Effects of Focused-Ultrasound-and-Microbubble-Induced Blood-Brain Barrier Disruption on Drug Transport under Liposome-Mediated Delivery in Brain Tumour: A Pilot Numerical Simulation Study


Abstract
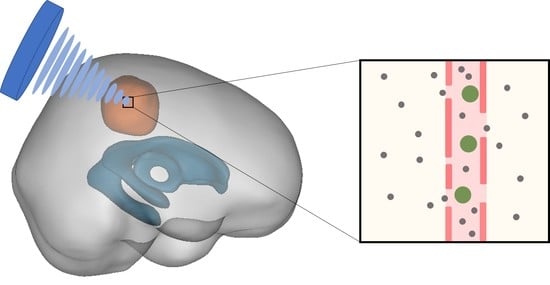
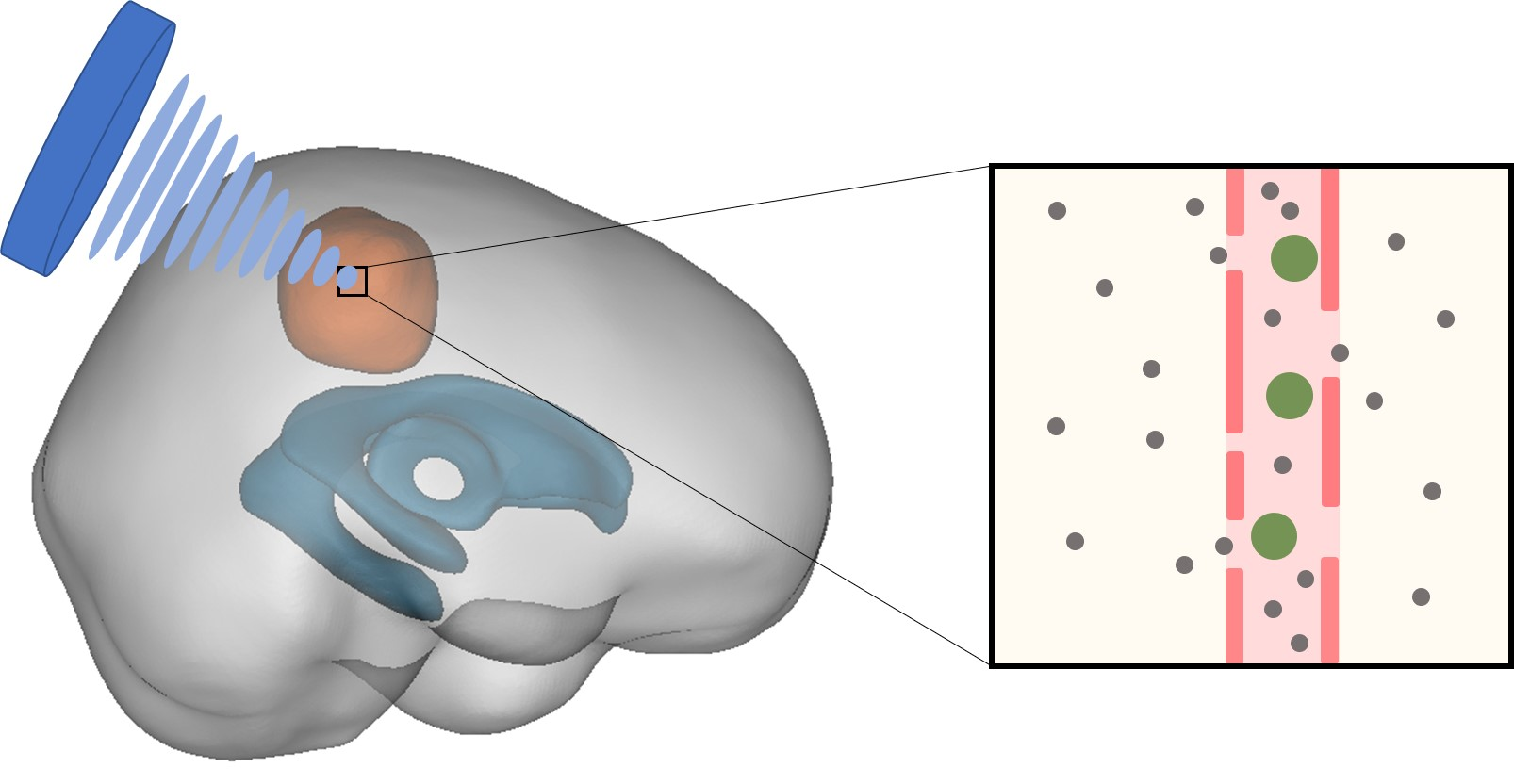
1. Introduction
2. Materials and Methods
2.1. Mathematical Model
2.1.1. Interstitial Fluid Flow
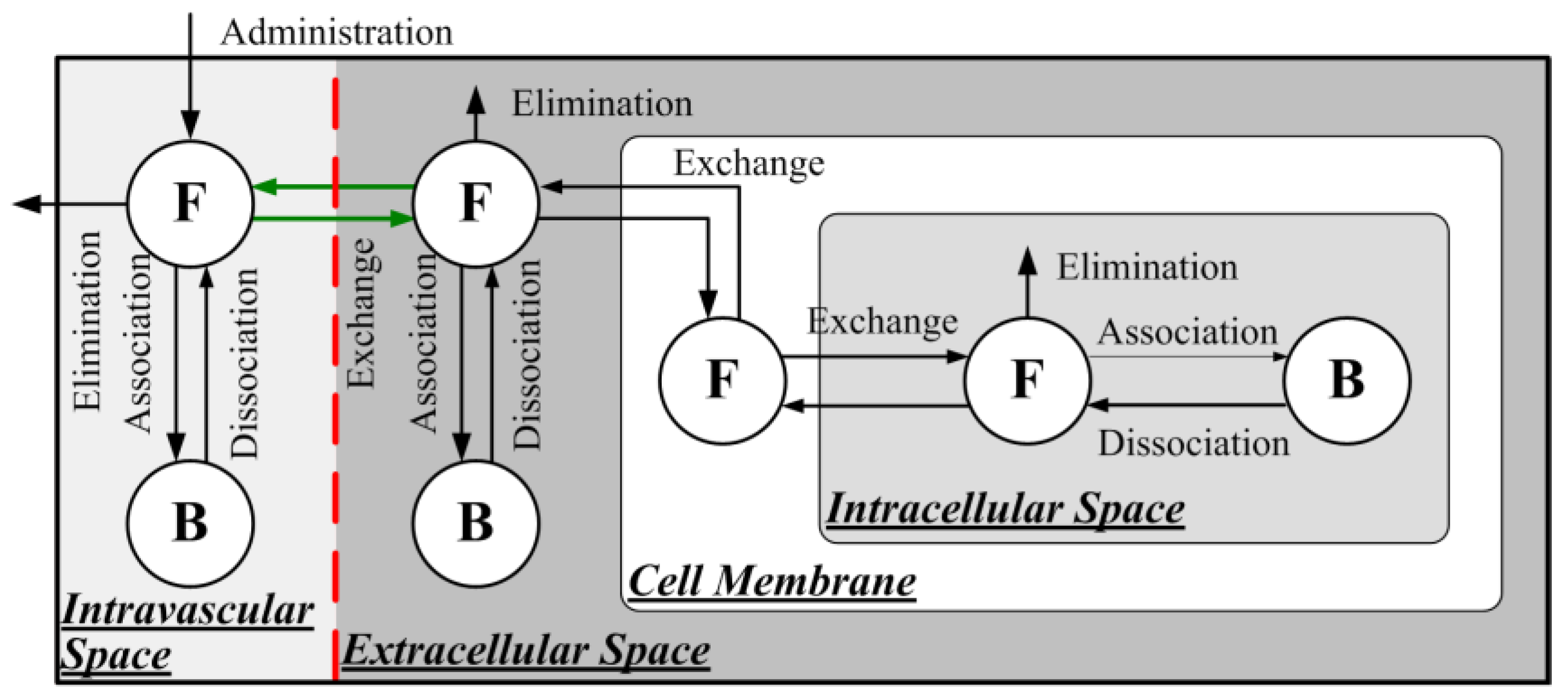
2.1.2. Direct Delivery of Free Doxorubicin
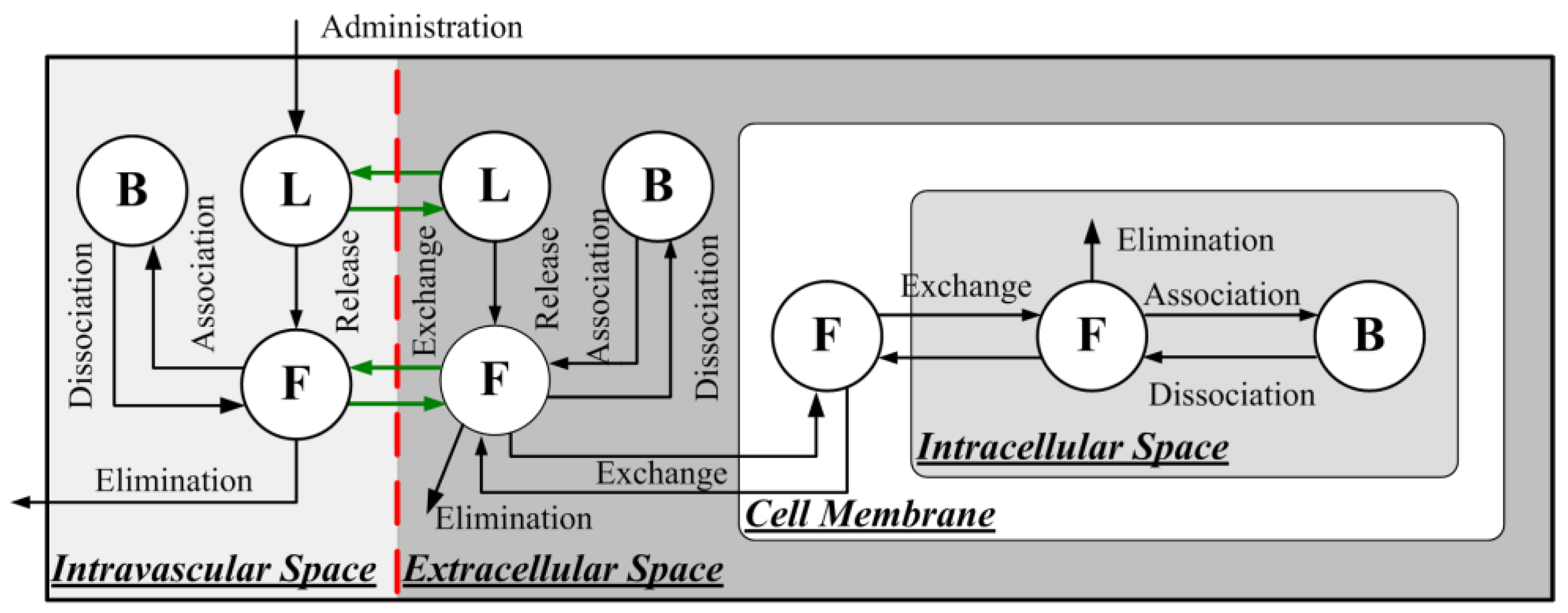
2.1.3. Delivery of Liposome-Encapsulated Doxorubicin
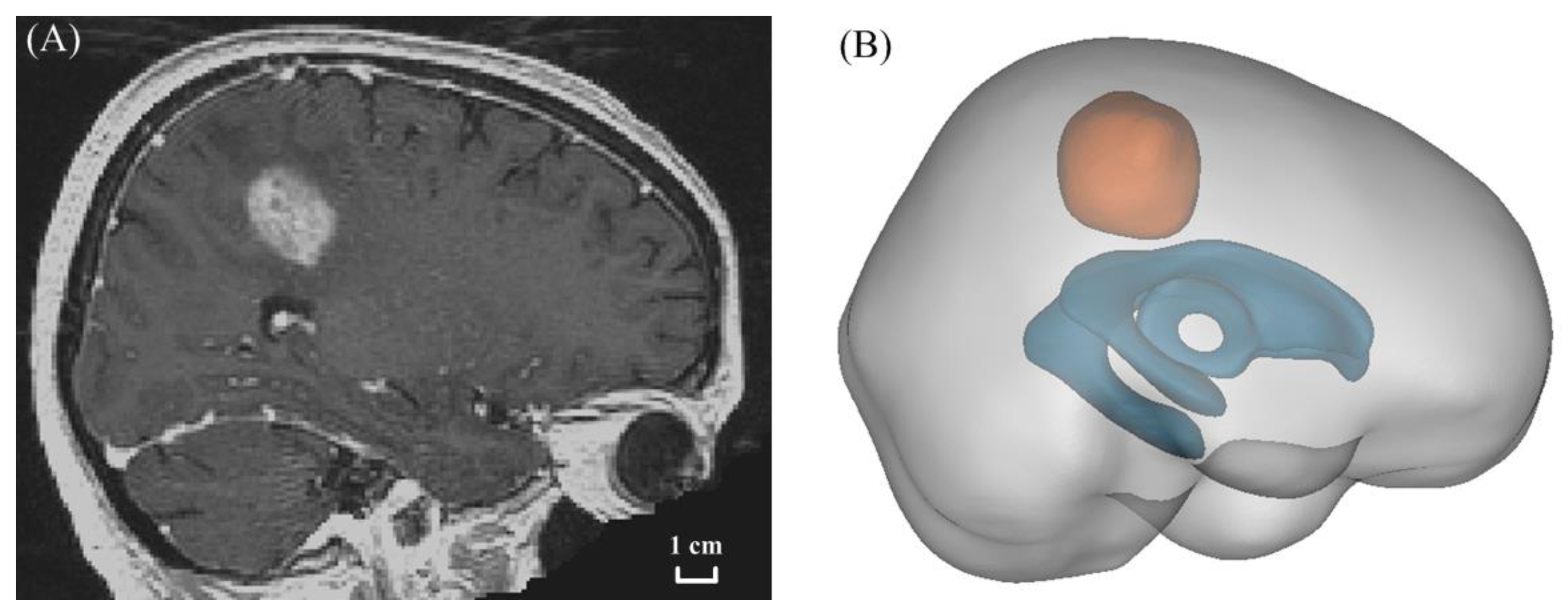
2.2. Model Geometry
2.3. Model Parameters
2.4. Numerical Methods
2.5. Boundary Conditions
2.6. Quantification of Delivery Outcomes
3. Results
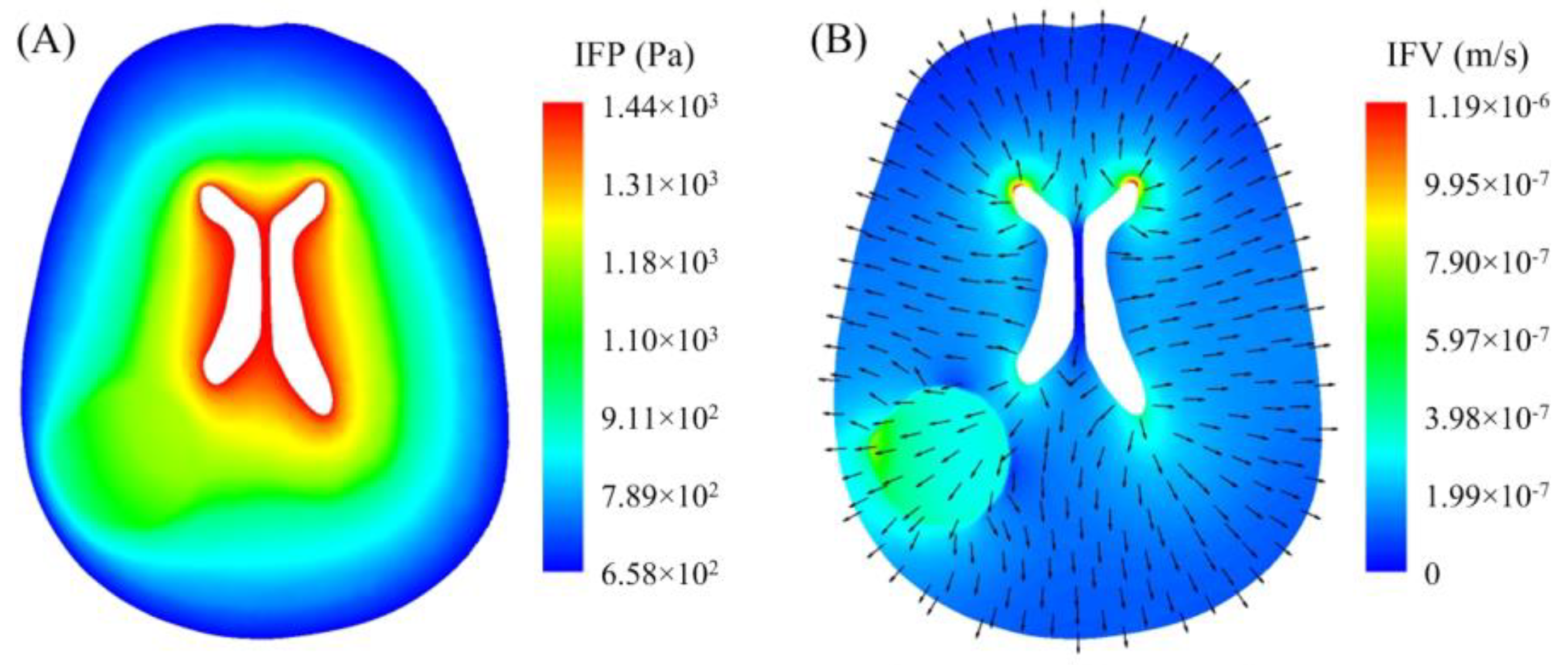
3.1. Interstitial Fluid Flow
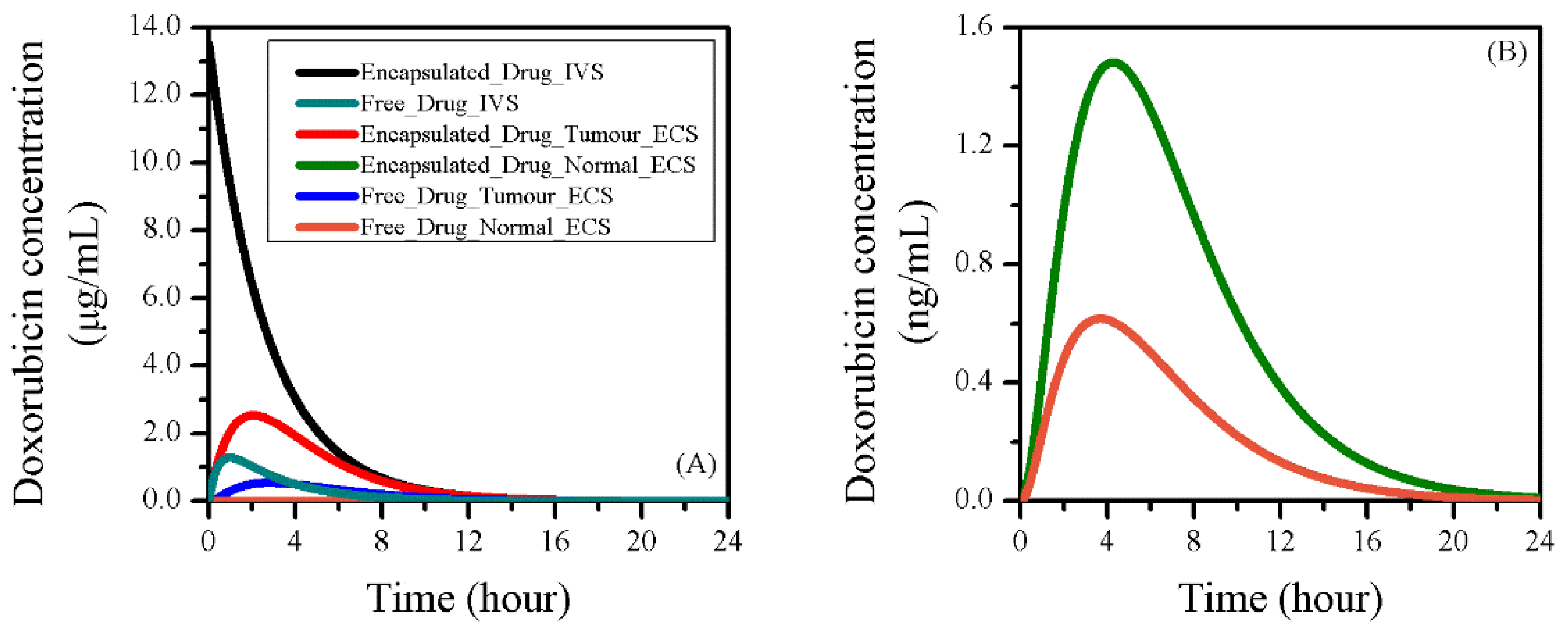
3.2. Baseline Study of Drug Transport and Accumulation
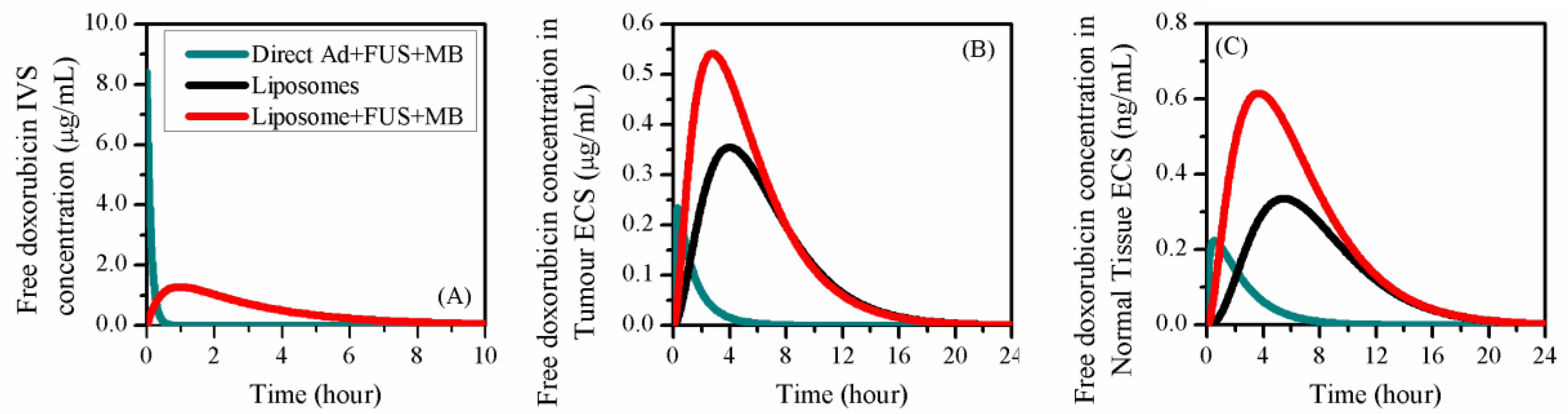
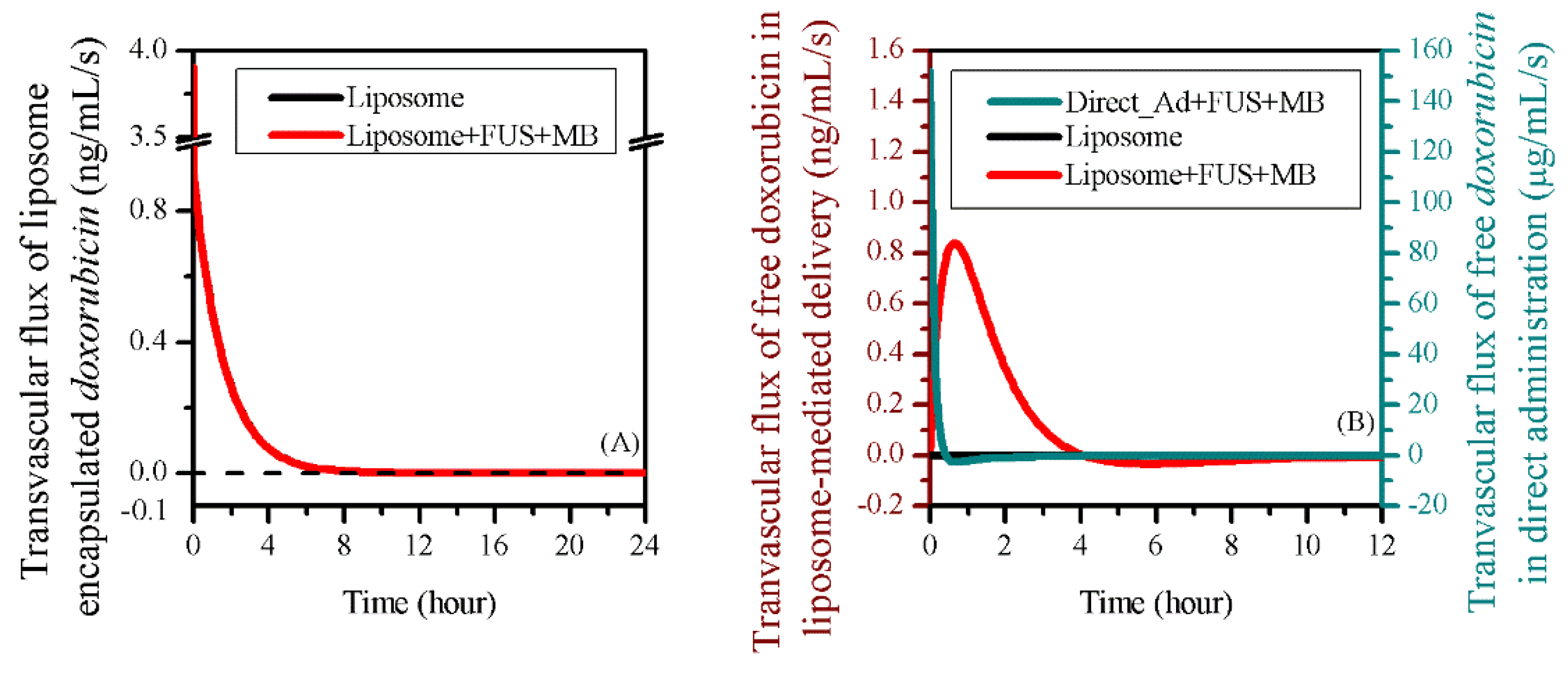
3.3. Comparisons to other Delivery Modes
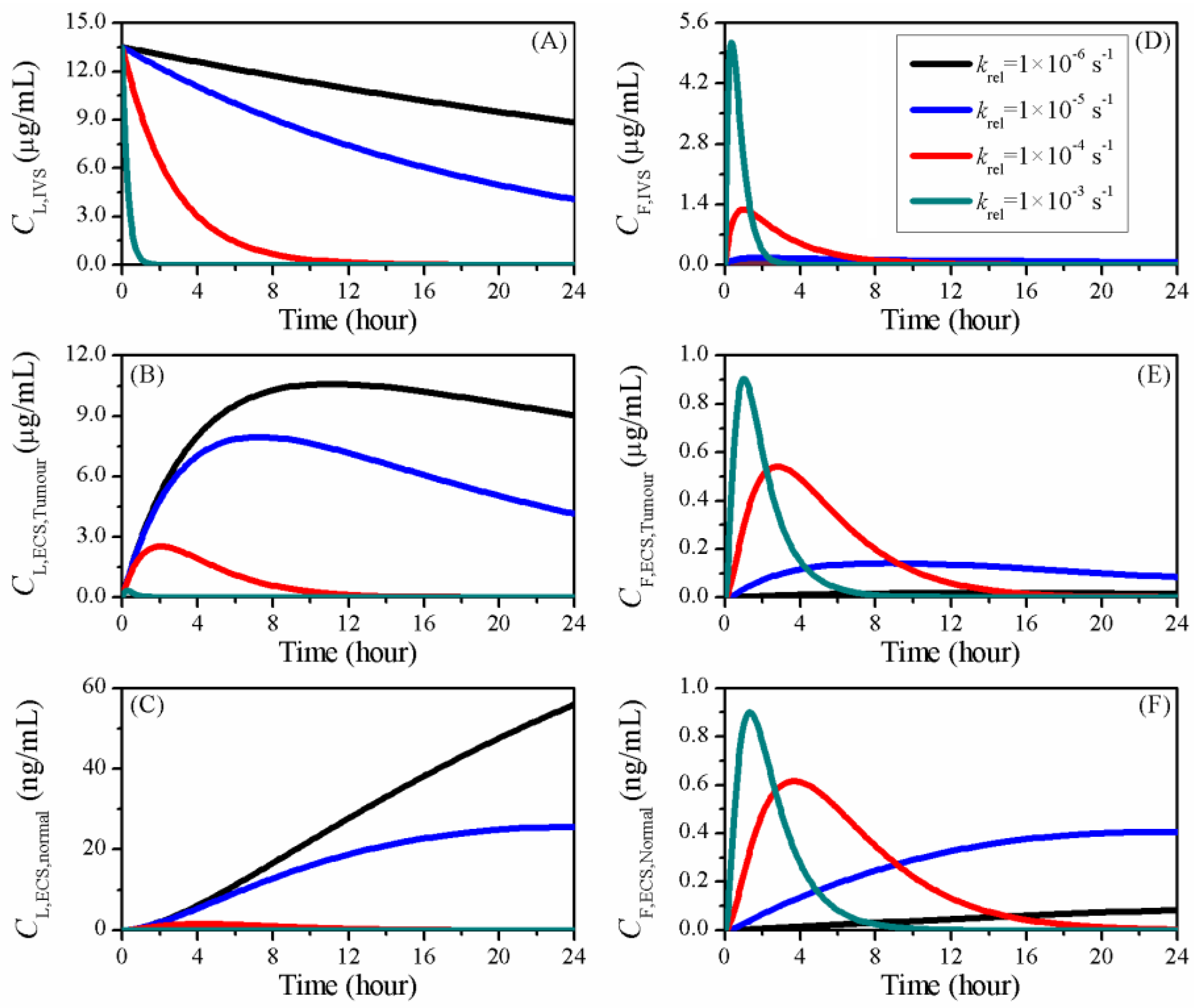
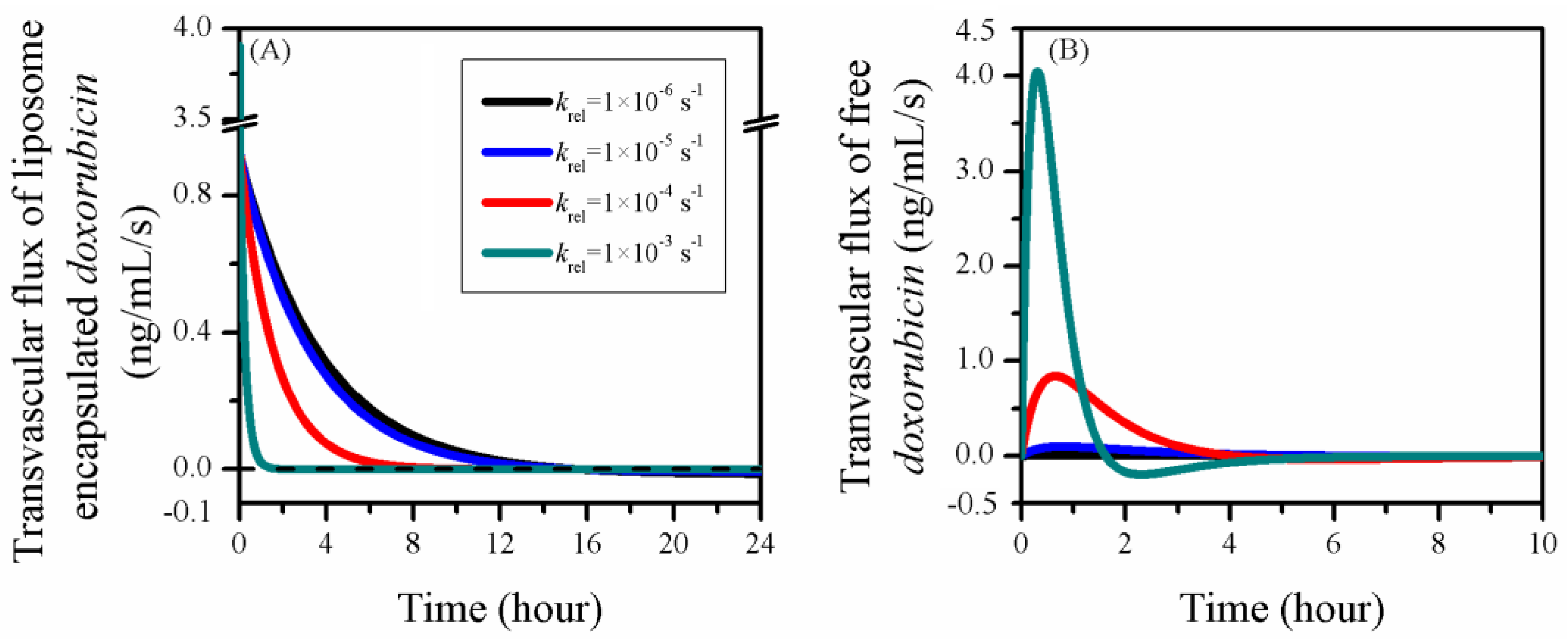
3.4. Impact of Release Rate
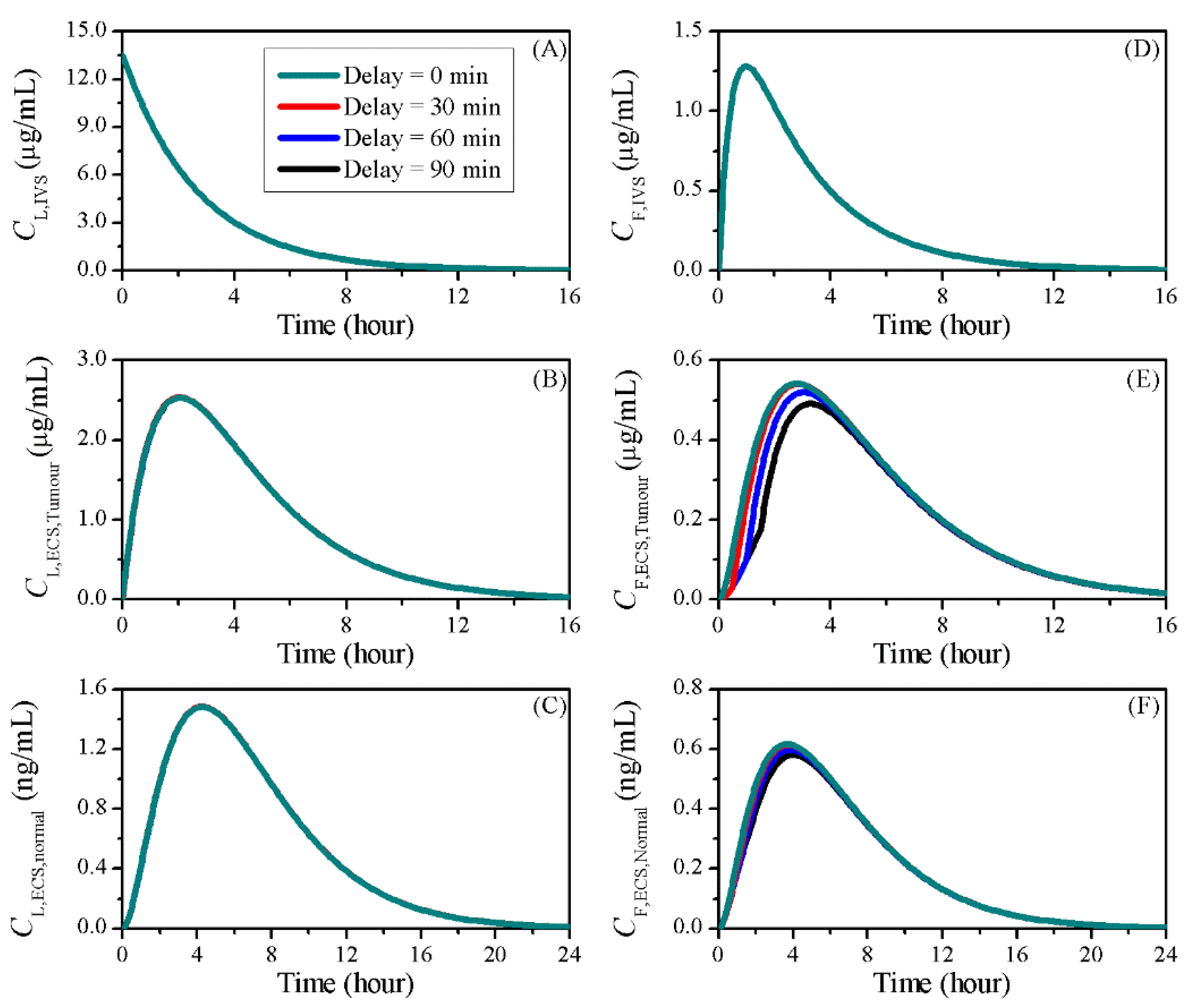
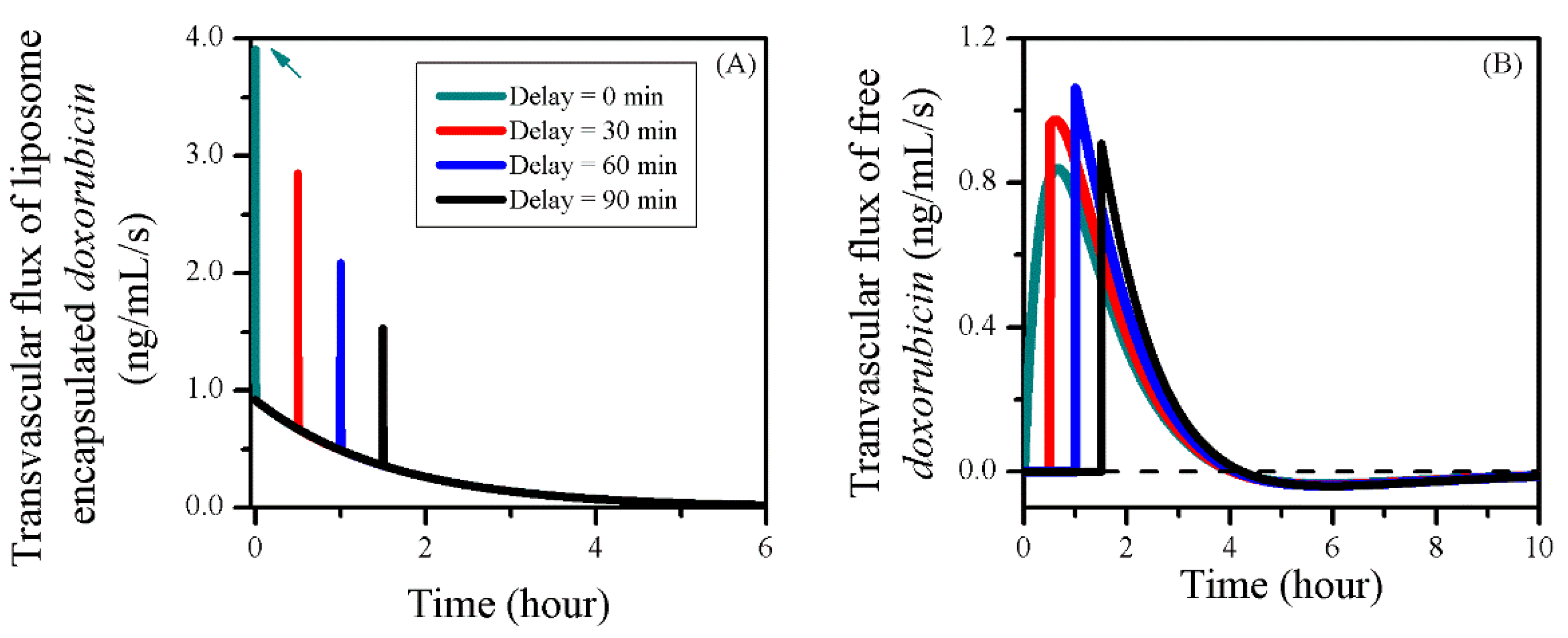
3.5. Impact of BBBD Timing
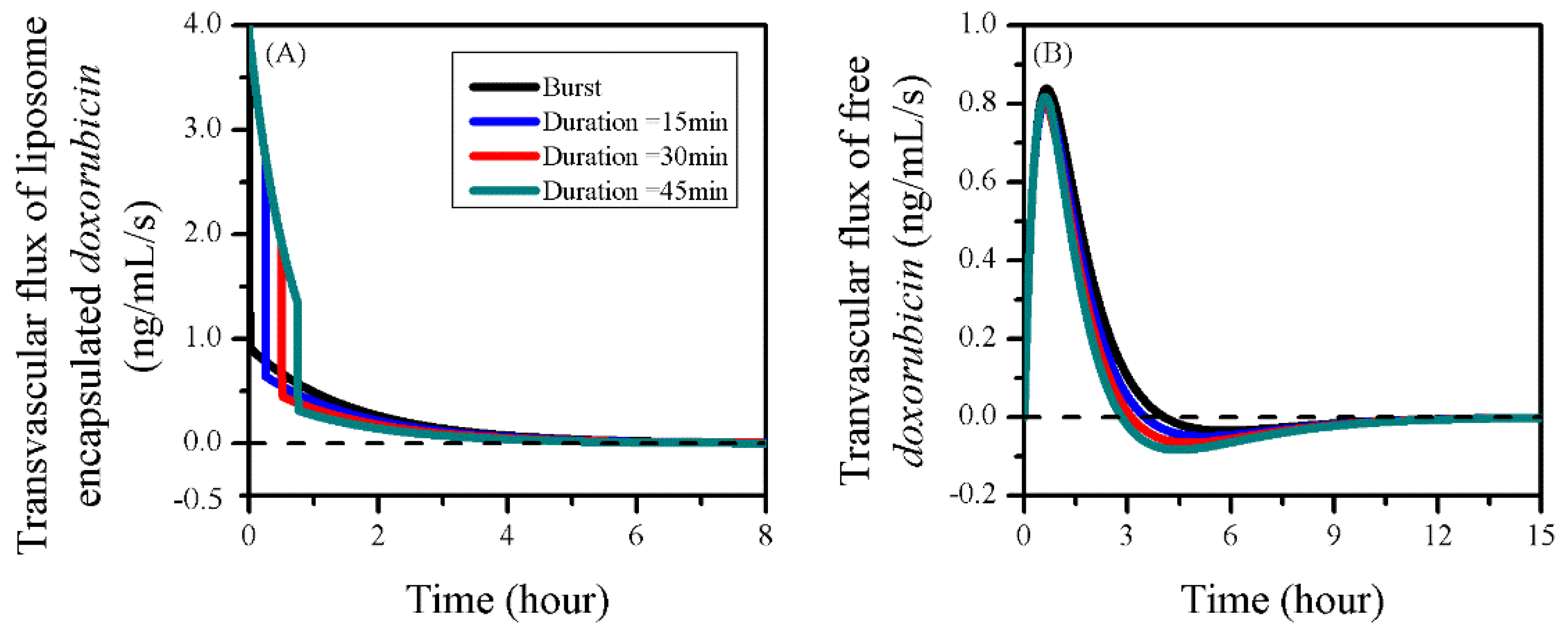
3.6. Impact of Sonication Duration
4. Discussion
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Mangiola, A.; Anile, C.; Pompucci, A.; Capone, G.; Rigante, L.; De Bonis, P. Glioblastoma therapy: Going beyond Hercules Columns. Expert Rev. Neurother. 2010, 10, 507–514. [Google Scholar] [CrossRef]
- Alam, M.I.; Beg, S.; Samad, A.; Baboota, S.; Kohli, K.; Ali, J.; Ahuja, A.; Akbar, M. Strategy for effective brain drug delivery. Eur. J. Pharm. Sci. 2010, 40, 385–403. [Google Scholar] [CrossRef]
- Zhou, J.; Patel, T.R.; Sirianni, R.W.; Strohbehn, G.; Zheng, M.-Q.; Duong, N.; Schafbauer, T.; Huttner, A.J.; Huang, Y.; Carson, R.E. Highly penetrative, drug-loaded nanocarriers improve treatment of glioblastoma. Proc. Natl. Acad. Sci. USA 2013, 110, 11751–11756. [Google Scholar] [CrossRef]
- Hynynen, K.; McDannold, N.; Vykhodtseva, N.; Jolesz, F.A. Noninvasive MR imaging–guided focal opening of the blood-brain barrier in rabbits. Radiology 2001, 220, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Samiotaki, G.; Konofagou, E.E. Dependence of the reversibility of focused-ultrasound-induced blood-brain barrier opening on pressure and pulse length in vivo. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 2013, 60, 2257–2265. [Google Scholar] [CrossRef] [PubMed]
- Bredlau, A.L.; Motamarry, A.; Chen, C.; McCrackin, M.; Helke, K.; Armeson, K.E.; Bynum, K.; Broome, A.-M.; Haemmerich, D. Localized delivery of therapeutic doxorubicin dose across the canine blood–brain barrier with hyperthermia and temperature sensitive liposomes. Drug Deliv. 2018, 25, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Veringa, S.J.; Biesmans, D.; van Vuurden, D.G.; Jansen, M.H.; Wedekind, L.E.; Horsman, I.; Wesseling, P.; Vandertop, W.P.; Noske, D.P.; Kaspers, G.J. In vitro drug response and efflux transporters associated with drug resistance in pediatric high grade glioma and diffuse intrinsic pontine glioma. PLoS ONE 2013, 8, e61512. [Google Scholar] [CrossRef]
