Mitoxantrone-Loaded Nanoparticles for Magnetically Controlled Tumor Therapy–Induction of Tumor Cell Death, Release of Danger Signals and Activation of Immune Cells


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Synthesis and Characterization of Superparamagnetic Iron Oxide Nanoparticles (SPIONs)
2.3. Endotoxin Content
2.4. Plasma Coagulation
2.5. Platelet Aggregation
2.6. Hemolysis
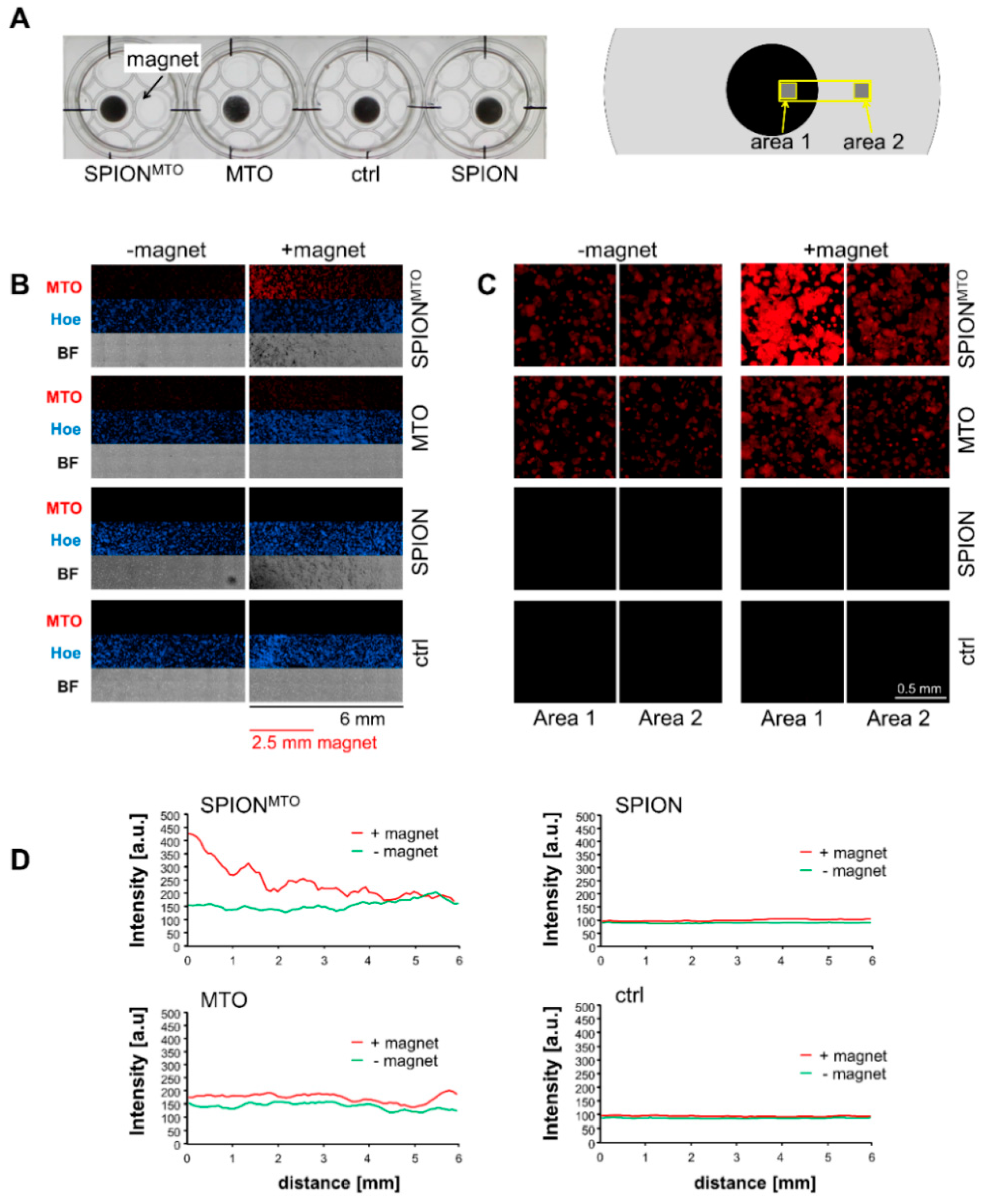
2.7. Magnetic Accumulation of SPIONMTO
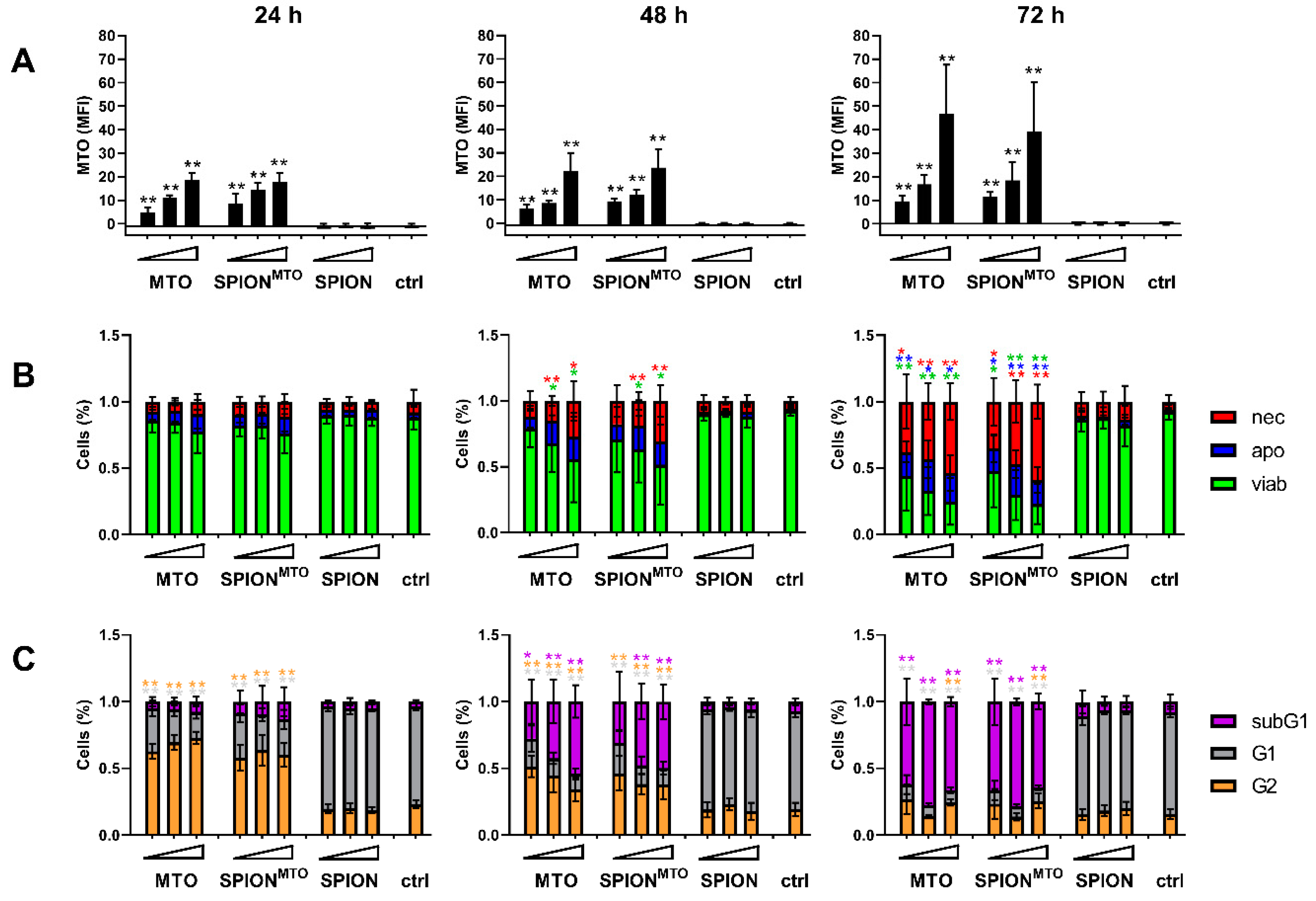
2.8. Determination of Cell Proliferation
2.9. Determination of Cell Cycle and Cell Death
2.10. Flow Cytometry
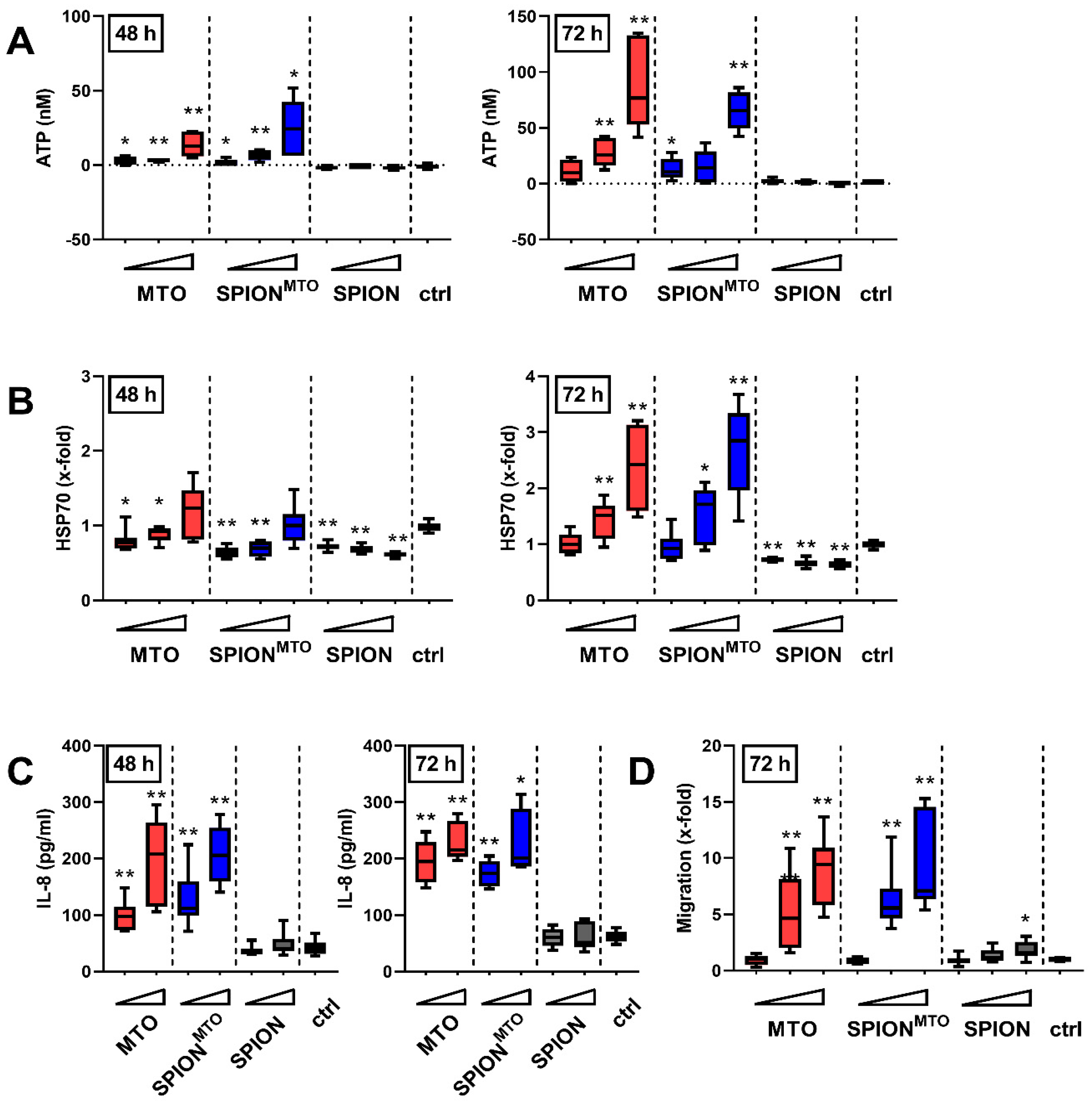
2.11. ATP Release
2.12. Heat Shock Protein 70 (HSP70) Release
2.13. Chemotaxis
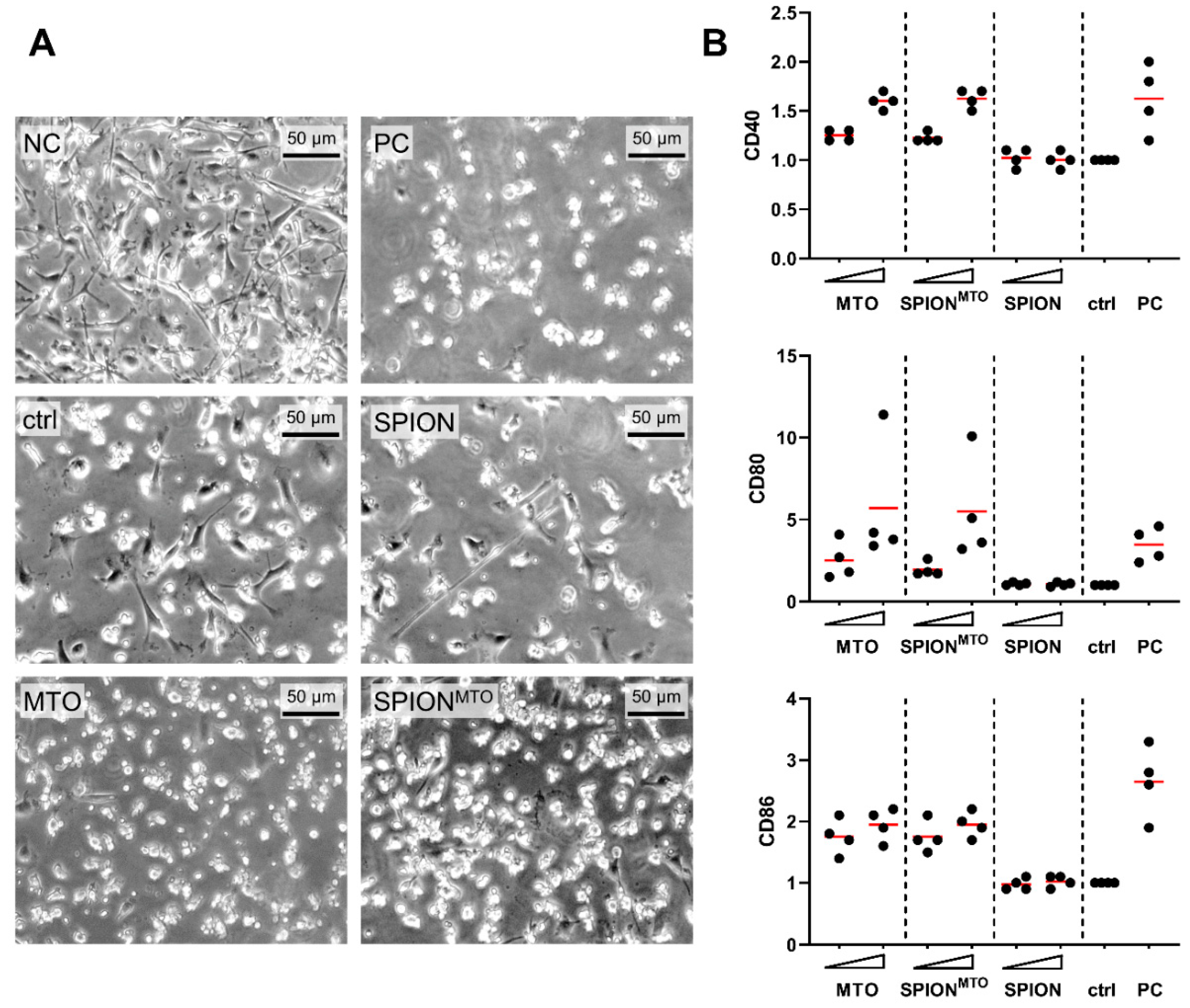
2.14. Generation of Human Immature Dendritic Cells (iDCs)
2.15. Maturation of Human Dendritic Cells with Supernatants of HT-29 Tumor Cells
2.16. Cytokine Analysis
2.17. Data Analysis and Statistics
3. Results
3.1. Biocompatibility
3.2. Magnetic Accumulation of SPIONMTO
3.3. Drug Uptake and Cell Death Induction
3.4. Release of Damage-Associated Molecular Patterns (DAMPs) and Activation of Immune Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef]
- Andrews, A. Treating with Checkpoint Inhibitors—Figure $1 Million per Patient. Am. Health Drug Benefits 2015, 8, 9. [Google Scholar] [PubMed]
- Tartari, F.; Santoni, M.; Burattini, L.; Mazzanti, P.; Onofri, A.; Berardi, R. Economic sustainability of anti-PD-1 agents nivolumab and pembrolizumab in cancer patients: Recent insights and future challenges. Cancer Treat. Rev. 2016, 48, 20–24. [Google Scholar] [CrossRef]
- Pages, F.; Galon, J.; Dieu-Nosjean, M.-C.; Tartour, E.; Sautès-Fridman, C.; Fridman, W.H. Immune infiltration in human tumors: A prognostic factor that should not be ignored. Oncogene 2009, 29, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Saito, K.; Shiiba, K.; Ohuchi, A.; Saigenji, K.; Nagura, H.; Ohtani, H. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998, 58, 3491–3494. [Google Scholar]
