Evaluation of Tumor-Targeting Properties of an Antagonistic Bombesin Analogue RM26 Conjugated with a Non-Residualizing Radioiodine Label Comparison with a Radiometal-Labelled Counterpart

 ,
, 

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
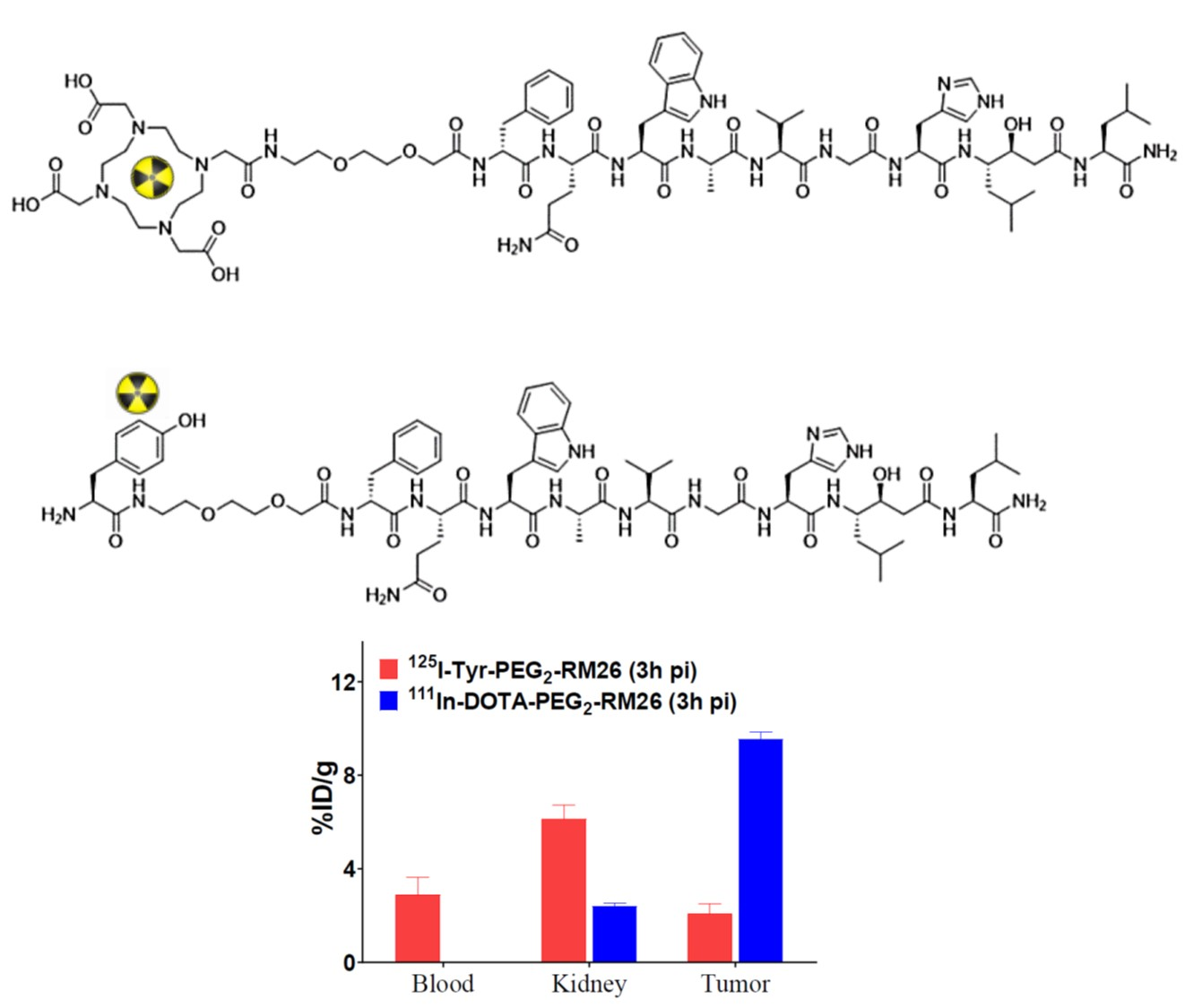
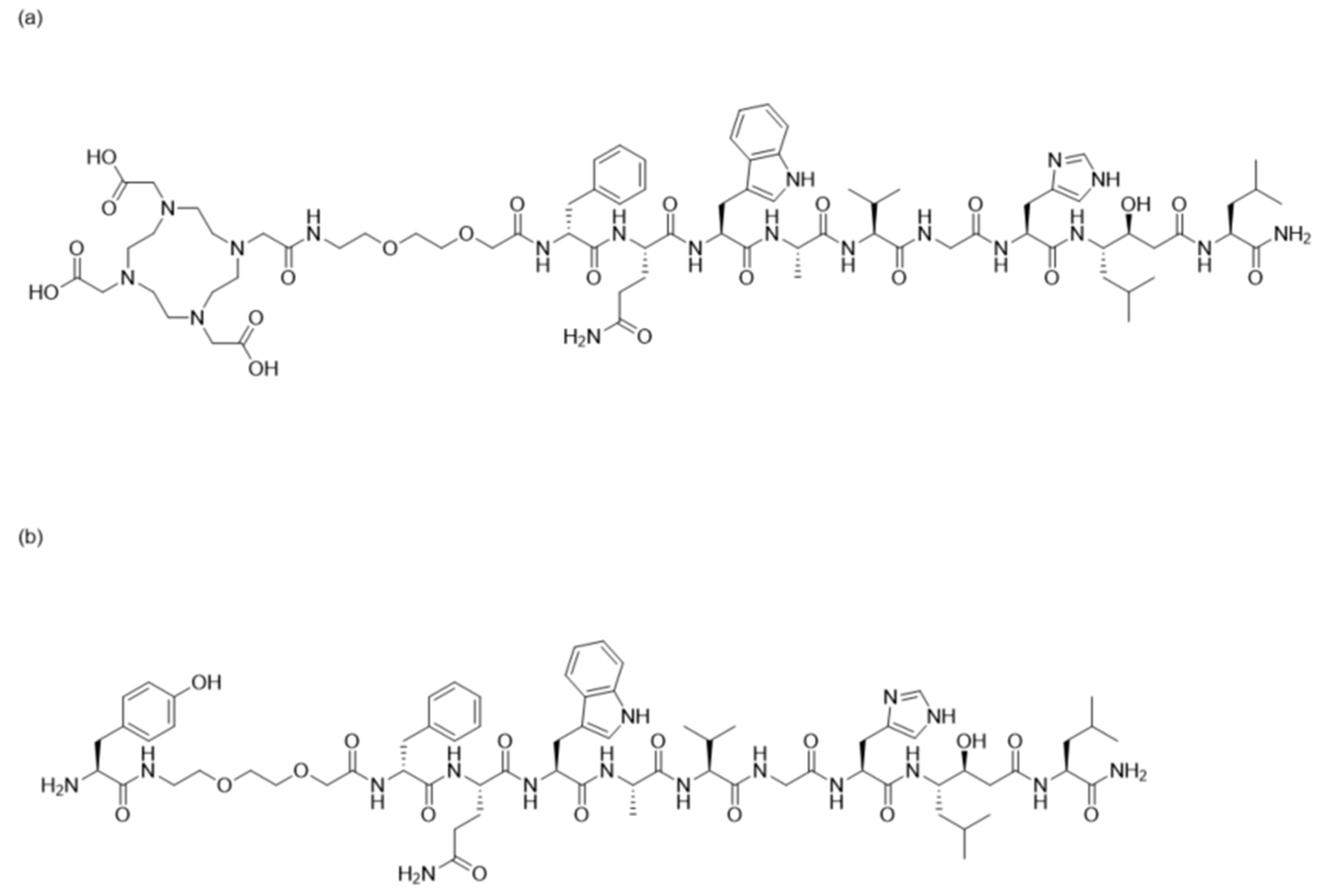
2.1. Peptide Synthesis and Characterization
2.2. Radiolabelling and In Vitro Stability Test
2.3. Lipophilicity Assay: LogP Measurements
2.4. In Vitro Studies
2.4.1. In Vitro Binding Specificity Assay
2.4.2. In Vitro Competitive Binding Assay: IC50 Determination
2.4.3. Cellular Processing Assay
2.5. In Vivo Studies
2.5.1. Biodistribution and In Vivo binding Specificity Test of [125I]I-Tyr-PEG2-RM26 and [111In]In-DOTA-PEG2-RM26 in BALB/c nu/nu Mice Bearing PC-3 Prostate Cancer Xenografts
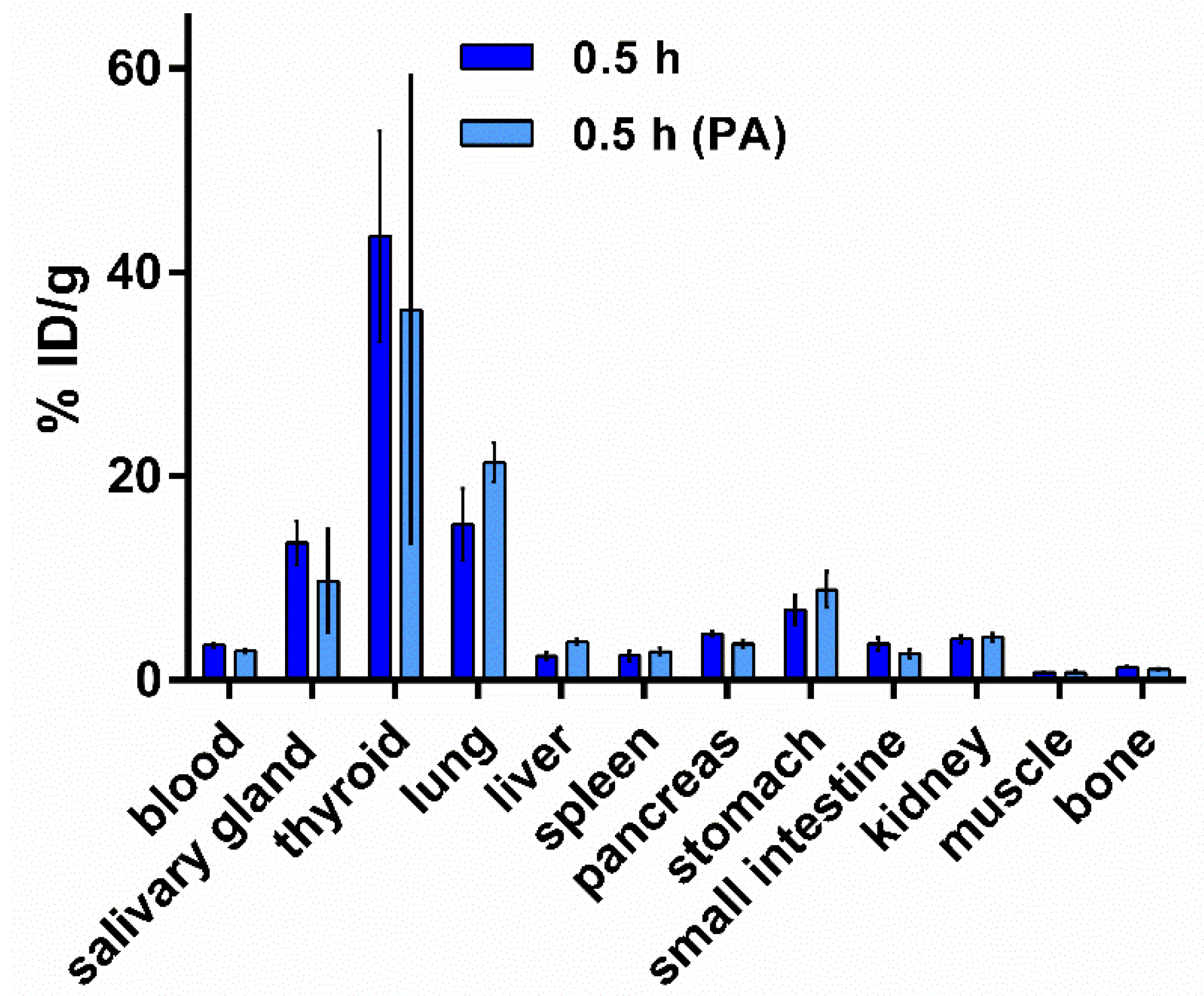
2.5.2. Biodistribution of [125I]I-Tyr-PEG2-RM26 in NMRI Mice by Co-Injection of Phosphoramidon (PA) as In Vivo Stabilizer
2.6. Statistical Analysis
3. Results
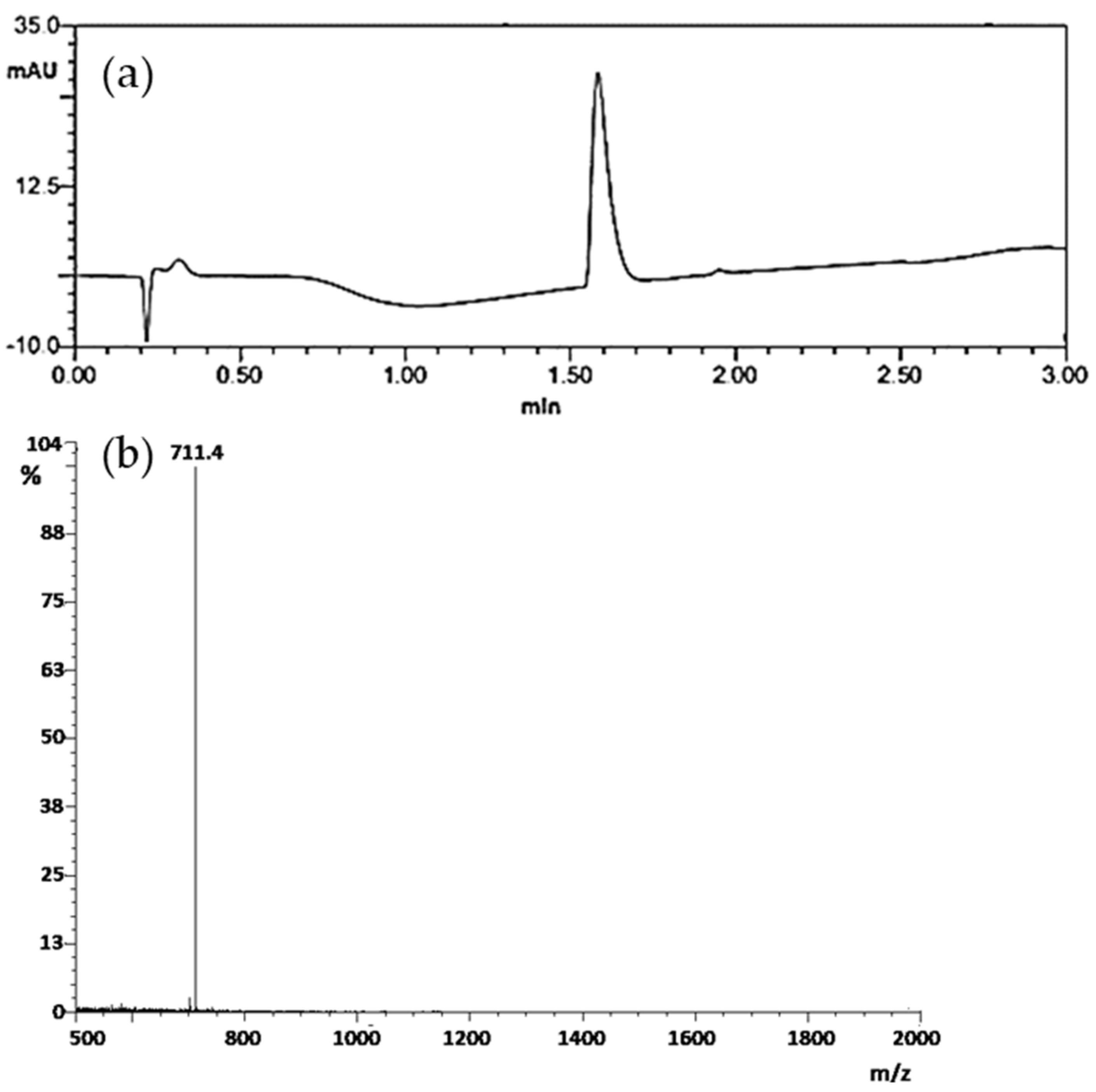
3.1. Peptide Synthesis and Characterization
3.2. Radiolabelling and In Vitro Stability Test
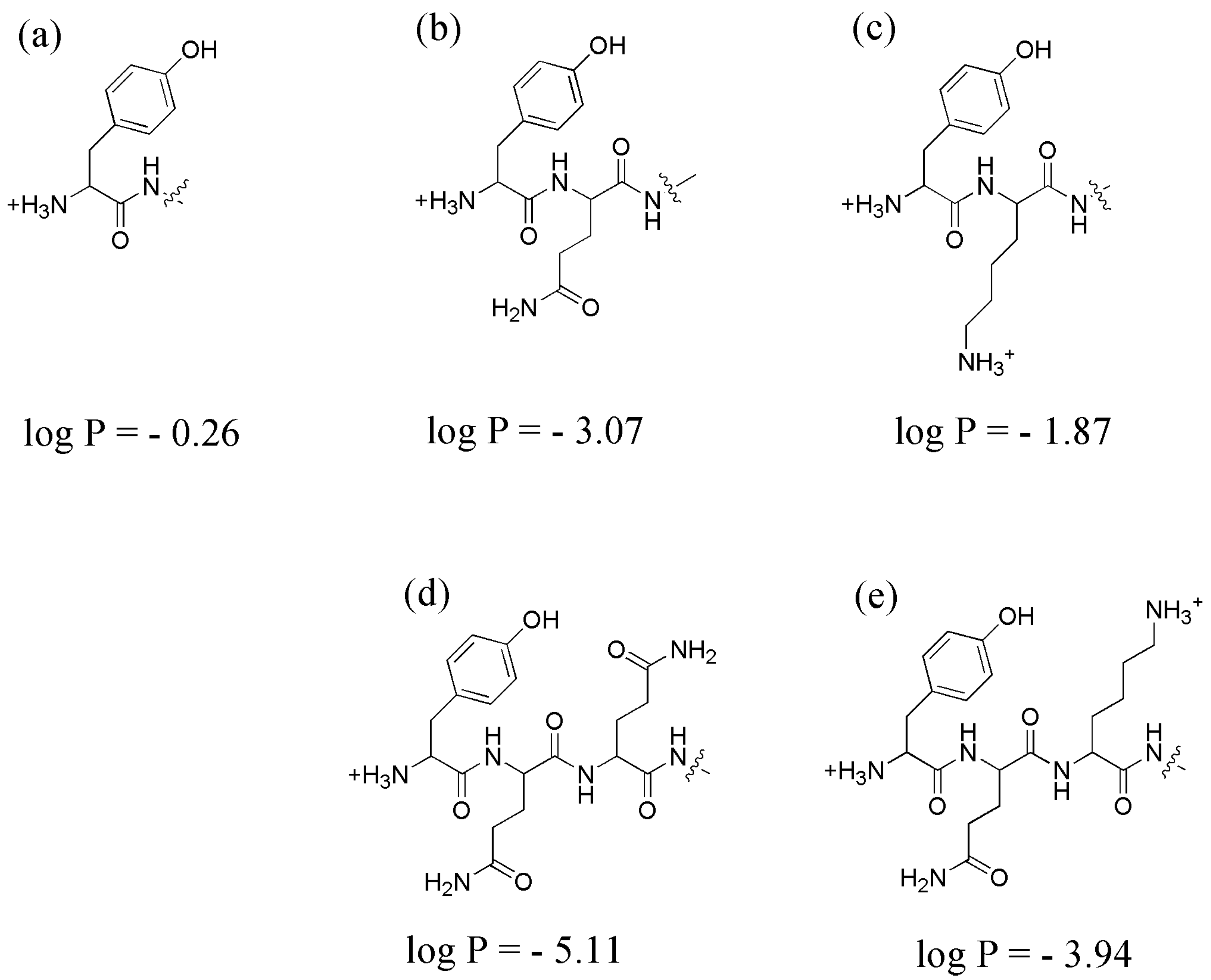
3.3. Lipophilicity Assay: LogP
3.4. In Vitro Studies
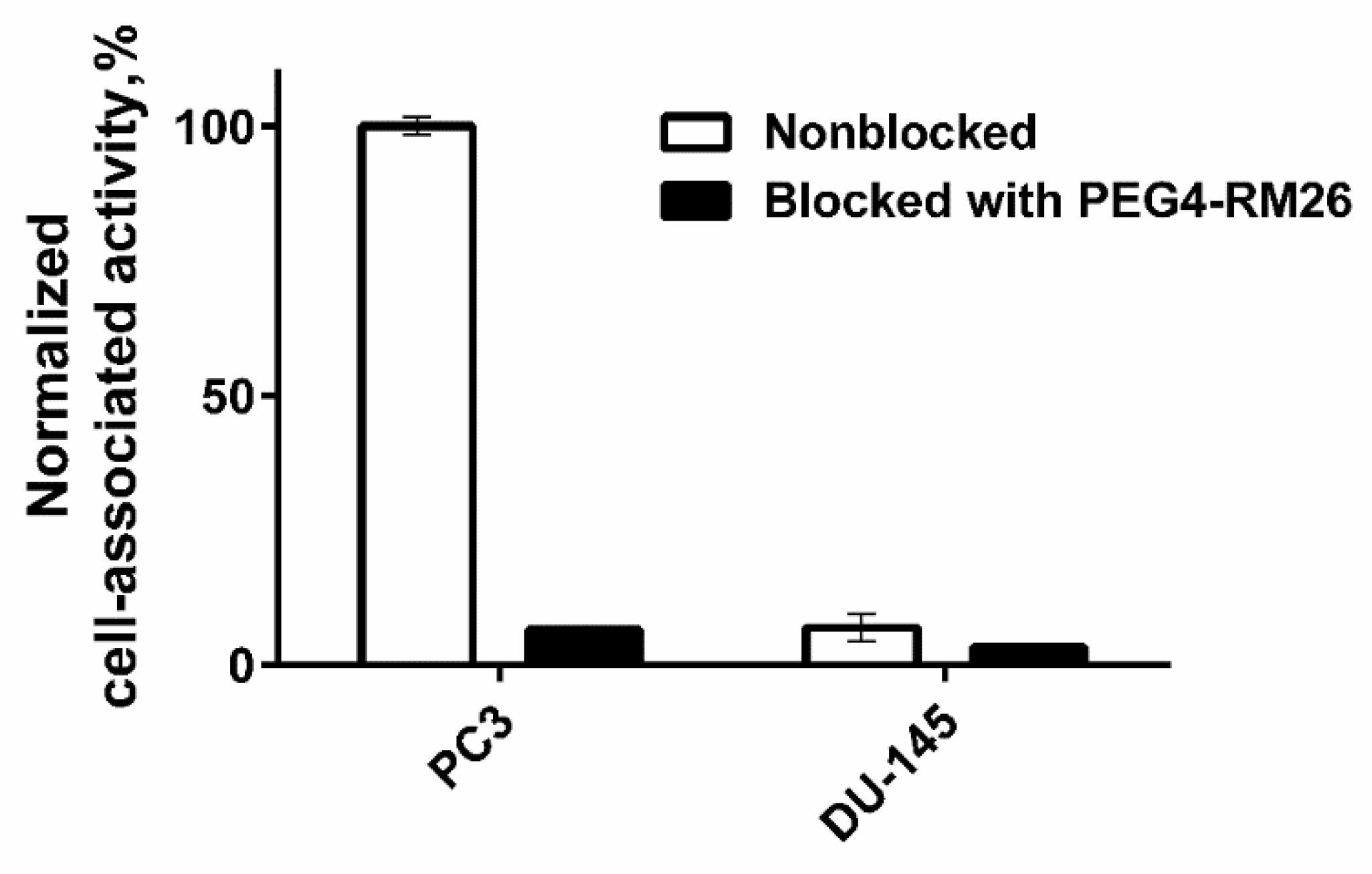
3.4.1. In Vitro Binding Specificity Assay
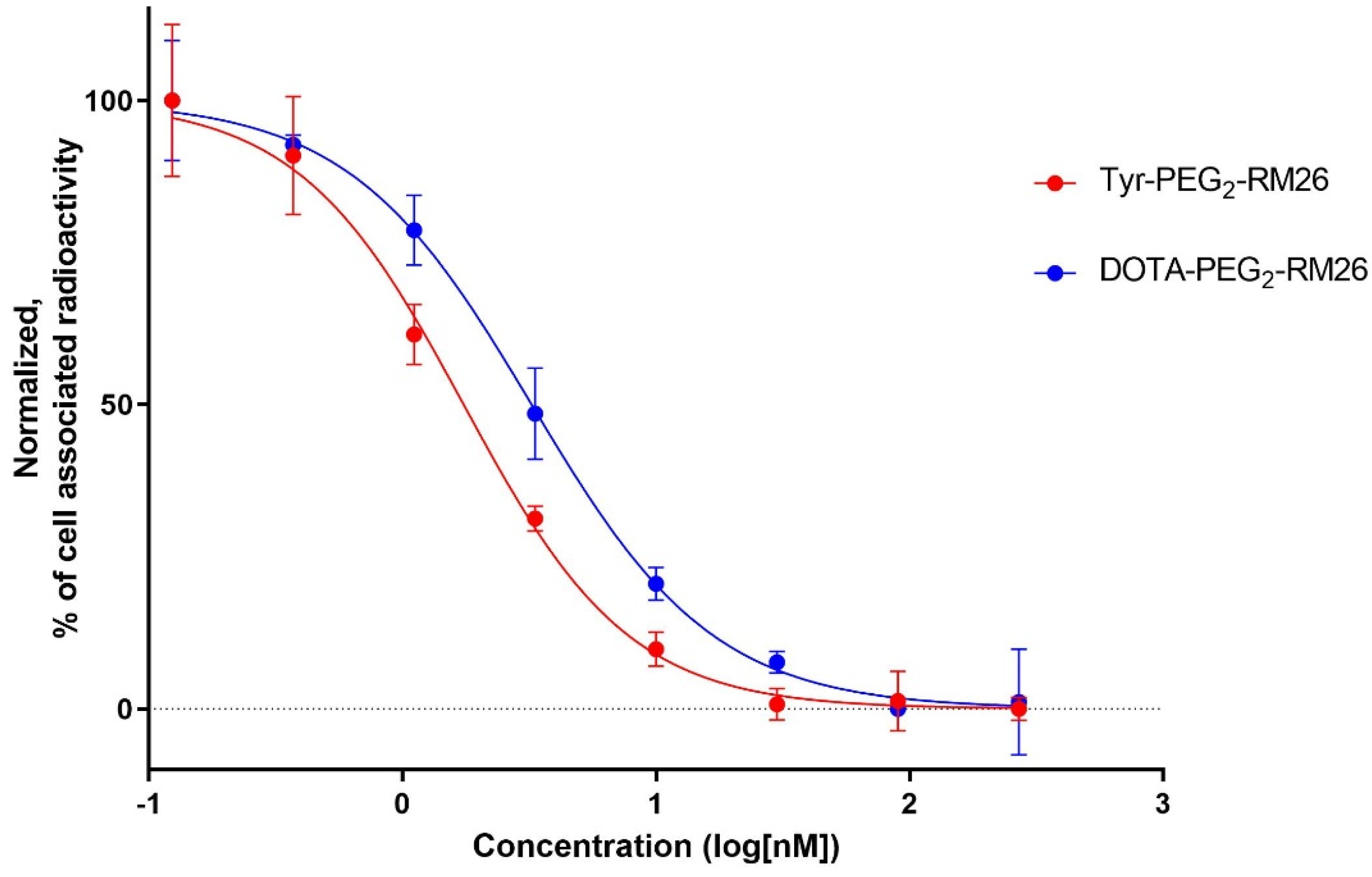
3.4.2. In Vitro Competitive Binding Assay: IC50 Determination
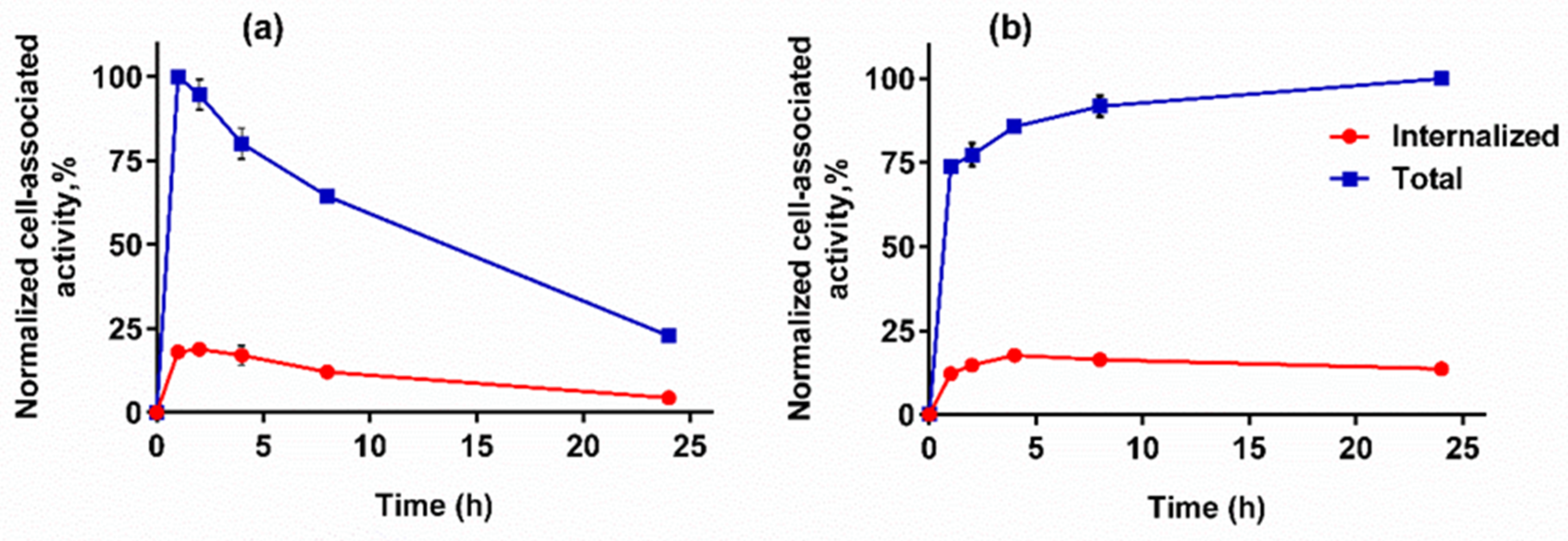
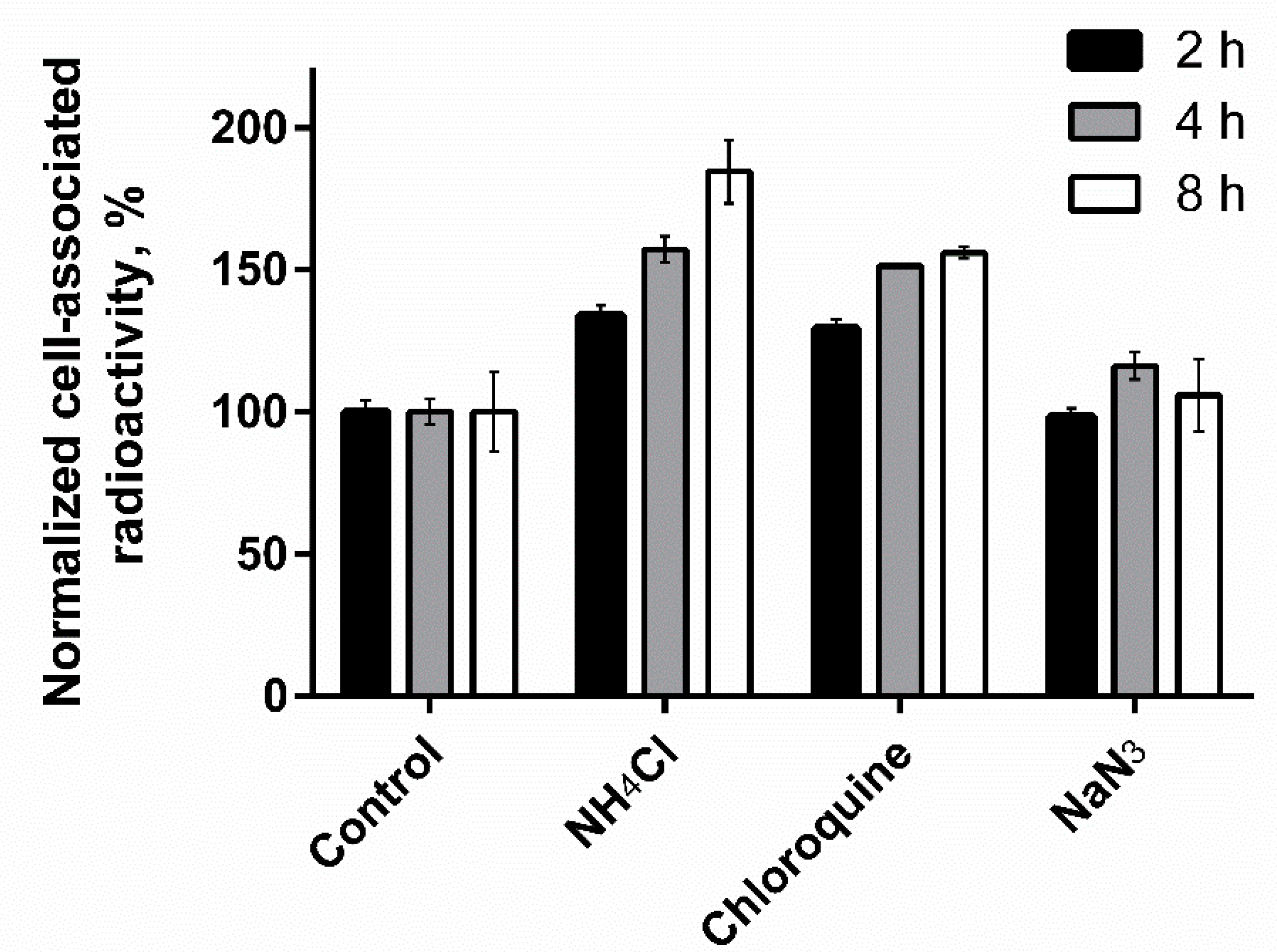
3.4.3. Cellular Processing Assay
3.5. In Vivo Studies
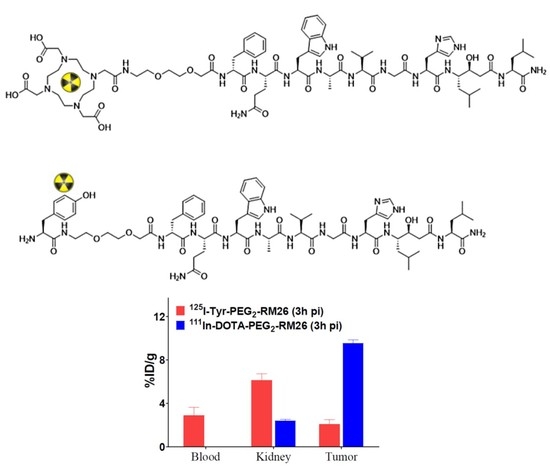
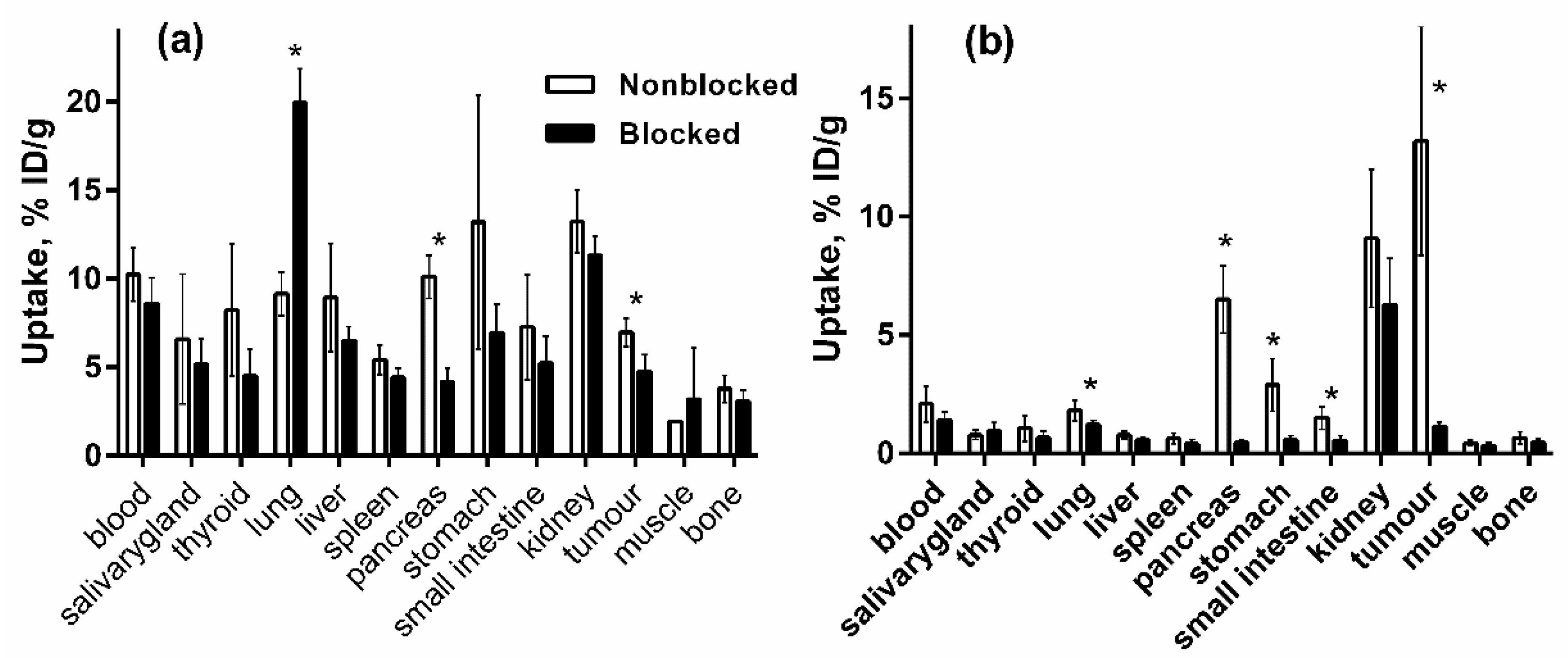
3.5.1. Biodistribution and In Vivo Binding Specificity Test of [125I]I-Tyr-PEG2-RM26 and [111In]In-DOTA-PEG2-RM26 in BALB/c nu/nu Mice Bearing PC-3 Prostate Cancer Xenografts
3.5.2. Biodistribution of [125I]I-Tyr-PEG2-RM26 in NMRI Mice by Co-Injection of Phosphoramidon (PA) as an In Vivo Stabilizer
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Aragon-Ching, J.B. The evolution of prostate cancer therapy: Targeting the androgen receptor. Front. Oncol. 2014, 4, 295. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yuan, X.; Cai, C.; Chen, S.; Yu, Z.; Balk, S.P. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene 2014, 33, 2815–2825. [Google Scholar] [CrossRef] [PubMed]
- Emmett, L.; Willowson, K.; Violet, J.; Shin, J.; Blanksby, A.; Lee, J. Lutetium 177 PSMA radionuclide therapy for men with prostate cancer: A review of the current literature and discussion of practical aspects of therapy. J. Med. Radiat. Sci. 2017, 64, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Hohenfellner, M.; Giesel, F.L.; Haberkorn, U.; Morgenstern, A. Targeted α-Therapy of Metastatic Castration-Resistant Prostate Cancer with 225Ac-PSMA-617: Dosimetry Estimate and Empiric Dose Finding. J. Nucl. Med. 2017, 58, 1624–1631. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar] [PubMed]