- Barenholz, Y.C. Doxil®—the first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Zhan, W.; Alamer, M.; Xu, X.Y. Computational modelling of drug delivery to solid tumour: Understanding the interplay between chemotherapeutics and biological system for optimised delivery system. Adv. Drug Deliv. Rev. 2018, 132, 81–103. [Google Scholar] [CrossRef]
- Baxter, L.T.; Jain, R.K. Transport of fluid and macromolecules in tumors: III. Role of binding and metabolism. Microvasc. Res. 1991, 41, 5–23. [Google Scholar] [CrossRef]
- Baxter, L.T.; Jain, R.K. Transport of fluid and macromolecules in tumors. II. Role of heterogeneous perfusion and lymphatics. Microvasc. Res. 1990, 40, 246–263. [Google Scholar] [CrossRef]
- Baxter, L.T.; Jain, R.K. Transport of fluid and macromolecules in tumors. IV. A microscopic model of the perivascular distribution. Microvasc. Res. 1991, 41, 252–272. [Google Scholar] [CrossRef]
- Tan, W.H.K.; Lee, T.; Wang, C.-H. Delivery of Etanidazole to Brain Tumor from PLGA Wafers: A Double Burst Release System. Biotechnol. Bioeng. 2003, 82, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Tzafriri, A.R.; Lerner, E.I.; Flashner-Barak, M.; Hinchcliffe, M.; Ratner, E.; Parnas, H. Mathematical modeling and optimization of drug delivery from intratumorally injected microspheres. Clin. Cancer Res. 2005, 11, 826–834. [Google Scholar]
- Lee, C.G.; Fu, Y.C.; Wang, C.H. Simulation of gentamicin delivery for the local treatment of osteomyelitis. Biotechnol. Bioeng. 2005, 91, 622–635. [Google Scholar] [CrossRef]
- Arifin, D.Y.; Lee, K.Y.T.; Wang, C.-H. Chemotherapeutic drug transport to brain tumor. J. Control. Release 2009, 137, 203–210. [Google Scholar] [CrossRef]
- Goh, Y.-M.F.; Kong, H.L.; Wang, C.-H. Simulation of the delivery of doxorubicin to hepatoma. Pharm. Res. 2001, 18, 761–770. [Google Scholar] [CrossRef]
- Liu, C.; Xu, X.Y. A systematic study of temperature sensitive liposomal delivery of doxorubicin using a mathematical model. Comput. Biol. Med. 2015, 60, 107–116. [Google Scholar] [CrossRef] [PubMed]
- El-Kareh, A.W.; Secomb, T.W. A mathematical model for comparison of bolus injection, continuous infusion, and liposomal delivery of doxorubicin to tumor cells. Neoplasia 2000, 2, 325–338. [Google Scholar] [CrossRef]
- Eikenberry, S. A tumor cord model for doxorubicin delivery and dose optimization in solid tumors. Theor. Biol. Med Model. 2009, 6, 16–35. [Google Scholar] [CrossRef]
- Nhan, T.; Burgess, A.; Lilge, L.; Hynynen, K. Modeling localized delivery of Doxorubicin to the brain following focused ultrasound enhanced blood-brain barrier permeability. Phys. Med. Biol. 2014, 59, 5987. [Google Scholar] [CrossRef] [PubMed]
- Dreher, M.R.; Liu, W.; Michelich, C.R.; Dewhirst, M.W.; Yuan, F.; Chilkoti, A. Tumor vascular permeability, accumulation, and penetration of macromolecular drug carriers. J. Natl. Cancer Inst. 2006, 98, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Zhan, W.; Wang, C.-H. Convection enhanced delivery of chemotherapeutic drugs into brain tumour. J. Control. Release 2018, 271, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Saltzman, W.M.; Radomsky, M.L. Drugs released from polymers: Diffusion and elimination in brain tissue. Chem. Eng. Sci. 1991, 46, 2429–2444. [Google Scholar] [CrossRef]