- Kroemer, G. Death, danger & immunity: Fundamental mechanisms linking pathogenic or iatrogenic cell death events to immune responses. Immunol. Rev. 2017, 280, 5–7. [Google Scholar]
- Rapoport, B.L.; Anderson, R. Realizing the Clinical Potential of Immunogenic Cell Death in Cancer Chemotherapy and Radiotherapy. Int. J. Mol. Sci. 2019, 20, 959. [Google Scholar] [CrossRef]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. OncoImmunology 2014, 3, e955691. [Google Scholar] [CrossRef]
- Tesniere, A.; Panaretakis, T.; Kepp, O.; Apetoh, L.; Ghiringhelli, F.; Zitvogel, L.; Kroemer, G. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2007, 15, 3–12. [Google Scholar] [CrossRef]
- Rossato, L.G.; Costa, V.M.; Dallegrave, E.; Arbo, M.D.; Dinis-Oliveira, R.J.; Santos-Silva, A.; Duarte, J.A.; Bastos, M.D.L.; Palmeira, C.M.; Remião, F. Cumulative Mitoxantrone-Induced Haematological and Hepatic Adverse Effects in a SubchronicIn vivoStudy. Basic Clin. Pharmacol. Toxicol. 2013, 114, 254–262. [Google Scholar] [CrossRef]
- Arranja, A.G.; Pathak, V.; Lammers, T.; Shi, Y. Tumor-targeted nanomedicines for cancer theranostics. Pharmacol. Res. 2017, 115, 87–95. [Google Scholar] [CrossRef]
- Minchinton, A.I.; Tannock, I.F. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar] [CrossRef]
- Robey, R.C.; Bower, M. Facing up to the ongoing challenge of Kaposi’s sarcoma. Curr. Opin. Infect. Dis. 2015, 28, 31–40. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Baruchel, S.; Stempak, D. Low-Dose Metronomic Chemotherapy: Myth or Truth? Oncol. Res. Treat. 2006, 29, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.; Dale, D.; Lyman, G.; Crawford, J.; Dale, D.C.; Lyman, G.H. Chemotherapy-induced neutropenia: Risks, consequences, and new directions for its management. Cancer 2004, 100, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.R. Immunotherapy of autoimmunity and cancer: The penalty for success. Nat. Rev. Immunol. 