90 | SIR-Spheres | IA | - | Metastatic liver tumors | 2002 | Active |
| Type of Therapy | Formulation Type | Mechanism of Action | Drug Substance | Trade Name | Administration Route | Dosing Frequency | Indications | First Approval | Current Status |
|---|---|---|---|---|---|---|---|---|---|
| Peptide-based therapy | Solid implant | Inhibition of gonadotropin secretion | Leuprolide acetate | Leptoprol | SC | 3 months | Prostate cancer | 2015 | Active |
| Polymer conjugate | l-asparaginase depletion | Calaspargase pegol | Asparlas | IV, IM | 3 weeks | Acute lymphoblastic leukemia | 2018 | Active | |
| Chemotherapy | Liposomes | Microtubule inhibition | Vincristine | Marqibo | IV | 1 week | Acute lymphoblastic leukemia | 2012 | Active |
| Microspheres | - | Various * | Embozene TANDEM | IA | - | Hepatocellular carcinoma | 2012 | Active | |
| Liposomes | Topoisomerase I inhibition | Irinotecan | Onivyde | IV | 2 weeks | Metastatic adenocarcinoma of the pancreas | 2015 | Active | |
| Microspheres | - | Various ** | Life Pearl | IA | - | Hepatocellular carcinoma | 2015 | Active | |
| Liposomes | DNA polymerase inhibition + topoisomerase-II inhibition, DNA intercalation | Cytarabine + daunorubicin | Vyxeos | IV | 2 days | Acute myeloid leukemia | 2017 | Active | |
| Microspheres | - | Various * | DC Bead LUMI | IA | - | Hepatocellular carcinoma | 2017 | Active | |
| Micelles | Microtubule inhibition | Paclitaxel | Apealea | IV | 3 weeks | Ovarian cancer, primary peritoneal cancer, fallopian tube cancer | 2018 | Active | |
| Radiotherapy | Microspheres | Beta particle emission | Holmium-166 | QuiremSpheres | IA | - | Hepatocellular carcinoma | 2015 | Active |
| Inorganic nanoparticles | Radio enhancer | Hafnium oxide | Hensify | IT | - | Soft-tissue sarcoma | 2019 | Active | |
| Other | Inorganic nanoparticles | Hyperthermia | Iron oxide | NanoTherm | IT | - | Glioblastoma | 2010 | Active |
| Type of Therapy | Formulation Type | Mechanism of Action | Drug Substance | Trade Name | Administration Route | Indications | Clinical Trial Number | Current Status |
|---|---|---|---|---|---|---|---|---|
| Peptide-based therapy | Liposomes | Immunomodulation | Tecemotide | Stimuvax | SC | Non-small-cell lung cancer | NCT00409188 | Completed |
| Polymer conjugate | Arginine depletion | Pegargiminase | ADI-PEG 20 | IM | Mesothelioma | NCT02709512 | Recruiting | |
| Polymer conjugate | Hyaluronan degradation | Pegvorhyaluronidase alfa | PEGPH20 | IV | Pancreatic cancer | NCT02715804 | Terminated | |
| Polymer conjugate | Immunomodulation | Pegylated IL-10 | Pegilodecakin | SC | Pancreatic cancer | NCT02923921 | Completed | |
| Chemotherapy | Liposomes | Topoisomerase-II inhibition, DNA intercalation | Doxorubicin | MM-302 | IV | Breast cancer | NCT02213744 | Terminated |
| Liposomes | Topoisomerase-II inhibition, DNA intercalation | Doxorubicin | Thermodox | IV | Hepatocellular carcinoma | NCT00617981 | Completed | |
| NCT02112656 | Completed | |||||||
| Liposomes | Microtubule inhibition | Paclitaxel | Endotag 1 | IV | Breast cancer | NCT03002103 | Recruiting | |
| Pancreatic cancer | NCT03126435 | Recruiting | ||||||
| Liposomes | DNA alkylation | Cisplatin | SPI-77 | IV | Pancreatic cancer | NCT00416507 | Completed | |
| Polymer conjugate | Topoisomerase I inhibition | Etirinotecan pegol | Onzeald | IV | Breast cancer | NCT01492101 | Completed | |
| NCT02915744 | Completed | |||||||
| Micelles | DNA alkylation | Cisplatin | Nanoplatin | IV | Pancreatic cancer | NCT02043288 | Completed | |
| Micelles | Microtubule inhibition | Paclitaxel | NK105 | IV | Breast cancer | NCT01644890 | Completed | |
| Polymeric nanoparticles | Topoisomerase-II inhibition, DNA intercalation | Doxorubicin | Livatag | IV | Hepatocellular carcinoma | NCT01655693 | Completed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-López, A.; Martín-Sabroso, C.; Torres-Suárez, A.I.; Aparicio-Blanco, J. Timeline of Translational Formulation Technologies for Cancer Therapy: Successes, Failures, and Lessons Learned Therefrom. Pharmaceutics 2020, 12, 1028. https://doi.org/10.3390/pharmaceutics12111028
Pérez-López A, Martín-Sabroso C, Torres-Suárez AI, Aparicio-Blanco J. Timeline of Translational Formulation Technologies for Cancer Therapy: Successes, Failures, and Lessons Learned Therefrom. Pharmaceutics. 2020; 12(11):1028. https://doi.org/10.3390/pharmaceutics12111028
Chicago/Turabian StylePérez-López, Alexandre, Cristina Martín-Sabroso, Ana Isabel Torres-Suárez, and Juan Aparicio-Blanco. 2020. "Timeline of Translational Formulation Technologies for Cancer Therapy: Successes, Failures, and Lessons Learned Therefrom" Pharmaceutics 12, no. 11: 1028. https://doi.org/10.3390/pharmaceutics12111028
APA StylePérez-López, A., Martín-Sabroso, C., Torres-Suárez, A. I., & Aparicio-Blanco, J. (2020). Timeline of Translational Formulation Technologies for Cancer Therapy: Successes, Failures, and Lessons Learned Therefrom. Pharmaceutics, 12(11), 1028. https://doi.org/10.3390/pharmaceutics12111028