Superhydrophobic Substrates for Ultrahigh Encapsulation of Hydrophilic Drug into Controlled-Release Polyelectrolyte Complex Beads: Statistical Optimization and In Vivo Evaluation


Abstract
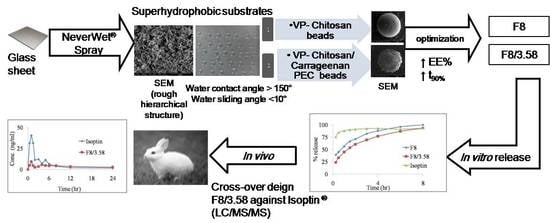
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation and Characterization of Superhydrophobic Substrates
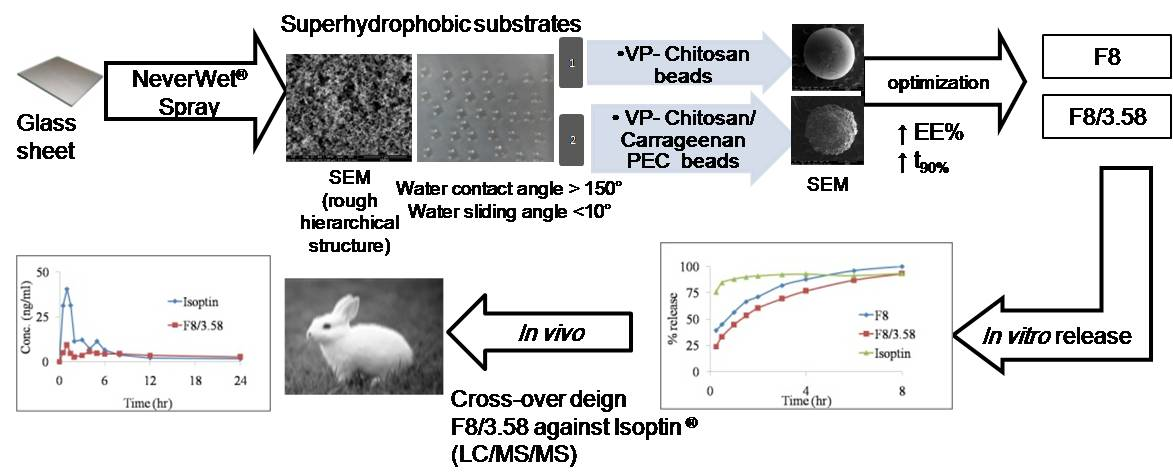
2.3. Preparation of VP-Loaded Ch-Based Beads
2.4. Preparation of VP-Loaded Ch/Kc-PEC-Based Beads
2.5. Evaluation of VP-Loaded Beads
2.5.1. Encapsulation Efficiency (EE%)
2.5.2. In Vitro Release Studies
2.6. Characterization of the Optimized Systems
2.6.1. Swelling Test
2.6.2. Scanning Electron Microscopy (SEM)
2.6.3. Fourier Transform Infrared (FTIR) Spectroscopy
2.7. In Vivo Pharmacokinetic Study
2.7.1. Study Design
2.7.2. Drug Administration and Dosing
2.7.3. Blood Sampling
2.7.4. Sample Preparation and LC-MS/MS Analysis
2.7.5. Pharmacokinetic Analysis
2.8. Statistical Analysis
3. Results and Discussion
3.1. Characterization of Superhydrophobic Substrates
3.2. Formulation of Ch- and Ch/Kc-Based Beads