2008, 8, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Mishchenko, T.; Mitroshina, E.; Balalaeva, I.V.; Krysko, O.; Vedunova, M.; Krysko, D.V. An emerging role for nanomaterials in increasing immunogenicity of cancer cell death. Biochim. Biophys. Acta BBA Rev. Cancer 2019, 1871, 99–108. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, K.; Zhao, R.; Ji, T.; Wang, X.; Yang, X.; Zhang, Y.; Cheng, K.; Liu, S.; Hao, J.; et al. Inducing enhanced immunogenic cell death with nanocarrier-based drug delivery systems for pancreatic cancer therapy. Biomaterials 2016, 102, 187–197. [Google Scholar] [CrossRef]
- He, C.; Duan, X.; Guo, N.; Chan, C.; Poon, C.; Weichselbaum, R.R.; Lin, W. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat. Commun. 2016, 7, 12499. [Google Scholar] [CrossRef] [PubMed]
- Kuai, R.; Yuan, W.; Son, S.; Nam, J.; Xu, Y.; Fan, Y.; Schwendeman, A.; Moon, J.J. Elimination of established tumors with nanodisc-based combination chemoimmunotherapy. Sci. Adv. 2018, 4, eaao1736. [Google Scholar] [CrossRef] [PubMed]
- Lü, J.; Liu, X.; Liao, Y.-P.; Salazar, F.; Sun, B.; Jiang, W.; Chang, C.H.; Jiang, J.; Wang, X.; Wu, A.M.; et al. Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression. Nat. Commun. 2017, 8, 1811. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Park, K. Targeted drug delivery to tumors: Myths, reality and possibility. J. Control. Release 2011, 153, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Al-Jamal, K.T.; Bai, J.; Wang, J.T.-W.; Protti, A.; Southern, P.; Bogart, L.; Heidari, H.; Li, X.; Cakebread, A.; Asker, D.; et al. Magnetic Drug Targeting: Preclinical in Vivo Studies, Mathematical Modeling, and Extrapolation to Humans. Nano Lett. 2016, 16, 5652–5660. [Google Scholar] [CrossRef]
- Tietze, R.; Lyer, S.; Dürr, S.; Struffert, T.; Engelhorn, T.; Schwarz, M.A.; Eckert, E.; Göen, T.; Vasylyev, S.; Peukert, W.; et al. Efficient drug-delivery using magnetic nanoparticles—biodistribution and therapeutic effects in tumour bearing rabbits. Nanomed. Nanotechnol. Boil. Med. 2013, 9, 961–971. [Google Scholar] [CrossRef]