- Barboriak, D. Data From RIDER_NEURO_MRI. Cancer Imaging Arch. 2015. [Google Scholar] [CrossRef]
- Clark, K.; Vendt, B.; Smith, K.; Freymann, J.; Kirby, J.; Koppel, P.; Moore, S.; Phillips, S.; Maffitt, D.; Pringle, M. The Cancer Imaging Archive (TCIA): Maintaining and operating a public information repository. J. Digit. Imaging 2013, 26, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.-Q.; Lv, Q.; Li, L.-M.; Tang, X.-J.; Li, F.-Z.; Hu, Y.-L.; Han, M. Glioma targeting and blood–brain barrier penetration by dual-targeting doxorubincin liposomes. Biomaterials 2013, 34, 5628–5639. [Google Scholar] [CrossRef]
- Qin, L.; Wang, C.Z.; Fan, H.J.; Zhang, C.J.; Zhang, H.W.; Lv, M.H.; Cui, S.D. A dual-targeting liposome conjugated with transferrin and arginine-glycine-aspartic acid peptide for glioma-targeting therapy. Oncol. Lett. 2014, 8, 2000–2006. [Google Scholar] [CrossRef]
- Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; Berk, D.A.; Torchilin, V.P.; Jain, R.K. Vascular permeability in a human tumor xenograft: Molecular size dependence and cutoff size. Cancer Res. 1995, 55, 3752–3756. [Google Scholar]
- Vlachos, F.; Tung, Y.S.; Konofagou, E. Permeability dependence study of the focused ultrasound-induced blood–brain barrier opening at distinct pressures and microbubble diameters using DCE-MRI. Magn. Reson. Med. 2011, 66, 821–830. [Google Scholar] [CrossRef]
- Shen, Y.; Pi, Z.; Yan, F.; Yeh, C.-K.; Zeng, X.; Diao, X.; Hu, Y.; Chen, S.; Chen, X.; Zheng, H. Enhanced delivery of paclitaxel liposomes using focused ultrasound with microbubbles for treating nude mice bearing intracranial glioblastoma xenografts. Int. J. Nanomed. 2017, 12, 5613–5629. [Google Scholar] [CrossRef]
- Marty, B.; Larrat, B.; Van Landeghem, M.; Robic, C.; Robert, P.; Port, M.; Le Bihan, D.; Pernot, M.; Tanter, M.; Lethimonnier, F. Dynamic study of blood–brain barrier closure after its disruption using ultrasound: A quantitative analysis. J. Cereb. Blood Flow Metab. 2012, 32, 1948–1958. [Google Scholar] [CrossRef]
- Fung, L.K.; Shin, M.; Tyler, B.; Brem, H.; Saltzman, W.M. Chemotherapeutic drugs released from polymers: Distribution of 1, 3-bis (2-chloroethyl)-l-nitrosourea in the rat brain. Pharm. Res. 1996, 13, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Formariz, T.; Sarmento, V.; Silva-Junior, A.; Scarpa, M.; Santilli, C.V.; Oliveira, A. Doxorubicin biocompatible O/W microemulsion stabilized by mixed surfactant containing soya phosphatidylcholine. Colloids Surf. B Biointerfaces 2006, 51, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Greene, R.F.; Collins, J.M.; Jenkins, J.F.; Speyer, J.L.; Myers, C.E. Plasma pharmacokinetics of adriamycin and adriamycinol: Implications for the design of in vitro experiments and treatment protocols. Cancer Res. 1983, 43, 3417–3421. [Google Scholar] [PubMed]
- Yuan, F.; Leunig, M.; Huang, S.K.; Berk, D.A.; Papahadjopoulos, D.; Jain, R.K. Mirovascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994, 54, 3352–3356. [Google Scholar]
- Zhan, W.; Gedroyc, W.; Xu, X.Y. Towards a multiphysics modelling framework for thermosensitive liposomal drug delivery to solid tumour combined with focused ultrasound hyperthermia. Biophys. Rep. 2019, 5, 43–59. [Google Scholar] [CrossRef]
- Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar]
- Robert, J.; Illiadis, A.; Hoerni, B.; Cano, J.-P.; Durand, M.; Lagarde, C. Pharmacokinetics of adriamycin in patients with breast cancer: Correlation between pharmacokinetic parameters and clinical short-term response. Eur. J. Cancer 1982, 18, 739–745. [Google Scholar] [CrossRef]
- Xu, H.; Hu, M.; Yu, X.; Li, Y.; Fu, Y.; Zhou, X.; Zhang, D.; Li, J. Design and evaluation of pH-sensitive liposomes constructed by poly (2-ethyl-2-oxazoline)-cholesterol hemisuccinate for doxorubicin delivery. Eur. J. Pharm. Biopharm. 2015, 91, 66–74. [Google Scholar] [CrossRef]
- Kalyanasundaram, S.; Calhoun, V.; Leong, K. A finite element model for predicting the distribution of drugs delivered intracranially to the brain. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1997, 273, R1810–R1821. [Google Scholar] [CrossRef]
- Green, D.W.; Perry, R.H. Perry’s Chemical Engineers’ Handbook/edición; The McGraw-Hill Companies, Inc.: Beijin, China, 1973; Volume 100, p. 660-28. [Google Scholar]
- Kimelberg, H. Water homeostasis in the brain: Basic concepts. Neuroscience 2004, 129, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Salmon, H.; Sarntinoranont, M. Effect of heterogeneous vasculature on interstitial transport within a solid tumor. Microvasc. Res. 2007, 73, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Gasselhuber, A.; Dreher, M.R.; Partanen, A.; Yarmolenko, P.S.; Woods, D.; Wood, B.J.; Haemmerich, D. Targeted drug delivery by high intensity focused ultrasound mediated hyperthermia combined with temperature-sensitive liposomes: Computational modelling and preliminary in vivo validation. Int. J. Hyperth. 2012, 28, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Soltani, M.; Chen, P. Numerical modeling of interstitial fluid flow coupled with blood flow through a remodeled solid tumor microvascular network. PLoS ONE 2013, 8, e67025. [Google Scholar] [CrossRef] [PubMed]
- Gross, J.F.; Popel, A.S. Mathematical Models of Transport Phenomena in Normal and Neoplastic Tissue; CRC Press: Boca Raton, FL, USA, 1979. [Google Scholar]
- Baxter, L.T.; Jain, R.K. Transport of fluid and macromolecules in tumors. I. Role of interstitial pressure and convection. Microvasc. Res. 1989, 37, 77–104. [Google Scholar] [CrossRef]
- Swabb, E.A.; Wei, J.; Gullino, P.M. Diffusion and convection in normal and neoplastic tissues. Cancer Res. 1974, 34, 2814–2822. [Google Scholar]
- Kinoshita, M.; McDannold, N.; Jolesz, F.A.; Hynynen, K. Noninvasive localized delivery of Herceptin to the mouse brain by MRI-guided focused ultrasound-induced blood–brain barrier disruption. Proc. Natl. Acad. Sci. USA 2006, 103, 11719–11723. [Google Scholar] [CrossRef]
- Yang, F.-Y.; Lin, Y.-S.; Kang, K.-H.; Chao, T.-K. Reversible blood–brain barrier disruption by repeated transcranial focused ultrasound allows enhanced extravasation. J. Control. Release 2011, 150, 111–116. [Google Scholar] [CrossRef]