3.3. Evaluation of VP-Loaded Ch-Based Beads
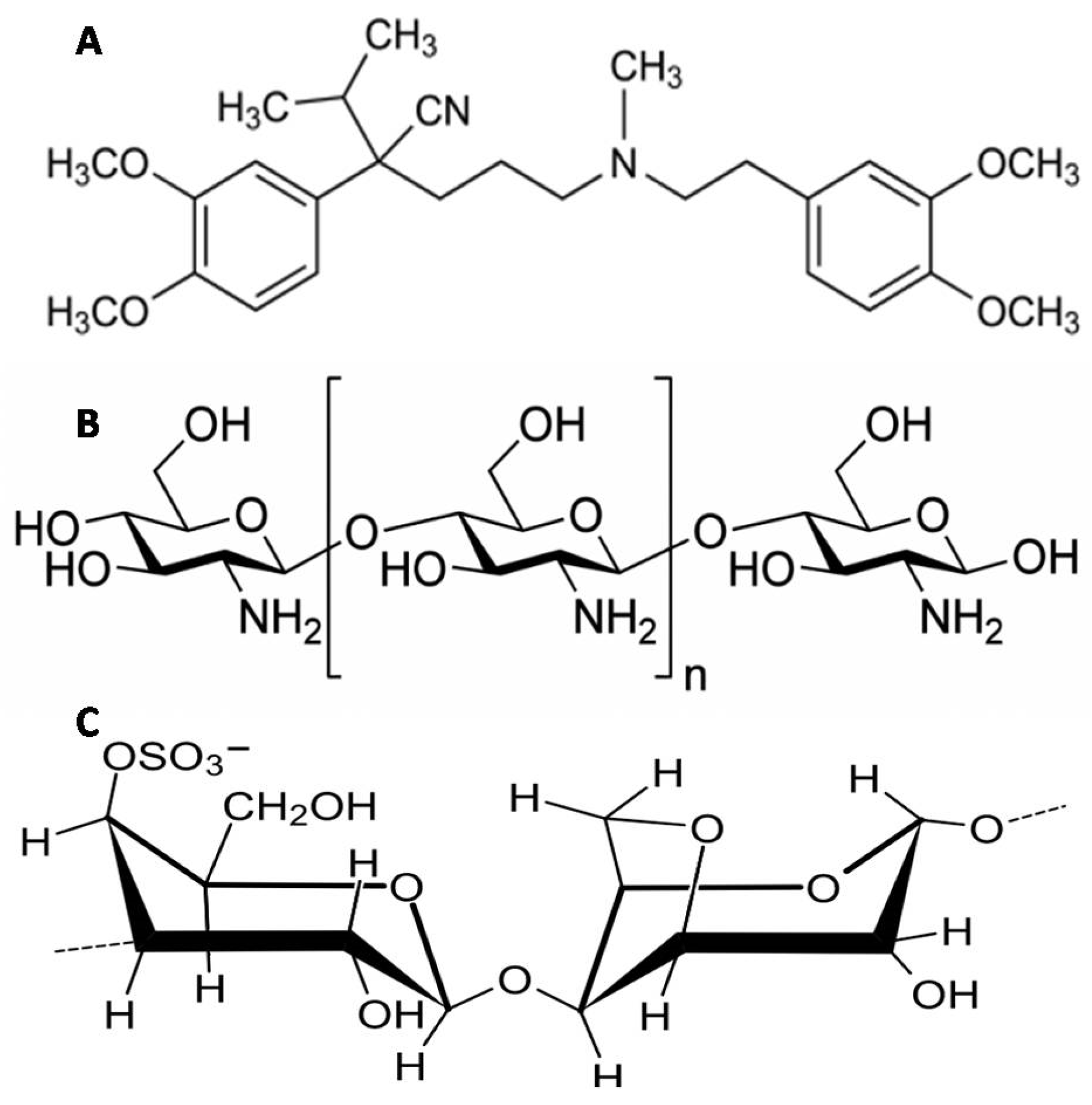
3.3.1. Encapsulation Efficiency (EE%)
3.3.2. In Vitro Release Studies
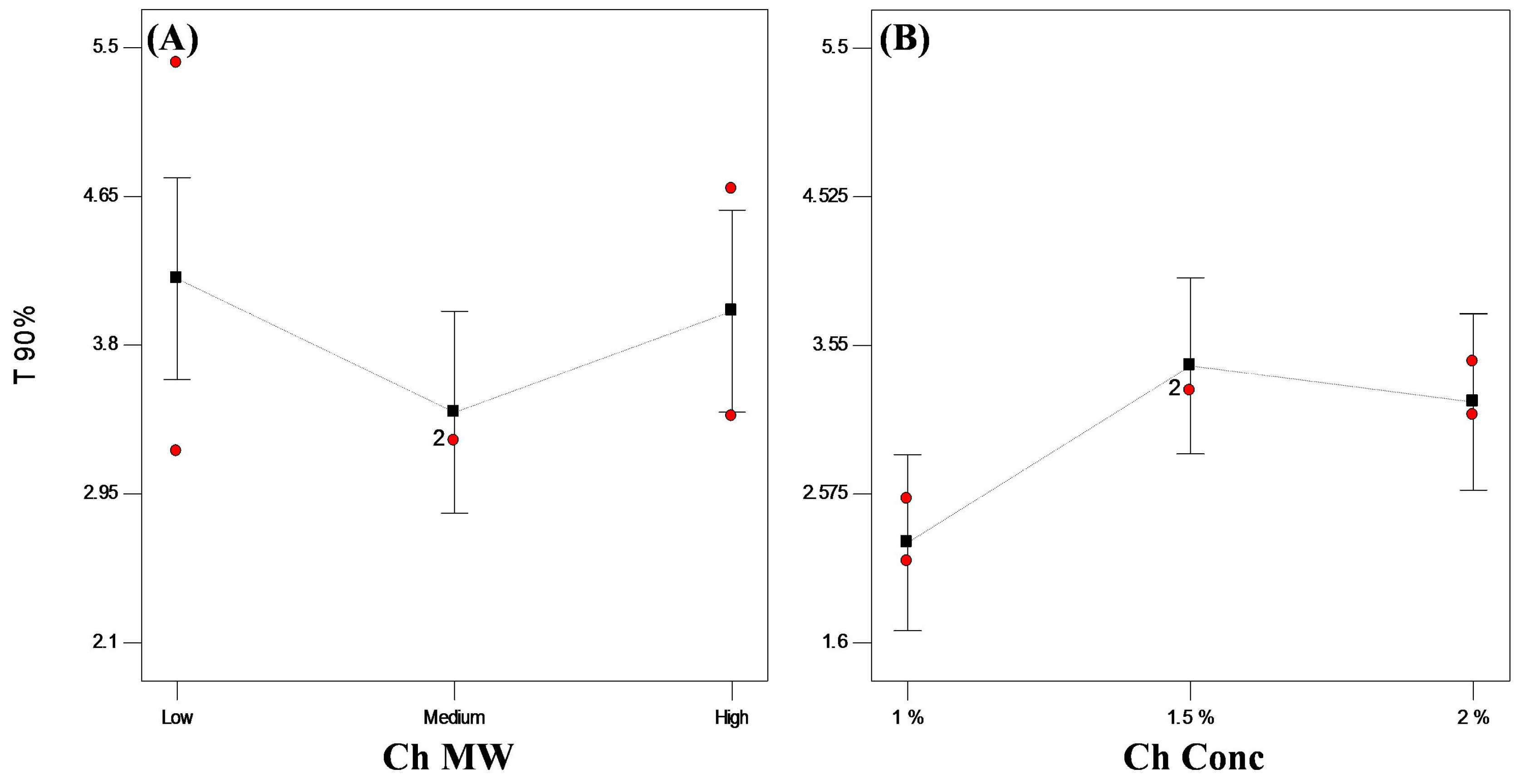
3.3.3. Statistical Optimization of VP-Loaded Ch-Based Beads
3.4. Evaluation of VP-Loaded Ch/Kc-PEC-Based Beads
3.4.1. Encapsulation Efficiency (EE%)
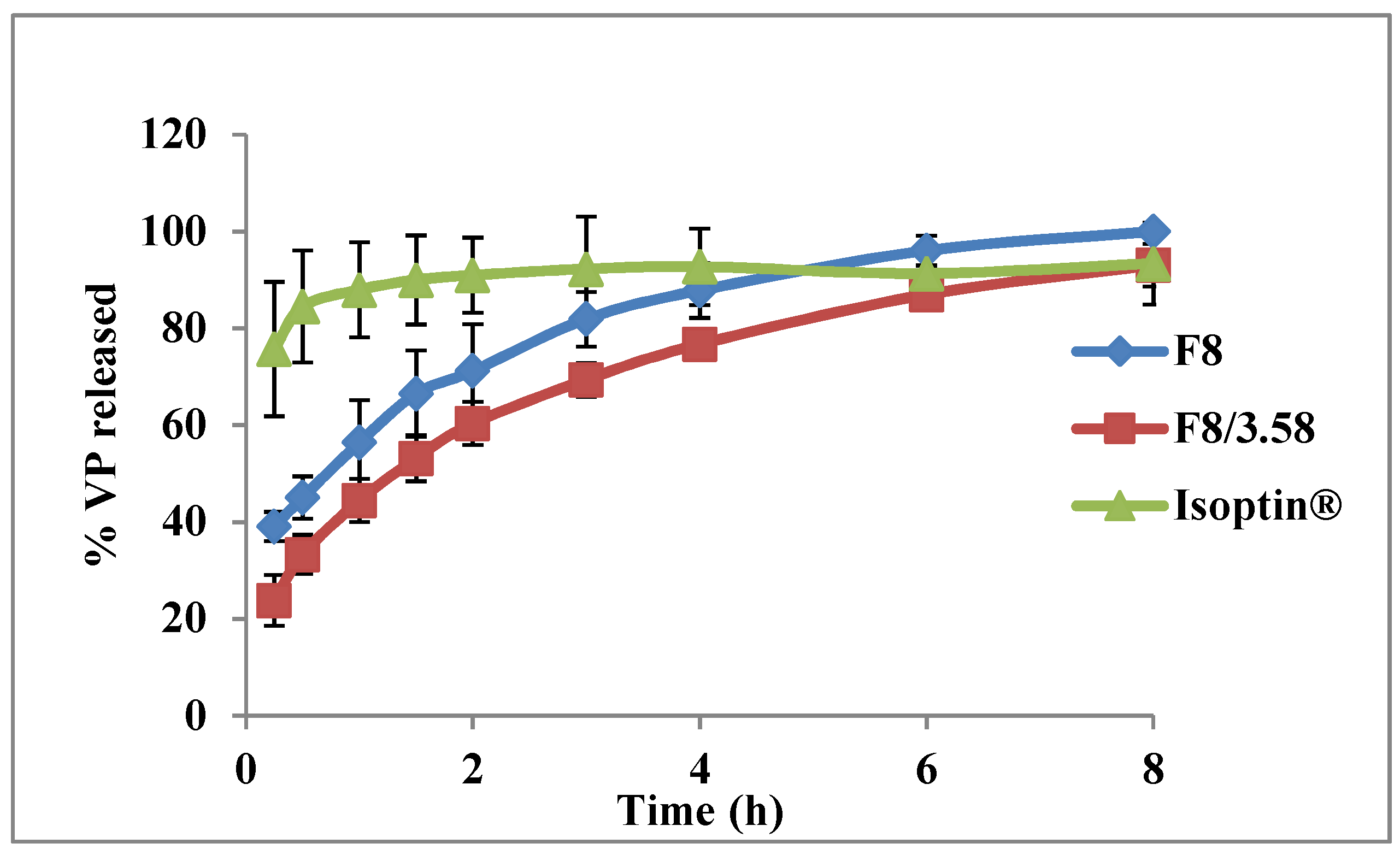
3.4.2. In Vitro Release Studies
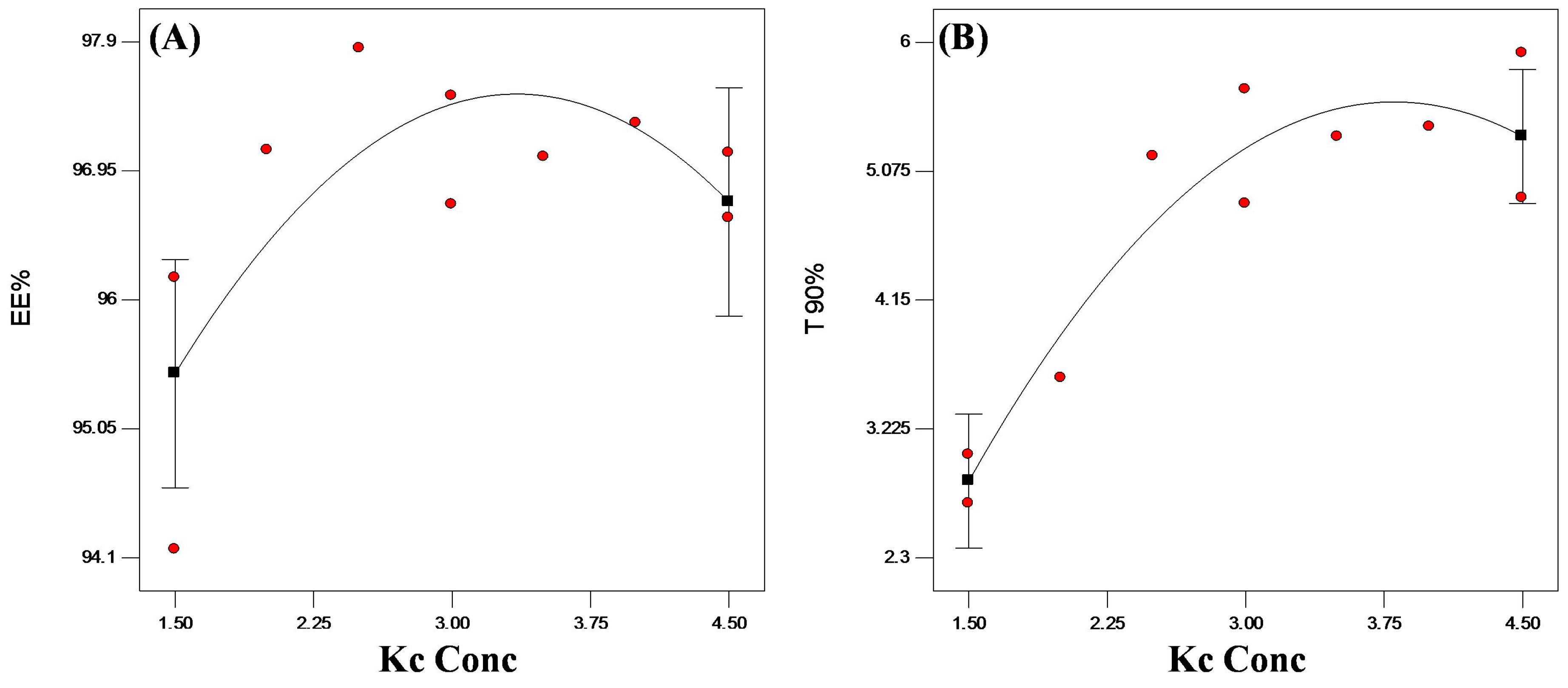
3.4.3. Statistical Optimization of VP-Loaded Ch/Kc-PEC-Based Beads
3.5. Characterization of Optimized PEC Beads
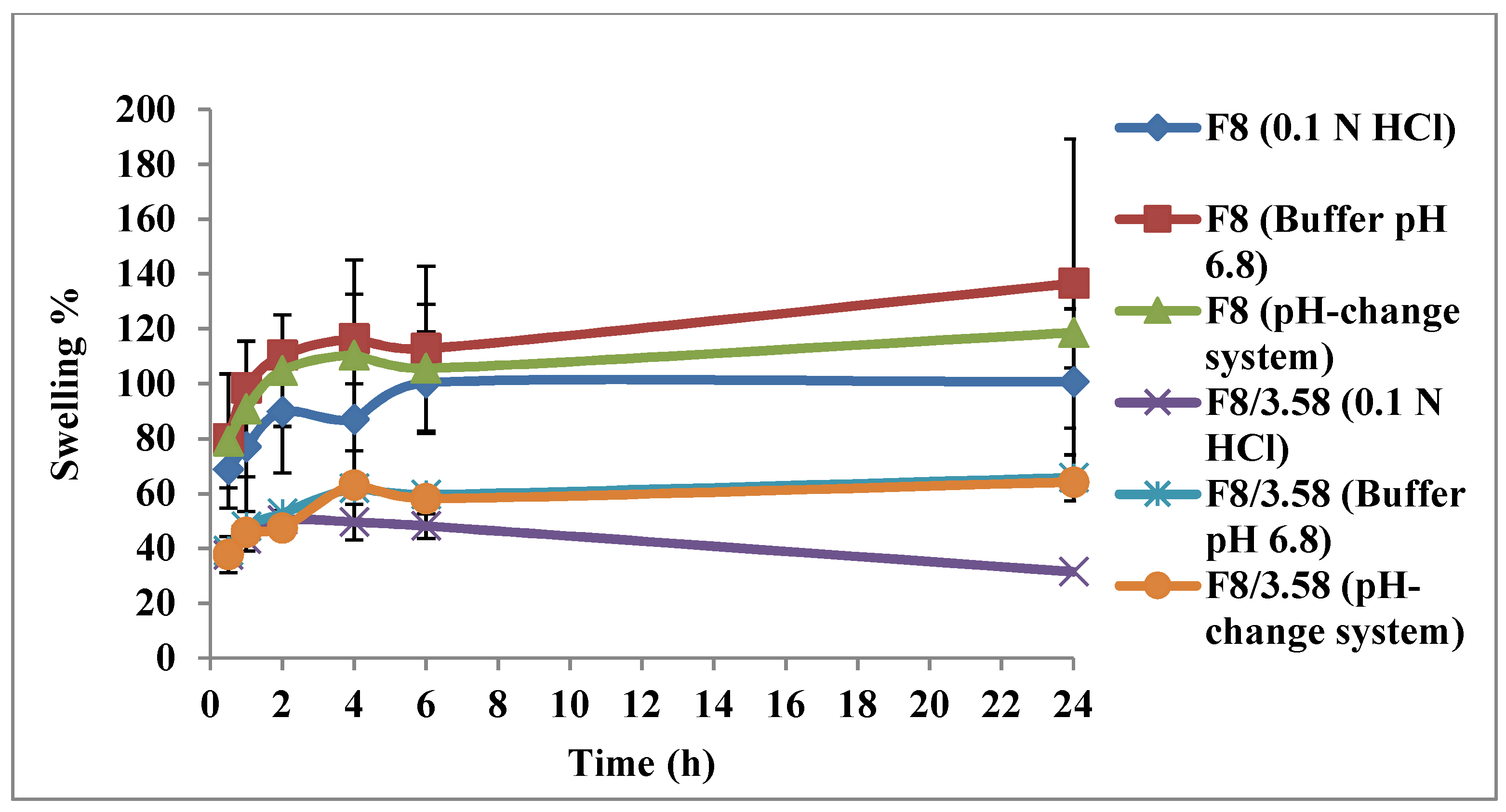
3.5.1. Swelling Test
3.5.2. Scanning Electron Microscope
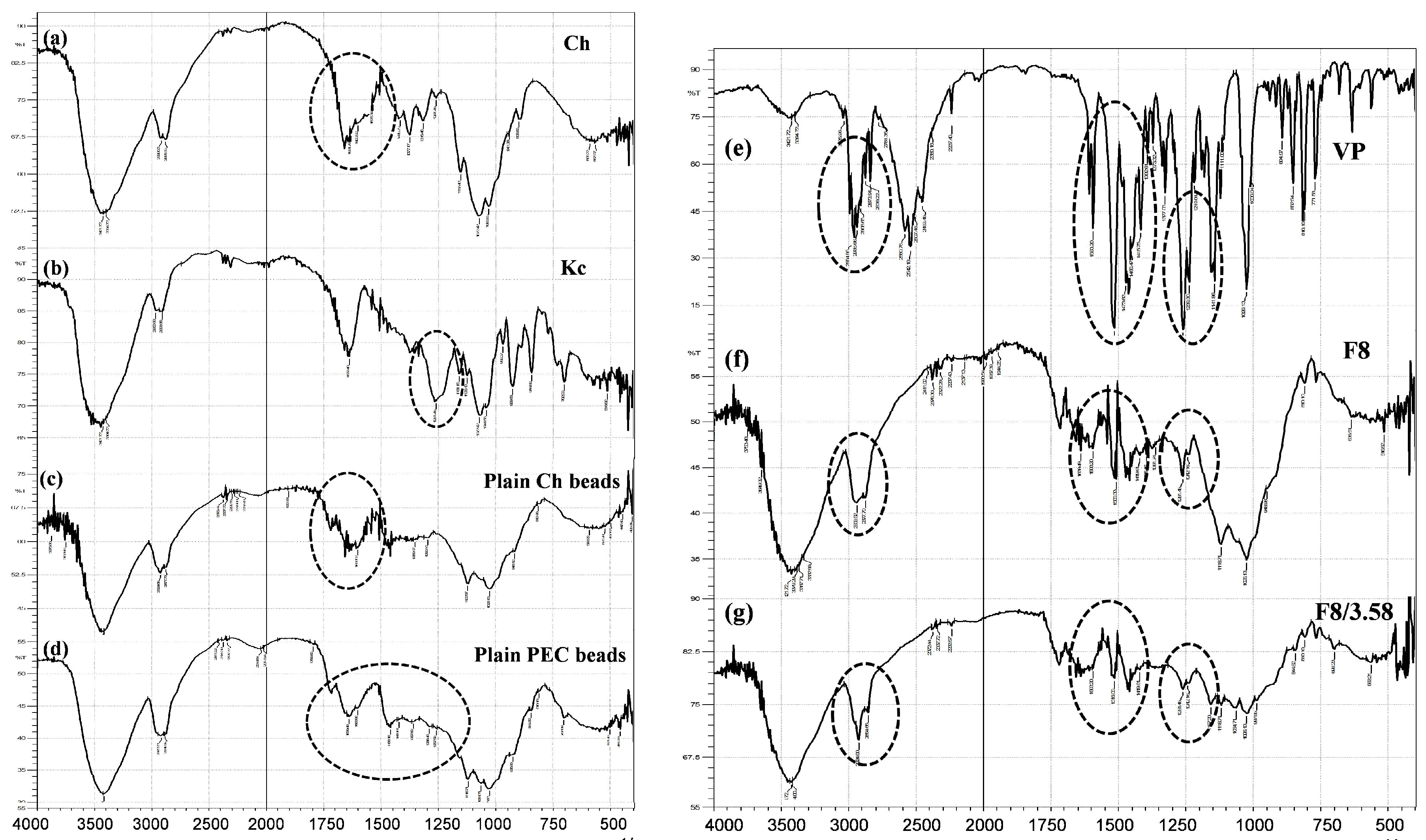
3.5.3. Fourier Transform Infrared (FTIR) Spectroscopy
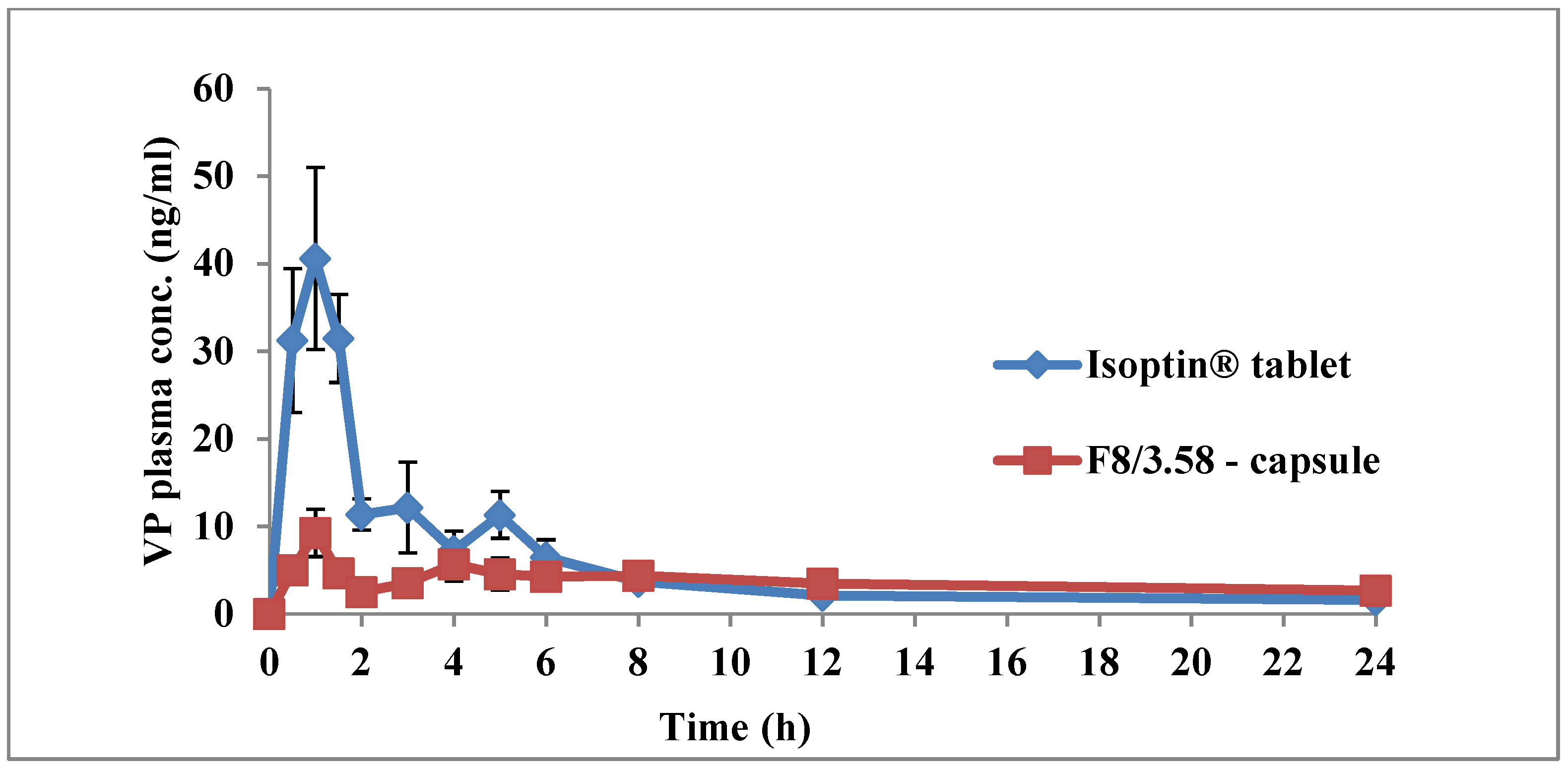
3.6. In Vivo Pharmacokinetic Study
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yassin, A.E.B.; Alsarra, I.A.; Al-Mohizea, A.M. Chitosan Beads as a New Gastroretentive System of Verapamil. Sci. Pharm. 2006, 74, 175–188. [Google Scholar] [CrossRef]