- Spatz, S.; Tolkach, Y.; Jung, K.; Stephan, C.; Busch, J.; Ralla, B.; Rabien, A.; Feldmann, G.; Brossart, P.; Bundschuh, R.A.; et al. Comprehensive evaluation of prostate-specific membrane antigen expression in the vasculature of renal tumors: implications for imaging studies and prognostic role. J. Urol. 2018, 199, 370–377. [Google Scholar]
- Sheikhbahaei, S.; Afshar-Oromieh, A.; Eiber, M.; Solnes, L.B.; Javadi, M.S.; Ross, A.E.; Pienta, K.J.; Allaf, M.E.; Haberkorn, U.; Pomper, M.G.; et al. Pearls and pitfalls in clinical interpretation of prostate-specific membrane antigen (PSMA)-targeted PET imaging. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 2117–2136. [Google Scholar] [CrossRef] [PubMed]
- Ananias, H.J.; van den Heuvel, M.C.; Helfrich, W.; de Jong, I.J. Expression of the gastrin-releasing peptide receptor, the prostate stem cell antigen and the prostate-specific membrane antigen in lymph node and bone metastases of prostate cancer. Prostate 2009, 69, 1101–1108. [Google Scholar] [CrossRef]
- Reubi, J.C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 2003, 2, 389–427. [Google Scholar] [CrossRef]
- Van de Wiele, C.; Dumont, F.; Vanden Broecke, R.; Oosterlinck, W.; Cocquyt, V.; Serreyn, R.; Peers, S.; Thornback, J.; Slegers, G.; Dierckx, R.A. Technetium-99m RP527, a GRP analogue for visualisation of GRP receptor-expressing malignancies: A feasibility study. Eur. J. Nucl. Med. 2000, 27, 1694–1699. [Google Scholar] [CrossRef]
- Jensen, R.T.; Battey, J.F.; Spindel, E.R.; Benya, R.V. International Union of Pharmacology. LXVIII. Mammalian bombesin receptors: Nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol. Rev. 2008, 60, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Nikolopoulou, A.; Galanis, A.; Cordopatis, P.; Waser, B.; Reubi, J.C.; Maina, T. Potent bombesin-like peptides for GRP-receptor targeting of tumors with 99mTc: A preclinical study. J. Med. Chem. 2005, 48, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cai, W.; Cao, F.; Schreibmann, E.; Wu, Y.; Wu, J.C.; Xing, L.; Chen, X. 18F-labeled bombesin analogs for targeting GRP receptor-expressing prostate cancer. J. Nucl. Med. 2006, 47, 492–501. [Google Scholar] [PubMed]
- Dimitrakopoulou-Strauss, A.; Hohenberger, P.; Haberkorn, U.; Macke, H.R.; Eisenhut, M.; Strauss, L.G. 68Ga-labeled bombesin studies in patients with gastrointestinal stromal tumors: Comparison with 18F-FDG. J. Nucl. Med. 2007, 48, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Däpp, S.; Müller, C.; Garayoa, E.G.; Bläuenstein, P.; Maes, V.; Brans, L.; Tourwé, D.A.; Schibli, R. PEGylation, increasing specific activity and multiple dosing as strategies to improve the risk-benefit profile of targeted radionuclide therapy with 177Lu-DOTA-bombesin analogues. EJNMMI Res. 2012, 2, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.; Frischknecht, M.; Zhang, H.; Morgenstern, A.; Bruchertseifer, F.; Boisclair, J.; Provencher-Bolliger, A.; Reubi, J.C.; Maecke, H.R. Alpha- versus beta-particle radiopeptide therapy in a human prostate cancer model (213Bi-DOTA-PESIN and 213Bi-AMBA versus177Lu-DOTA-PESIN). Cancer Res. 2011, 71, 1009–1018. [Google Scholar] [CrossRef]