- Janko, C.; Dürr, S.; Munoz, L.; Lyer, S.; Chaurio, R.; Tietze, R.; Von Löhneysen, S.; Schorn, C.; Herrmann, M.; Alexiou, C. Magnetic Drug Targeting Reduces the Chemotherapeutic Burden on Circulating Leukocytes. Int. J. Mol. Sci. 2013, 14, 7341–7355. [Google Scholar] [CrossRef]
- Alev, M.; Egenberger, L.; Mühleisen, L.; Weigel, B.; Frey, B.; Friedrich, R.P.; Pöttler, M.; Alexiou, C.; Janko, C. Targeting of drug-loaded nanoparticles to tumor sites increases cell death and release of danger signals. J. Control. Release 2018, 285, 67–80. [Google Scholar] [CrossRef]
- Zaloga, J.; Pöttler, M.; Leitinger, G.; Friedrich, R.P.; Almer, G.; Lyer, S.; Baum, E.; Tietze, R.; Heimke-Brinck, R.; Mangge, H.; et al. Pharmaceutical formulation of HSA hybrid coated iron oxide nanoparticles for magnetic drug targeting. Eur. J. Pharm. Biopharm. 2016, 101, 152–162. [Google Scholar] [CrossRef]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat. Protoc. 2006, 1, 1458–1461. [Google Scholar] [CrossRef]
- Schaft, N.; Dorrie, J.; Thumann, P.; Beck, V.E.; Muller, I.; Schultz, E.S.; Kampgen, E.; Dieckmann, D.; Schuler, G. Generation of an optimized polyvalent monocyte-derived dendritic cell vaccine by transfecting defined RNAs after rather than before maturation. J. Immunol. 2005, 174, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Rother, R.P.; Bell, L.; Hillmen, P.; Gladwin, M.T. The Clinical Sequelae of Intravascular Hemolysis and Extracellular Plasma Hemoglobin. JAMA 2005, 293, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Janko, C.; Zaloga, J.; Pöttler, M.; Dürr, S.; Eberbeck, D.; Tietze, R.; Lyer, S.; Alexiou, C. Strategies to optimize the biocompatibility of iron oxide nanoparticles–“SPIONs safe by design”. J. Magnet. Magnet. Mater. 2017, 431, 281–284. [Google Scholar] [CrossRef]