- El-Gazayerly, O.N.; Rakkanka, V.; Ayres, J.W. Novel Chewable Sustained-Release Tablet Containing Verapamil Hydrochloride. Pharm. Dev. Technol. 2004, 9, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.; Singla, A.; Wadhawan, S.; Kaushik, R.; Kumria, R.; Bansal, K.; Dhawan, S. Chitosan microspheres as a potential carrier for drugs. Int. J. Pharm. 2004, 274, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Puga, A.M.; Lima, A.C.; Mano, J.F.; Concheiro, A.; Alvarez-Lorenzo, C. Pectin-coated chitosan microgels crosslinked on superhydrophobic surfaces for 5-fluorouracil encapsulation. Carbohydr. Polym. 2013, 98, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Yousry, C.; Elkheshen, S.A.; El-Laithy, H.M.; Essam, T.; Fahmy, R.H. Studying the influence of formulation and process variables on Vancomycin-loaded polymeric nanoparticles as potential carrier for enhanced ophthalmic delivery. Eur. J. Pharm. Sci. 2017, 100, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Mobarak, D.H.; Salah, S.; Elkheshen, S.A. Elkheshen, Formulation of ciprofloxacin hydrochloride loaded biodegradable nanoparticles: optimization of technique and process variables. Pharm. Dev. Technol. 