- Park, J.; Zhang, Y.; Vykhodtseva, N.; Jolesz, F.A.; McDannold, N.J. The kinetics of blood brain barrier permeability and targeted doxorubicin delivery into brain induced by focused ultrasound. J. Control. Release 2012, 162, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Mayer, L.D.; Tai, L.C.; Ko, D.S.; Masin, D.; Ginsberg, R.S.; Cullis, P.R.; Bally, M.B. Influence of vesicle size, lipid composition, and drug-to-lipid ratio on the biological activity of liposomal doxorubicin in mice. Cancer Res. 1989, 49, 5922–5930. [Google Scholar] [PubMed]
- Bally, M.B.; Nayar, R.; Masin, D.; Cullis, P.R.; Mayer, L.D. Studies on the myelosuppressive activity of doxorubicin entrapped in liposomes. Cancer Chemother. Pharmacol. 1990, 27, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Mehra, T.; Hansen, C.; Chin, Y.C. Stealth liposomes: An improved sustained release system for 1-β-D-arabinofuranosylcytosine. Cancer Res. 1992, 52, 2431–2439. [Google Scholar] [PubMed]
- Tagami, T.; Ernsting, M.J.; Li, S.-D. Optimization of a novel and improved thermosensitive liposome formulated with DPPC and a Brij surfactant using a robust in vitro system. J. Control. Release 2011, 154, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.; Yatvin, M.B.; Huang, L. pH-sensitive liposomes: Acid-induced liposome fusion. Proc. Natl. Acad. Sci. USA 1984, 81, 1715–1718. [Google Scholar] [CrossRef]
- Garcion, E.; Lamprecht, A.; Heurtault, B.; Paillard, A.; Aubert-Pouessel, A.; Denizot, B.; Menei, P.; Benoît, J.-P. A new generation of anticancer, drug-loaded, colloidal vectors reverses multidrug resistance in glioma and reduces tumor progression in rats. Mol. Cancer Ther. 2006, 5, 1710–1722. [Google Scholar] [CrossRef]
- Tagami, T.; May, J.P.; Ernsting, M.J.; Li, S.-D. A thermosensitive liposome prepared with a Cu 2+ gradient demonstrates improved pharmacokinetics, drug delivery and antitumor efficacy. J. Control. Release 2012, 161, 142–149. [Google Scholar] [CrossRef]
- Treat, L.H.; McDannold, N.; Vykhodtseva, N.; Zhang, Y.; Tam, K.; Hynynen, K. Targeted delivery of doxorubicin to the rat brain at therapeutic levels using MRI-guided focused ultrasound. Int. J. Cancer 2007, 121, 901–907. [Google Scholar] [CrossRef]
- Hwang, J.H.; Brayman, A.A.; Reidy, M.A.; Matula, T.J.; Kimmey, M.B.; Crum, L.A. Vascular effects induced by combined 1-MHz ultrasound and microbubble contrast agent treatments in vivo. Ultrasound Med. Biol. 2005, 31, 553–564. [Google Scholar] [CrossRef]
- McDannold, N.; Vykhodtseva, N.; Hynynen, K. Effects of acoustic parameters and ultrasound contrast agent dose on focused-ultrasound induced blood-brain barrier disruption. Ultrasound Med. Biol. 2008, 34, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Konofagou, E.E. The size of blood–brain barrier opening induced by focused ultrasound is dictated by the acoustic pressure. J. Cereb. Blood Flow Metab. 2014, 34, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.-Y.; Liu, S.-H.; Ho, F.-M.; Chang, C.-H. Effect of ultrasound contrast agent dose on the duration of focused-ultrasound-induced blood-brain barrier disruption. J. Acoust. Soc. Am. 2009, 126, 3344–3349. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.-Y.; Fu, W.-M.; Chen, W.-S.; Yeh, W.-L.; Lin, W.-L. Quantitative evaluation of the use of microbubbles with transcranial focused ultrasound on blood–brain-barrier disruption. Ultrason. Sonochem. 2008, 15, 636–643. [Google Scholar] [CrossRef]