- Smith, C.J.; Sieckman, G.L.; Owen, N.K.; Hayes, D.L.; Mazuru, D.G.; Volkert, W.A.; Hoffman, T.J. Radiochemical investigations of [188Re(H2O)(CO)3-diaminopropionic acid-SSS-bombesin(7-14)NH2]: Syntheses, radiolabeling and in vitro/in vivo GRP receptor targeting studies. Anticancer Res. 2003, 23, 63–70. [Google Scholar]
- Lantry, L.E.; Cappelletti, E.; Maddalena, M.E.; Fox, J.S.; Feng, W.; Chen, J.; Thomas, R.; Eaton, S.M.; Bogdan, N.J.; Arunachalam, T.; et al. 177Lu-AMBA: Synthesis and characterization of a selective 177Lu-labeled GRP-R agonist for systemic radiotherapy of prostate cancer. J. Nucl. Med. 2006, 47, 1144–1152. [Google Scholar]
- Koumarianou, E.; Mikołajczak, R.; Pawlak, D.; Zikos, X.; Bouziotis, P.; Garnuszek, P.; Karczmarczyk, U.; Maurin, M.; Archimandritis, S.C. Comparative study on DOTA-derivatized bombesin analog labeled with 90Y and 177Lu: In vitro and in vivo evaluation. Nucl. Med. Biol. 2009, 36, 591–603. [Google Scholar] [CrossRef]
- Nock, B.; Nikolopoulou, A.; Chiotellis, E.; Loudos, G.; Maintas, D.; Reubi, J.C.; Maina, T. [99mTc]Demobesin 1, a novel potent bombesin analogue for GRP receptor-targeted tumour imaging. Eur. J. Nucl. Med. 2003, 30, 247–258. [Google Scholar] [CrossRef]
- Abiraj, K.; Mansi, R.; Tamma, M.L.; Fani, M.; Forrer, F.; Nicolas, G.; Cescato, R.; Reubi, J.C.; Maecke, H.R. Bombesin antagonist-based radioligands for translational nuclear imaging of gastrin-releasing peptide receptor-positive tumors. J. Nucl. Med. 2011, 52, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Cescato, R.; Maina, T.; Nock, B.; Nikolopoulou, A.; Charalambidis, D.; Piccand, V.; Reubi, J.C. Bombesin Receptor Antagonists May Be Preferable to Agonists for Tumor Targeting. J. Nucl. Med. 2008, 49, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Mansi, R.; Wang, X.; Forrer, F.; Kneifel, S.; Tamma, M.L.; Waser, B.; Cescato, R.; Reubi, J.C.; Maecke, H.R. Evaluation of a 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid-conjugated bombesin-based radioantagonist for the labeling with single-photon emission computed tomography, positron emission tomography, and therapeutic radionuclides. Clin. Cancer Res. 2009, 15, 5240–5249. [Google Scholar] [CrossRef] [PubMed]