2014, 19, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.C.; Sher, P.; Mano, J.F. Production methodologies of polymeric and hydrogel particles for drug delivery applications. Expert Opin. Drug Deliv. 2012, 9, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Liu, W.; Su, B.-L. Superhydrophobic surfaces: From natural to biomimetic to functional. J. Colloid Interface Sci. 2011, 353, 335–355. [Google Scholar] [CrossRef]
- Song, W.; Lima, A.C.; Mano, J.F. Bioinspired methodology to fabricate hydrogel spheres for multi-applications using superhydrophobic substrates. Soft Matter 2010, 6, 5868–5871. [Google Scholar] [CrossRef]
- Lima, A.C.; Song, W.; Blanco-Fernandez, B.; Alvarez-Lorenzo, C.; Mano, J.F. Synthesis of Temperature-Responsive Dextran-MA/PNIPAAm Particles for Controlled Drug Delivery Using Superhydrophobic Surfaces. Pharm. Res. 2011, 28, 1294–1305. [Google Scholar] [CrossRef]
- Giri, T.K.; Thakur, A.; Alexander, A.; Badwaik, H.; Tripathi, D.K. Modified chitosan hydrogels as drug delivery and tissue engineering systems: present status and applications. Acta Pharm. Sin. B 2012, 2, 439–449. [Google Scholar] [CrossRef]