- Choi, J.J.; Feshitan, J.A.; Baseri, B.; Wang, S.; Tung, Y.-S.; Borden, M.A.; Konofagou, E.E. Microbubble-Size Dependence of Focused Ultrasound-Induced Blood–Brain Barrier Opening in MiceIn Vivo. IEEE Trans. Biomed. Eng. 2009, 57, 145–154. [Google Scholar] [CrossRef]
- Nhan, T.; Burgess, A.; Cho, E.E.; Stefanovic, B.; Lilge, L.; Hynynen, K. Drug delivery to the brain by focused ultrasound induced blood–brain barrier disruption: Quantitative evaluation of enhanced permeability of cerebral vasculature using two-photon microscopy. J. Control. Release 2013, 172, 274–280. [Google Scholar] [CrossRef]
- Hynynen, K.; McDannold, N.; Sheikov, N.A.; Jolesz, F.A.; Vykhodtseva, N. Local and reversible blood–brain barrier disruption by noninvasive focused ultrasound at frequencies suitable for trans-skull sonications. Neuroimage 2005, 24, 12–20. [Google Scholar] [CrossRef]
- McDannold, N.; Vykhodtseva, N.; Hynynen, K. Targeted disruption of the blood–brain barrier with focused ultrasound: Association with cavitation activity. Phys. Med. Biol. 2006, 51, 793–807. [Google Scholar] [CrossRef]
- Todd, N.; Zhang, Y.; Arcaro, M.; Becerra, L.; Borsook, D.; Livingstone, M.; McDannold, N. Focused ultrasound induced opening of the blood-brain barrier disrupts inter-hemispheric resting state functional connectivity in the rat brain. Neuroimage 2018, 178, 414–422. [Google Scholar] [CrossRef]
- Roux, E.; Passirani, C.; Scheffold, S.; Benoit, J.-P.; Leroux, J.-C. Serum-stable and long-circulating, PEGylated, pH-sensitive liposomes. J. Control. Release 2004, 94, 447–451. [Google Scholar] [CrossRef]
- McDannold, N.; Arvanitis, C.D.; Vykhodtseva, N.; Livingstone, M.S. Temporary disruption of the blood–brain barrier by use of ultrasound and microbubbles: Safety and efficacy evaluation in rhesus macaques. Cancer Res. 2012, 72, 3652–3663. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Nyborg, W.L. Ultrasound, cavitation bubbles and their interaction with cells. Adv. Drug Deliv. Rev. 2008, 60, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.-Y.; Lin, G.-L.; Horng, S.-C.; Chang, T.-K.; Wu, S.-Y.; Wong, T.-T.; Wang, H.-E. Pulsed high-intensity focused ultrasound enhances the relative permeability of the blood–tumor barrier in a glioma-bearing rat model. IEEE Trans. Ultrason. Ferroelectr. Freq. Control 2011, 58, 964–970. [Google Scholar] [CrossRef]
- Carpentier, A.; Canney, M.; Vignot, A.; Reina, V.; Beccaria, K.; Horodyckid, C.; Karachi, C.; Leclercq, D.; Lafon, C.; Chapelon, J.-Y. Clinical trial of blood-brain barrier disruption by pulsed ultrasound. Sci. Transl. Med. 2016, 8, re342–re343. [Google Scholar] [CrossRef] [PubMed]
- Aryal, M.; Park, J.; Vykhodtseva, N.; Zhang, Y.-Z.; McDannold, N. Enhancement in blood-tumor barrier permeability and delivery of liposomal doxorubicin using focused ultrasound and microbubbles: Evaluation during tumor progression in a rat glioma model. Phys. Med. Biol. 2015, 60, 2511. [Google Scholar] [CrossRef] [PubMed]
- Zhan, W.; Gedroyc, W.; Yun Xu, X. Mathematical modelling of drug transport and uptake in a realistic model of solid tumour. Protein Pept. Lett. 2014, 21, 1146–1156. [Google Scholar] [CrossRef] [PubMed]