- Dumont, R.A.; Tamma, M.; Braun, F.; Borkowski, S.; Reubi, J.C.; Maecke, H.; Weber, W.A.; Mansi, R. Targeted radiotherapy of prostate cancer with a gastrin-releasing peptide receptor antagonist is effective as monotherapy and in combination with rapamycin. J. Nucl. Med. 2013, 54, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Chatalic, K.L.; Konijnenberg, M.; Nonnekens, J.; de Blois, E.; Hoeben, S.; de Ridder, C.; Brunel, L.; Fehrentz, J.A.; Martinez, J.; van Gent, D.C.; et al. In Vivo Stabilization of a Gastrin-Releasing Peptide Receptor Antagonist Enhances PET Imaging and Radionuclide Therapy of Prostate Cancer in Preclinical Studies. Theranostics 2016, 6, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Kaloudi, A.; Lymperis, E.; Giarika, A.; Kulkarni, H.R.; Klette, I.; Singh, A.; Krenning, E.P.; de Jong, M.; Maina, T.; et al. Theranostic Perspectives in Prostate Cancer with the Gastrin-Releasing Peptide Receptor Antagonist NeoBOMB1: Preclinical and First Clinical Results. J. Nucl. Med. 2017, 58, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Tolmachev, V.; Mume, E.; Sjöberg, S.; Frejd, F.Y.; Orlova, A. Influence of valency and labelling chemistry on in vivo targeting using radioiodinated HER2-binding Affibody molecules. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 692–701. [Google Scholar] [CrossRef]
- Lindbo, S.; Garousi, J.; Mitran, B.; Altai, M.; Buijs, J.; Orlova, A.; Hober, S.; Tolmachev, V. Radionuclide Tumor Targeting Using ADAPT Scaffold Proteins: Aspects of Label Positioning and Residualizing Properties of the Label. J. Nucl. Med. 2018, 59, 93–99. [Google Scholar] [CrossRef]
- Wilbur, S. Radiohalogenation of proteins: An overview of radionuclides, labeling methods and reagents for conjugate labeling. Bioconjug. Chem. 1992, 3, 433–470. [Google Scholar] [CrossRef]
- Llinares, M.; Devin, C.; Chaloin, O.; Azay, J.; Noel-Artis, A.M.; Bernad, N.; Fehrentz, J.A.; Martinez, J. Syntheses and biological activities of potent bombesin receptor antagonists. J. Pept. Res. 1999, 53, 275–283. [Google Scholar] [CrossRef]
- Varasteh, Z.; Velikyan, I.; Lindeberg, G.; Sörensen, J.; Larhed, M.; Sandström, M.; Selvaraju, R.K.; Malmberg, J.; Tolmachev, V.; Orlova, A. Synthesis and characterization of a high-affinity NOTA-conjugated bombesin antagonist for GRPR-targeted tumour imaging. Bioconjug. Chem. 2013, 24, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Varasteh, Z.; Rosenström, U.; Velikyan, I.; Mitran, B.; Altai, M.; Honarvar, H.; Rosestedt, M.; Lindeberg, G.; Sörensen, J.; Larhed, M.; et al. The effect of mini-PEG-based spacer length on binding and pharmacokinetic properties of a 68Ga-labeled NOTA-conjugated antagonistic analog of bombesin. Molecules 2014, 19, 10455–10472. [Google Scholar] [CrossRef] [PubMed]
- Varasteh, Z.; Mitran, B.; Rosenström, U.; Velikyan, I.; Rosestedt, M.; Lindeberg, G.; Sörensen, J.; Larhed, M.; Tolmachev, V.; Orlova, A. The effect of macrocyclic chelators on the targeting properties of the 68Ga-labeled gastrin releasing peptide receptor antagonist PEG2-RM26. Nucl. Med. Biol. 2015, 42, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Varasteh, Z.; Aberg, O.; Velikyan, I.; Lindeberg, G.; Sörensen, J.; Larhed, M.; Antoni, G.; Sandström, M.; Tolmachev, V.; Orlova, A. In vitro and in vivo evaluation of a (18)F-labeled high affinity NOTA conjugated bombesin antagonist as a PET ligand for GRPR-targeted tumor imaging. PLoS ONE 2013, 8, e81932. [Google Scholar] [CrossRef] [PubMed]