- Fröhlich, E. Action of Nanoparticles on Platelet Activation and Plasmatic Coagulation. Curr. Med. Chem. 2016, 23, 408–430. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, R.P.; Janko, C.; Pöttler, M.; Tripal, P.; Zaloga, J.; Cicha, I.; Dürr, S.; Nowak, J.; Odenbach, S.; Slabu, I.; et al. Flow cytometry for intracellular SPION quantification: Specificity and sensitivity in comparison with spectroscopic methods. Int. J. Nanomed. 2015, 10, 4185–4201. [Google Scholar] [CrossRef] [PubMed]
- Pietkiewicz, S.; Schmidt, J.H.; Lavrik, I.N. Quantification of apoptosis and necroptosis at the single cell level by a combination of Imaging Flow Cytometry with classical Annexin V/propidium iodide staining. J. Immunol. Methods 2015, 423, 99–103. [Google Scholar] [CrossRef]
- Zaloga, J.; Janko, C.; Agarwal, R.; Nowak, J.; Müller, R.; Boccaccini, A.R.; Lee, G.; Odenbach, S.; Lyer, S.; Alexiou, C. Different Storage Conditions Influence Biocompatibility and Physicochemical Properties of Iron Oxide Nanoparticles. Int. J. Mol. Sci. 2015, 16, 9368–9384. [Google Scholar] [CrossRef]
- Zaloga, J.; Janko, C.; Nowak, J.; Matuszak, J.; Knaup, S.; Eberbeck, D.; Tietze, R.; Unterweger, H.; Friedrich, R.P.; Duerr, S.; et al. Development of a lauric acid/albumin hybrid iron oxide nanoparticle system with improved biocompatibility. Int. J. Nanomed. 2014, 9, 4847–4866. [Google Scholar] [CrossRef]
- Bilyy, R.; Unterweger, H.; Weigel, B.; Dumych, T.; Paryzhak, S.; Vovk, V.; Liao, Z.; Alexiou, C.; Herrmann, M.; Janko, C. Inert Coats of Magnetic Nanoparticles Prevent Formation of Occlusive Intravascular Co-aggregates With Neutrophil Extracellular Traps. Front. Immunol. 2018, 9, 2266. [Google Scholar] [CrossRef]
- Lyer, S.; Singh, R.; Tietze, R.; Alexiou, C. Magnetic nanoparticles for magnetic drug targeting. Biomed. Tech. Eng. 2015, 60, 465–475. [Google Scholar] [CrossRef]