- Boucher, Y.; Jain, R.K. Microvascular pressure is the principal driving force for interstitial hypertension in solid tumors: Implications for vascular collapse. Cancer Res. 1992, 52, 5110–5114. [Google Scholar]
- Butler, T.P.; Grantham, F.H.; Gullino, P.M. Bulk transfer of fluid in the interstitial compartment of mammary tumors. Cancer Res. 1975, 35, 3084–3088. [Google Scholar]
- Neeves, K.; Lo, C.; Foley, C.; Saltzman, W.; Olbricht, W. Fabrication and characterization of microfluidic probes for convection enhanced drug delivery. J. Control. Release 2006, 111, 252–262. [Google Scholar] [CrossRef]
- Bhandari, A.; Bansal, A.; Singh, A.; Sinha, N. Perfusion kinetics in human brain tumor with DCE-MRI derived model and CFD analysis. J. Biomech. 2017, 59, 80–89. [Google Scholar] [CrossRef]
- Ranganath, S.H.; Fu, Y.; Arifin, D.Y.; Kee, I.; Zheng, L.; Lee, H.-S.; Chow, P.K.-H.; Wang, C.-H. The use of submicron/nanoscale PLGA implants to deliver paclitaxel with enhanced pharmacokinetics and therapeutic efficacy in intracranial glioblastoma in mice. Biomaterials 2010, 31, 5199–5207. [Google Scholar] [CrossRef] [PubMed]
- Zhan, W.; Wang, C.-H. Convection enhanced delivery of liposome encapsulated doxorubicin for brain tumour therapy. J. Control. Release 2018, 285, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Maurer, N.; Fenske, D.B.; Cullis, P.R. Developments in liposomal drug delivery systems. Expert Opin. Biol. Ther. 2001, 1, 923–947. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.R.; Bondurant, B.; McLean, S.D.; McGovern, K.A.; O’Brien, D.F. Liposome—cell interactions in vitro: Effect of liposome surface charge on the binding and endocytosis of conventional and sterically stabilized liposomes. Biochemistry 1998, 37, 12875–12883. [Google Scholar] [CrossRef]
- Vertut-Doï, A.; Ishiwata, H.; Miyajima, K. Binding and uptake of liposomes containing a poly (ethylene glycol) derivative of cholesterol (stealth liposomes) by the macrophage cell line J774: Influence of PEG content and its molecular weight. Biochim. et Biophys. Acta Biomembr. 1996, 1278, 19–28. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Parameter | Unit | Liposome | Doxorubicin |
|---|---|---|---|---|
| PICS-ECS | Partition coefficient between ICS and ECS | - | - | 1.0 [33] |
| PCM-ECS | Partition coefficient between CM and ECS | - | - | 0.3 [34] |
| KIVS, KECS, KICS | Binding constant in IVS, ECS and ICS | - | - | 3.0 [35] |
| DECS | Diffusion coefficient in tissue ECS | m2/s | 9.0 × 10−12 (T) [36] 5.8 × 10−12 (N) [36] | 3.4 × 10−10 (T) [17] 1.6 × 10−10 (N) [17] |
| P0 | Transvascular permeability with BBBD | m/s | 3.4 × 10−9 (T) [29] 0.0 (N) | 0.0 (T) 0.0 (N) |
| σ | Drug osmotic reflection coefficient | - | 0.95 (T) [37] 1.0 (N) [37] | 0.15 (T) [17] 0.15 (N) [17] |
| ke | Drug elimination rate in tissue | s−1 | - | 5.8 × 10−4 [17] |
| kc | Drug clearance rate in blood | s−1 | 3.9 × 10−6 [38] | 2.4 × 10−3 [39] |
| krel | Drug release rate from liposomes | s−1 | 1.0 × 10−4 [40] | - |
| Vd | Distribution volume | m3 | 6.4 × 10−3 [36] | 7.7 × 10−3 [39] |
| Symbol | Parameter | Unit | Brain Tumour | Normal Tissue |
|---|---|---|---|---|
| α | Volume fraction of ECS | - | 0.35 [41] | 0.20 [33] |