- Mitran, B.; Thisgaard, H.; Rosenström, U.; Dam, J.H.; Larhed, M.; Tolmachev, V.; Orlova, A. High Contrast PET Imaging of GRPR Expression in Prostate Cancer Using Cobalt-Labeled Bombesin Antagonist RM26. Contrast Media Mol. Imaging 2017, 2017, 6873684. [Google Scholar] [CrossRef] [PubMed]
- Mitran, B.; Rinne, S.S.; Konijnenberg, M.W.; Maina, T.; Nock, B.A.; Altai, M.; Vorobyeva, A.; Larhed, M.; Tolmachev, V.; de Jong, M.; et al. Trastuzumab co-treatment improves survival of mice with PC-3 prostate cancer xenografts treated with the GRPR antagonist 177Lu-DOTAGA-PEG2-RM26. Int. J. Cancer. 2019. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Crawley, J.N.; Jensen, R.T. Pharmacology and neurochemistry of bombesin like peptides. Peptides 1982, 3, 559–563. [Google Scholar] [CrossRef]
- Biddlecombe, G.B.; Rogers, B.E.; de Visser, M.; Parry, J.J.; de Jong, M.; Erion, J.L.; Lewis, J.S. Molecular imaging of gastrin-releasing peptide receptor-positive tumors in mice using 64Cu- and 86Y-DOTA-(Pro1,Tyr4)-bombesin(1–14). Bioconjug. Chem. 2007, 18, 724–730. [Google Scholar] [CrossRef]
- Hoffman, T.J.; Gali, H.; Smith, C.J.; Sieckman, G.L.; Hayes, D.L.; Owen, N.K.; Volkert, W.A. Novel series of 111In-labeled bombesin analogs as potential radiopharmaceuticals for specific targeting of gastrin-releasing peptide receptors expressed on human prostate cancer cells. J. Nucl. Med. 2003, 44, 823–831. [Google Scholar]
- Mitran, B.; Varasteh, Z.; Selvaraju, R.K.; Lindeberg, G.; Sörensen, J.; Larhed, M.; Tolmachev, V.; Rosenström, U.; Orlova, A. Selection of optimal chelator improves the contrast of GRPR imaging using bombesin analogue RM26. Int. J. Oncol. 2016, 48, 2124–2134. [Google Scholar] [CrossRef]
- Láznícek, M.; Kvĕtina, J.; Mazák, J.; Krch, V. Plasma protein binding—lipophilicity relationships: Interspecies comparison of some organic acids. J. Pharm. Pharmacol. 1987, 39, 79–83. [Google Scholar] [CrossRef]
- Wållberg, H.; Orlova, A. Slow intenalisation of anti-HER2 synthetic affibody monomer 111In-DOTA-ZHER2:342-pep2: Implications for development of labeled tracers. Cancer Biother. Radiopharm. 2008, 23, 435–442. [Google Scholar] [CrossRef]
- Maddalena, M.E.; Fox, J.; Chen, J.; Feng, W.; Cagnolini, A.; Linder, K.E.; Tweedle, M.F.; Nunn, A.D.; Lantry, L.E. 177Lu-AMBA biodistribution, radiotherapeutic efficacy, imaging, and autoradiography in prostate cancer models with low GRP-R expression. J. Nucl. Med. 2009, 50, 2017–2024. [Google Scholar] [CrossRef]