- Rani, M.; Agarwal, A.; Negi, Y.S. Chitosan based hydrogel polymeric beads—As drug delivery system. BioResources 2010, 5, 2765–2807. [Google Scholar]
- Saleem, M.A.; Kotadia, D.R.; Kulkarni, R.V. Effect of Formulation Variables on Dissolution of Water-Soluble Drug from Polyelectrolyte Complex Beads. Dissolution Technol. 2012, 19, 21–28. [Google Scholar] [CrossRef]
- Piyakulawat, P.; Praphairaksit, N.; Chantarasiri, N.; Muangsin, N. Preparation and evaluation of chitosan/carrageenan beads for controlled release of sodium diclofenac. AAPS PharmSciTech 2007, 8, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Tapia, C.; Escobar, Z.; Costa, E.; Sapag-Hagar, J.; Valenzuela, F.; Basualto, C.; Gai, M.N.; Yazdani-Pedram, M. Comparative studies on polyelectrolyte complexes and mixtures of chitosan–alginate and chitosan–carrageenan as prolonged diltiazem clorhydrate release systems. Eur. J. Pharm. Biopharm. 2004, 57, 65–75. [Google Scholar] [CrossRef]
- Park, S.-H.; Chun, M.-K.; Choi, H.-K. Preparation of an extended-release matrix tablet using chitosan/Carbopol interpolymer complex. Int. J. Pharm. 2008, 347, 39–44. [Google Scholar] [CrossRef]
- Moustafine, R.I.; Margulis, E.B.; Sibgatullina, L.F.; Kemenova, V.A.; Mooter, G.V.D. Comparative evaluation of interpolyelectrolyte complexes of chitosan with Eudragit® L100 and Eudragit® L100-55 as potential carriers for oral controlled drug delivery. Eur. J. Pharm. Biopharm. 2008, 70, 215–225. [Google Scholar] [CrossRef]
- McTavish, D.; Sorkin, E.M. Verapamil: An updated review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in hypertension. Drugs 1989, 38, 19–76. [Google Scholar] [CrossRef]
- Jankowski, A.; Marzec, A.; Lamparczyk, H. Comparative bioavailability of verapamil from rapidly absorbed and slow release preparations. J. Pharm. Biomed. Anal. 1992, 10, 1101–1103. [Google Scholar] [CrossRef]
- Mouez, M.A.; Nasr, M.; Abdel-Mottaleb, M.; Geneidi, A.S.; Mansour, S. Composite chitosan-transfersomal vesicles for improved transnasal permeation and bioavailability of verapamil. Int. J. Boil. Macromol. 2016, 93, 591–599. [Google Scholar] [CrossRef]
- Mouez, M.A.; Zaki, N.M.; Mansour, S.; Geneidi, A.S. Bioavailability enhancement of verapamil HCl via intranasal chitosan microspheres. Eur. J. Pharm. Sci. 2014, 51, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Deng, W. A simple method to prepare superamphiphobic aluminum surface with excellent stability. Colloids Surf. A Physicochem. Eng. Asp. 2015, 481, 143–150. [Google Scholar] [CrossRef]
- Cassie, A.B.D.; Baxter, S. Wettability Of porous surfaces. Trans. Faraday Soc. 1944, 40, 546–551. [Google Scholar] [CrossRef]
- Yousry, C.; Amin, M.M.; Elshafeey, A.H.; El Gazayerly, O.N. Ultrahigh verapamil-loaded controlled release polymeric beads using superamphiphobic substrate: D-optimal statistical design, in vitro and in vivo performance. Drug Deliv. 2018, 25, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- 〈711〉 Dissolution. The United States Pharmacopeial Convention. 2011. Available online: https://www.usp.org/sites/default/files/usp/document/harmonization/gen-method/stage_6_monograph_25_feb_2011.pdf (accessed on 27 May 2019).