- Nigam, S.; Bahadur, D. Doxorubicin-loaded dendritic-Fe3O4 supramolecular nanoparticles for magnetic drug targeting and tumor regression in spheroid murine melanoma model. Nanomed. Nanotechnol. Boil. Med. 2018, 14, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Nowicka, A.; Kowalczyk, A.; Jarzebinska, A.; Donten, M.; Krysinski, P.; Stojek, Z.; Augustin, E.; Mazerska, Z. Progress in Targeting Tumor Cells by Using Drug-Magnetic Nanoparticles Conjugate. Biomacromolecules 2013, 14, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.; Nuß, B.; Mühlberger, M.; Unterweger, H.; Friedrich, R.P.; Alexiou, C.; Janko, C. Superparamagnetic Iron Oxide Nanoparticles Carrying Chemotherapeutics Improve Drug Efficacy in Monolayer and Spheroid Cell Culture by Enabling Active Accumulation. Nanomaterial 2020, 10, 1577. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; DeMaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef]
- Martins, I.; Tesniere, A.; Kepp, O.; Michaud, M.; Schlemmer, F.; Senovilla, L.; Séror, C.; Métivier, D.; Perfettini, J.-L.; Zitvogel, L.; et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle 2009, 8, 3723–3728. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2006, 13, 54–61. [Google Scholar] [CrossRef]
- Panaretakis, T.; Joza, N.; Modjtahedi, N.; Tesniere, A.; Vitale, I.; Durchschlag, M.; Fimia, G.M.; Kepp, O.; Piacentini, M.; Froehlich, K.-U.; et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008, 15, 1499–1509. [Google Scholar] [CrossRef]
- Dürr, S.; Lyer, S.; Mann, J.; Janko, C.; Tietze, R.; Schreiber, E.; Herrmann, M.; Alexiou, C. Real-time cell analysis of human cancer cell lines after chemotherapy with functionalized magnetic nanoparticles. Anticancer Res. 2012, 32, 1983–1989. [Google Scholar]
- Hornung, A.; Pöttler, M.; Friedrich, R.P.; Zaloga, J.; Unterweger, H.; Lyer, S.; Nowak, J.; Odenbach, S.; Alexiou, C.; Janko, C. Treatment Efficiency of Free and Nanoparticle-Loaded Mitoxantrone for Magnetic Drug Targeting in Multicellular Tumor Spheroids. Molecules 2015, 20, 18016–18030. [Google Scholar] [CrossRef]