- Grady, E.F.; Slice, L.W.; Brant, W.O.; Walsh, J.H.; Payan, D.G.; Bunnett, N.W. Direct observation of endocytosis of gastrin releasing peptide and its receptor. J. Biol. Chem. 1995, 270, 4603–4611. [Google Scholar] [CrossRef]
- La Bella, R.; Garcia-Garayoa, E.; Bahler, M.; Bläuenstein, P.; Schibli, R.; Conrath, P.; Tourwé, D.; Schubiger, P.A. A 99mTc(I)-postlabeled high affinity bombesin analogue as a potential tumor imaging agent. Bioconjug. Chem. 2002, 13, 599–604. [Google Scholar] [CrossRef]
- García Garayoa, E.; Schweinsberg, C.; Maes, V.; Rüegg, D.; Blanc, A.; Bläuenstein, P.; Tourwé, D.A.; Beck-Sickinger, A.G.; Schubiger, P.A. New [99mTc]bombesin analogues with improved biodistribution for targeting gastrin releasing-peptide receptor-positive tumors. J. Nucl. Med. Mol. Imaging 2007, 51, 42–50. [Google Scholar]
- Zhang, H.; Chen, J.; Waldherr, C.; Hinni, K.; Waser, B.; Reubi, J.C.; Maecke, H.R. Synthesis and evaluation of bombesin derivatives on the basis of pan-bombesin peptides labeled with indium-111, lutetium-177, and yttrium-90 for targeting bombesin receptor-expressing tumors. Cancer Res. 2004, 64, 6707–6715. [Google Scholar] [CrossRef]
- García Garayoa, E.; Schweinsberg, C.; Maes, V.; Brans, L.; Bläuenstein, P.; Tourwe, D.A.; Schibli, R.; Schubiger, P.A. Influence of the molecular charge on the biodistribution of bombesin analogues labeled with the [99mTc(CO)3]-core. Bioconjug. Chem. 2008, 19, 2409–2416. [Google Scholar] [CrossRef]
- Sundberg, A.L.; Steffen, A. C Enhancing the effect of radionuclide tumor targeting, using lysosomotropic weak bases. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 279–287. [Google Scholar] [CrossRef]
- Reubi, J.C.; Erchegyi, J.; Cescato, R.; Waser, B.; Rivier, J.E. Switch from antagonist to agonist after addition of a DOTA chelator to a somatostatin analog. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1551–1558. [Google Scholar] [CrossRef]
- Shipp, M.A.; Tarr, G.E.; Chen, C.Y.; Switzer, S.N.; Hersh, L.B.; Stein, H.; Sunday, M.E.; Reinherz, E.L. CD10/neutral endopeptidase 24.11 hydrolyzes bombesin-like peptides and regulates the growth of small cell carcinomas of the lung. Proc. Natl. Acad. Sci. USA 1991, 88, 10662–10666. [Google Scholar] [CrossRef]
- Roques, B.P.; Noble, F.; Dauge, V.; Fournié-Zaluski, M.C.; Beaumont, A. Neutral endopeptidase 24.11: Structure, inhibition, and experimental and clinical pharmacology. Pharmacol. Rev. 1993, 45, 87–146. [Google Scholar]
- Nock, B.A.; Maina, T.; Krenning, E.P.; de Jong, M. “To serve and protect”: Enzyme inhibitors as radiopeptide escorts promote tumor targeting. J. Nucl. Med. 2014, 55, 121–127. [Google Scholar] [CrossRef]
- Wittendorf, R.W.; Swagzdis, J.E.; Gifford, R.; Mico, B.A. Protein binding of glycopeptide antibiotics with diverse physical-chemical properties in mouse, rat, and human serum. J. Pharmacokinet. Biopharm. 1987, 15, 5–13. [Google Scholar] [CrossRef]