| β | Volume fraction of ICS | - | 0.55 [41] | 0.65 [33] |
| ρ | Density of interstitial fluid | kg/m3 | 1.0 × 103 [42] | 1.0 × 103 [42] |
| μ | Viscosity of interstitial fluid | kg/m/s | 7.8 × 10−4 [42] | 7.8 × 10−4 [42] |
| πb | Osmotic pressure of blood | Pa | 3.4 × 103 [43] | 3.4 × 103 [43] |
| πi | Osmotic pressure of interstitial fluid | Pa | 1.1 × 103 [12] | 7.4 × 102 [12] |
| pb | Pressure in intravascular space | Pa | 4.6 × 103 [43] | 4.6 × 103 [43] |
| S/V | Area of blood vessel surface per tissue volume | m−1 | 2.0 × 104 [12] | 7.0 × 103 [12] |
| σT | Osmotic reflection coefficient of tissue | - | 0.82 [12] | 0.91 [12] |
| Kb | Hydraulic conductivity of blood vessel wall | m/Pa/s | 1.1 × 10−12 [16] | 1.4 × 10−13 [16] |
| κ | Tissue Darctian permeability | m2 | 6.4 × 10−14 [16] | 6.5 × 10−15 [16] |
| Tissue Type | ||
|---|---|---|
| Brain tumour | 1071.79 | 0.43 |
| Normal tissue | 876.03 | 0.13 |
| Delivery Mode | IVS | Tumour ECS | Normal Tissue ECS |
|---|---|---|---|
| Direct administration + BBBD | 3.47 | 1.37 | 2.54 × 10−3 |
| Liposomes | 17.78 | 9.68 | 1.07 × 10−2 |
| Liposomes + BBBD | 17.78 | 12.95 | 1.79 × 10−2 |
| Release Rate (s−1) | IVS | Tumour ECS | Normal Tissue ECS |
|---|---|---|---|
| krel = 1.0 × 10−6 | 1.28 × 10−3 | 1.39 × 10−3 | 3.83 × 10−6 |
| krel = 1.0 × 10−5 | 9.20 × 10−3 | 9.69 × 10−3 | 2.44 × 10−5 |
| krel = 1.0 × 10−4 | 1.78 × 10−2 | 1.29 × 10−2 | 1.79 × 10−5 |
| krel = 1.0 × 10−3 | 1.83 × 10−2 | 7.98 × 10−3 | 9.96 × 10−6 |
| Delay (min) | IVS | Tumour ECS | Normal Tissue ECS |
|---|---|---|---|
| 0 min | 1.78 × 10−2 | 1.29 × 10−2 | 1.79 × 10−5 |
| 30 min | 1.78 × 10−2 | 1.27 × 10−2 | 1.77 × 10−5 |
| 60 min | 1.78 × 10−2 | 1.19 × 10−2 | 1.73 × 10−5 |
| 90 min | 1.78 × 10−2 | 1.12 × 10−2 | 1.69 × 10−5 |
| Sonication Duration (min) | IVS | Tumour ECS | Normal Tissue ECS |
|---|---|---|---|
| Burst | 1.78 × 10−2 | 1.29 × 10−2 | 1.79 × 10−5 |
| 15 min | 1.78 × 10−2 | 1.48 × 10−2 | 2.02 × 10−5 |
| 30 min | 1.78 × 10−2 | 1.61 × 10−2 | 2.19 × 10−5 |
| 45 min | 1.78 × 10−2 | 1.70 × 10−2 | 2.31 × 10−5 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhan, W. Effects of Focused-Ultrasound-and-Microbubble-Induced Blood-Brain Barrier Disruption on Drug Transport under Liposome-Mediated Delivery in Brain Tumour: A Pilot Numerical Simulation Study. Pharmaceutics 2020, 12, 69. https://doi.org/10.3390/pharmaceutics12010069
Zhan W. Effects of Focused-Ultrasound-and-Microbubble-Induced Blood-Brain Barrier Disruption on Drug Transport under Liposome-Mediated Delivery in Brain Tumour: A Pilot Numerical Simulation Study. Pharmaceutics. 2020; 12(1):69. https://doi.org/10.3390/pharmaceutics12010069
Chicago/Turabian StyleZhan, Wenbo. 2020. "Effects of Focused-Ultrasound-and-Microbubble-Induced Blood-Brain Barrier Disruption on Drug Transport under Liposome-Mediated Delivery in Brain Tumour: A Pilot Numerical Simulation Study" Pharmaceutics 12, no. 1: 69. https://doi.org/10.3390/pharmaceutics12010069
APA StyleZhan, W. (2020). Effects of Focused-Ultrasound-and-Microbubble-Induced Blood-Brain Barrier Disruption on Drug Transport under Liposome-Mediated Delivery in Brain Tumour: A Pilot Numerical Simulation Study. Pharmaceutics, 12(1), 69. https://doi.org/10.3390/pharmaceutics12010069