- Mei, L.; Liu, Y.; Rao, J.; Tang, X.; Li, M.; Zhang, Z.-R.; He, Q. Enhanced Tumor Retention Effect by Click Chemistry for Improved Cancer Immunochemotherapy. ACS Appl. Mater. Interfaces 2018, 10, 17582–17593. [Google Scholar] [CrossRef]
- Kather, J.N.; Suarez-Carmona, M.; Charoentong, P.; Weis, C.-A.; Hirsch, D.; Bankhead, P.; Horning, M.; Ferber, D.; Kel, I.; Herpel, E.; et al. Topography of cancer-associated immune cells in human solid tumors. eLife 2018, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Van Der Woude, L.L.; Gorris, M.; Halilovic, A.; Figdor, C.G.; De Vries, I.J.M. Migrating into the Tumor: A Roadmap for T Cells. Trends Cancer 2017, 3, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Amini, M.A.; Abbasi, A.Z.; Cai, P.; Lip, H.; Gordijo, C.R.; Li, J.; Chen, B.; Zhang, L.; Rauth, A.M.; Wu, X.Y. Combining Tumor Microenvironment Modulating Nanoparticles with Doxorubicin to Enhance Chemotherapeutic Efficacy and Boost Antitumor Immunity. J. Natl. Cancer Inst. 2018, 111, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Janko, C.; Ratschker, T.; Nguyen, K.; Zschiesche, L.; Tietze, R.; Lyer, S.; Alexiou, C. Functionalized Superparamagnetic Iron Oxide Nanoparticles (SPIONs) as Platform for the Targeted Multimodal Tumor Therapy. Front. Oncol. 2019, 9, 59. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical Properties | Method | SPION | SPIONMTO |
|---|---|---|---|
| Hydrodynamic size (nm) and polydispersity index | Dynamic light scattering (DLS) in RPMI 1640 (Malvern Zetasizer NanoZS) | 52.7 ± 0.8 nm; 0.202 ± 0.026 | 55.7 ± 1.0 nm; 0.239 ± 0.019 |
| Core size (nm) | Freeze fracture TEM | 10 nm iron core size with 2 nm organic layer | n.d. |
| Zeta potential (mV) | Malvern Zetasizer NanoZS; in RPMI 1640 medium | −11.9 ± 0.8 | −12.4 ± 0.5 |
| Drug loading efficacy (%) (200 µg mitoxantrone (MTO) per 1 mL SPIONs) | High-performance liquid chromotograpy (HPLC)-UV of supernatant to detect unbound drug | 98.9 ± 0.2% | |
| MTO release (%) after 72 h at 37 °C in RPMI 1640 medium | (1) Dialysis bag method (2) Magnetic assay | 11.6 ± 0.1% 23.7 ± 0.4% | |