- Decristoforo, C.; Mather, S.J. 99m-Technetium-labelled peptide-HYNIC conjugates: Effects of lipophilicity and stability on biodistribution. Nucl. Med. Biol. 1999, 26, 389–396. [Google Scholar] [CrossRef]
- Engfeldt, T.; Tran, T.; Orlova, A.; Widström, C.; Feldwisch, J.; Abrahmsen, L.; Wennborg, A.; Karlström, A.E.; Tolmachev, V. 99mTc-chelator engineering to improve tumour targeting properties of a HER2-specific Affibody molecule. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1843–1853. [Google Scholar] [CrossRef]
- Tran, T.; Engfeldt, T.; Orlova, A.; Sandström, M.; Feldwisch, J.; Abrahmsén, L.; Wennborg, A.; Tolmachev, V.; Karlström, A.E. 99mTc-maEEE-ZHER2:342, an Affibody molecule-based tracer for the detection of HER2 expression in malignant tumors. Bioconjug. Chem. 2007, 18, 1956–1964. [Google Scholar] [CrossRef]
- Rusckowski, M.; Qu, T.; Gupta, S.; Ley, A.; Hnatowich, D.J. A comparison in monkeys of 99mTc labeled to a peptide by 4 methods. J. Nucl. Med. 2001, 42, 1870–1877. [Google Scholar]
- Rosik, D.; Thibblin, A.; Antoni, G.; Honarvar, H.; Strand, J.; Selvaraju, R.K.; Altai, M.; Orlova, A.; Eriksson Karlström, A.; Tolmachev, V. Incorporation of a triglutamyl spacer improves the biodistribution of synthetic affibody molecules radiofluorinated at the N-terminus via oxime formation with (18)F-4-fluorobenzaldehyde. Bioconjug. Chem. 2014, 25, 82–92. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uptake, % ID/g | ||||||
|---|---|---|---|---|---|---|
| 0.5 h | 3 h | 24 h | ||||
| [125I]I-Tyr-PEG2-RM26 | [111In]In-DOTA-PEG2-RM26 | [125I]I-Tyr-PEG2-RM26 | [111In]In-DOTA-PEG2-RM26 | [125I]I-Tyr-PEG2-RM26 | [111In]In-DOTA-PEG2-RM26 | |
| blood | 10.2 ± 1.5 a,d,e | 2.1 ± 0.8 a,g,h | 2.9 ± 1.5 b,d,f | 0.03 ± 0.00 b,g,i | 0.02 ± 0.00 c, e, f | 0.006 ± 0.002 c,h,i |
| salivary gland | 6.6 ± 3.7 a,d | 0.8 ± 0.2 a,g | 2.0 ± 1.2 b,d | 0.07 ± 0.02 b,g | NM | NM |
| thyroid | 8.2 ± 3.7 a,d | 1.1 ± 0.5 a,g | 2.2 ± 1.6 b,d | 0.1 ± 0.0 b,g | NM | NM |
| lung | 9.1 ± 1.2 a,d | 1.8 ± 0.4 a,g | 3.5 ± 1.4 b,d,f | 0.1 ± 0.0 b,g | 0.2 ± 0.1 f | NM |
| liver | 8.9 ± 3.1 a,d,e | 0.8 ± 0.2 a,g,h | 2.6 ± 1.1 b,d,f | 0.2 ± 0.0 b,g,i | 0.2 ± 0.0 c,e,f | 0.1 ± 0.0 c,h,i |
| spleen | 5.4 ± 0.8 a,d | 0.6 ± 0.2 a,g | 2.0 ± 0.8 b,d | 0.1 ± 0.0 b,g | NM | NM |
| pancreas | 10.1 ± 1.2 a,d | 6.5 ± 1.4 a,g | 2.3 ± 0.6 b,d | 0.2 ± 0.0 b,g | NM | NM |
| stomach | 13.2 ± 7.2 a,d | 2.9 ± 1.1 a,g | 4.6 ± 2.7 b,d,f | 0.4 ± 0.1 b,g,h | 0.1 ± 0.0 f | NM |
| small intestine | 7.3 ± 3.0 a,d | 1.5 ± 0.5 a,g | 1.8 ± 0.7 b,d | 0.2 ± 0.1 b,g | NM | NM |
| kidney | 13.2 ± 1.8 d,e | 9.1 ± 2.9 g,h | 6.1 ± 1.1 b,d | 2.4 ± 0.3 b,f,g,i | 1.5 ± 0.3 c,e,f | 0.8 ± 0.2 c,h,i |
| tumour | 7.0 ± 0.8 a,d | 13.2 ± 4.8 a,h | 2.1 ± 0.8 b,d | 9.6 ± 0.6 b,i | NM | 3.6 ± 1.3 h,,i |
| muscle | 2.0 ± 0.0a,d | 0.4 ± 0.1 a,g | 0.6 ± 0.3 b,d | 0.02 ± 0.00 b,g | NM | NM |
| bone | 3.8 ± 0.8 a,d,h | 0.6 ± 0.3 a,g | 1.1 ± 0.5 b,d | 0.05 ± 0.02 b,g | NM | NM |