| Substance | Free MTO | SPIONMTO | SPION | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MTO [µM] | 0.5 | 1 | 2 | 5 | 0.5 | 1 | 2 | 5 | - | - | - | - |
| MTO [µg/mL] | 0.22 | 0.44 | 0.88 | 2.2 | 0.22 | 0.44 | 0.88 | 2.2 | - | - | - | - |
| Fe [µg/mL] | - | - | - | 5.32 | 10.64 | 21.28 | 53.2 | 5.32 | 10.64 | 21.28 | 53.2 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ratschker, T.; Egenberger, L.; Alev, M.; Zschiesche, L.; Band, J.; Schreiber, E.; Frey, B.; Derer, A.; Alexiou, C.; Janko, C. Mitoxantrone-Loaded Nanoparticles for Magnetically Controlled Tumor Therapy–Induction of Tumor Cell Death, Release of Danger Signals and Activation of Immune Cells. Pharmaceutics 2020, 12, 923. https://doi.org/10.3390/pharmaceutics12100923
Ratschker T, Egenberger L, Alev M, Zschiesche L, Band J, Schreiber E, Frey B, Derer A, Alexiou C, Janko C. Mitoxantrone-Loaded Nanoparticles for Magnetically Controlled Tumor Therapy–Induction of Tumor Cell Death, Release of Danger Signals and Activation of Immune Cells. Pharmaceutics. 2020; 12(10):923. https://doi.org/10.3390/pharmaceutics12100923
Chicago/Turabian StyleRatschker, Teresa, Laura Egenberger, Magdalena Alev, Lisa Zschiesche, Julia Band, Eveline Schreiber, Benjamin Frey, Anja Derer, Christoph Alexiou, and Christina Janko. 2020. "Mitoxantrone-Loaded Nanoparticles for Magnetically Controlled Tumor Therapy–Induction of Tumor Cell Death, Release of Danger Signals and Activation of Immune Cells" Pharmaceutics 12, no. 10: 923. https://doi.org/10.3390/pharmaceutics12100923
APA StyleRatschker, T., Egenberger, L., Alev, M., Zschiesche, L., Band, J., Schreiber, E., Frey, B., Derer, A., Alexiou, C., & Janko, C. (2020). Mitoxantrone-Loaded Nanoparticles for Magnetically Controlled Tumor Therapy–Induction of Tumor Cell Death, Release of Danger Signals and Activation of Immune Cells. Pharmaceutics, 12(10), 923. https://doi.org/10.3390/pharmaceutics12100923