| Tumor-to-Organ Ratio | ||||||
|---|---|---|---|---|---|---|
| 0.5 h | 3 h | 24 h | ||||
| [125I]I-Tyr-PEG2-RM26 | [111In]In-DOTA-PEG2-RM26 | [125I]I-Tyr-PEG2-RM26 | [111In]In-DOTA-PEG2-RM26 | [125I]I-Tyr-PEG2-RM26 | [111In]In-DOTA-PEG2-RM26 | |
| blood | 0.7 ± 0.0 a,e | 6.3 ± 1.1 a,g,h | 0.8 ± 0.1 b,f | 373 ± 47 b,g,i | 6 ± 4 c,e,f | 672 ± 41 c,h,i |
| salivary gland | 1.3 ± 0.6 a | 16.5 ± 3.6 a,g | 1.2 ± 0.3 b | 146 ± 36 b,g | NM | NM |
| thyroid | 1.0 ± 0.5 a | 14.6 ± 5.8 a,g | 1.2 ± 0.6 b | 105 ± 40 b,g | NM | NM |
| lung | 0.8 ± 0.0 a,d | 7.1 ± 1.8 a,g | 0.6 ± 0.1 b,d | 123 ± 7 b,g | 1.0 ± 0.6 | NM |
| liver | 0.8 ± 0.2 a | 16.4 ± 3.8 a,g,h | 0.8 ± 0.2 b | 61 ± 3 b,g,i | 0.6 ± 0.5 c | 45 ± 8 c,h,i |
| spleen | 1.30 ± 0.2 a,d | 20.8 ± 2.5 a,g | 1.1 ± 0.1 b,d | 119 ± 7 b,g | NM | NM |
| pancreas | 0.7 ± 0.1 a | 2.0 ± 0.4 a,g | 0.9 ± 0.1 b | 50 ± 6 b,g | NM | NM |
| stomach | 0.6 ± 0.2 a | 4.6 ± 1.1 a,g | 0.5 ± 0.1 b | 27 ± 6 b,g | NM | NM |
| small intestine | 1.1 ± 0.6 a | 9.0 ± 3.3 a,g | 1.3 ± 0.4 b | 68 ± 35 b,g | NM | NM |
| kidney | 0.5 ± 0.0 a,d,e | 1.4 ± 0.3 a,g,h | 0.3 ± 0.1 b,d,f | 4 ± 0 b,g | 0.10 ± 0.0 c,e,f,i | 5 ± 1 c,h,i |
| muscle | 3.6 ± 0.4 a | 29.9 ± 6.6 a,g | 3.7 ± 0.6 b | 428 ± 41 b,g | NM | NM |
| bone | 1.9 ± 0.2 a | 22.8 ± 12.8 a,g | 1.9 ± 0.3 b | 221 ± 110 b,g | NM | NM |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oroujeni, M.; Abouzayed, A.; Lundmark, F.; Mitran, B.; Orlova, A.; Tolmachev, V.; Rosenström, U. Evaluation of Tumor-Targeting Properties of an Antagonistic Bombesin Analogue RM26 Conjugated with a Non-Residualizing Radioiodine Label Comparison with a Radiometal-Labelled Counterpart. Pharmaceutics 2019, 11, 380. https://doi.org/10.3390/pharmaceutics11080380
Oroujeni M, Abouzayed A, Lundmark F, Mitran B, Orlova A, Tolmachev V, Rosenström U. Evaluation of Tumor-Targeting Properties of an Antagonistic Bombesin Analogue RM26 Conjugated with a Non-Residualizing Radioiodine Label Comparison with a Radiometal-Labelled Counterpart. Pharmaceutics. 2019; 11(8):380. https://doi.org/10.3390/pharmaceutics11080380
Chicago/Turabian StyleOroujeni, Maryam, Ayman Abouzayed, Fanny Lundmark, Bogdan Mitran, Anna Orlova, Vladimir Tolmachev, and Ulrika Rosenström. 2019. "Evaluation of Tumor-Targeting Properties of an Antagonistic Bombesin Analogue RM26 Conjugated with a Non-Residualizing Radioiodine Label Comparison with a Radiometal-Labelled Counterpart" Pharmaceutics 11, no. 8: 380. https://doi.org/10.3390/pharmaceutics11080380
APA StyleOroujeni, M., Abouzayed, A., Lundmark, F., Mitran, B., Orlova, A., Tolmachev, V., & Rosenström, U. (2019). Evaluation of Tumor-Targeting Properties of an Antagonistic Bombesin Analogue RM26 Conjugated with a Non-Residualizing Radioiodine Label Comparison with a Radiometal-Labelled Counterpart. Pharmaceutics, 11(8), 380. https://doi.org/10.3390/pharmaceutics11080380