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar] [PubMed]
- Lima, A.C.; Correia, C.R.; Oliveira, M.B.; Mano, J.F. Sequential ionic and thermogelation of chitosan spherical hydrogels prepared using superhydrophobic surfaces to immobilize cells and drugs. J. Bioact. Compat. Polym. 2014, 29, 50–65. [Google Scholar] [CrossRef]
- Choi, J.S.; Burm, J.P. Pharmacokinetics of verapamil and its major metabolite, nor-verapamil from oral administration of verapamil in rabbits with hepatic failure induced by carbon tetrachloride. Arch. Pharmacal 2005, 28, 483–487. [Google Scholar] [CrossRef]
- Meier, J.; Schmidt, R. Pharmacokinetic criteria for the evaluation of retard formulations. Eur. J. Clin. Pharmacol. 1974, 7, 429–432. [Google Scholar] [CrossRef]
- Deng, R.; Hu, Y.; Wang, L.; Li, Z.; Shen, T.; Zhu, Y.; Xiang, J. An easy and environmentally-friendly approach to superamphiphobicity of aluminum surfaces. Appl. Surf. Sci. 2017, 402, 301–307. [Google Scholar] [CrossRef]
- Yousry, C.; Fahmy, R.H.; Essam, T.; El-Laithy, H.M.; Elkheshen, S.A. Nanoparticles as tool for enhanced ophthalmic delivery of vancomycin: A multidistrict-based microbiological study, solid lipid nanoparticles formulation and evaluation. Drug Dev. Ind. Pharm. 2016, 42, 1–34. [Google Scholar] [CrossRef]
- Bodmeier, R.; Oh, K.-H.; Pramar, Y. Preparation and Evaluation of Drug-Containing Chitosan Beads. Drug Dev. Ind. Pharm. 1989, 15, 1475–1494. [Google Scholar] [CrossRef]
- Campos-Aldrete, M.E.; Villafuerte-Robles, L. Influence of the viscosity grade and the particle size of HPMC on metronidazole release from matrix tablets. Eur. J. Pharm. Biopharm. 1997, 43, 173–178. [Google Scholar] [CrossRef]
- Alvarado, H.L.; Abrego, G.; Garduño-Ramirez, M.L.; Clares, B.; Calpena, A.C.; Garcia, M.L. Design and optimization of oleanolic/ursolic acid-loaded nanoplatforms for ocular anti-inflammatory applications. Nanomed. Nanotechnol. Boil. Med. 2015, 11, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Hardy, I.J.; Windberg-Baarup, A.; Neri, C.; Byway, P.V.; Booth, S.W.; Fitzpatrick, S. Modulation of drug release kinetics from hydroxypropyl methyl cellulose matrix tablets using polyvinyl pyrrolidone. Int. J. Pharm. 2007, 337, 246–253. [Google Scholar] [CrossRef]
- Huang, X.; Brazel, C.S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J. Control. Release 2001, 73, 121–136. [Google Scholar] [CrossRef]
- Yang, L.; Fassihi, R. Examination of drug solubility, polymer types, hydrodynamics and loading dose on drug release behavior from a triple-layer asymmetric configuration delivery system. Int. J. Pharm. 1997, 155, 219–229. [Google Scholar] [CrossRef]
- Reza, M.S.; Quadir, M.A.; Haider, S.S. Comparative evaluation of plastic, hydrophobic and hydrophilic polymers as matrices for controlled-release drug delivery. J. Pharm. Pharm. Sci. 2003, 6, 282–291. [Google Scholar]
- De Yao, K.; Yin, Y.J.; Xu, M.X.; Wang, Y.F. Investigation of pH-sensitive drug delivery system of chitosan/gelatin hybrid polymer network. Polym. Int. 1995, 38, 77–82. [Google Scholar] [CrossRef]
- Maderuelo, C.; Zarzuelo, A.; Lanao, J.M. Critical factors in the release of drugs from sustained release hydrophilic matrices. J. Control. Release 2011, 154, 2–19. [Google Scholar] [CrossRef]
- Bettini, R.; Catellani, P.; Santi, P.; Massimo, G.; Peppas, N.; Colombo, P. Translocation of drug particles in HPMC matrix gel layer: Effect of drug solubility and influence on release rate. J. Control. Release 2001, 70, 383–391. [Google Scholar] [CrossRef]
- Mitchell, K.; Ford, J.; Armstrong, D.; Elliott, P.; Rostron, C.; Hogan, J. The influence of concentration on the release of drugs from gels and matrices containing Methocel®. Int. J. Pharm. 1993, 100, 155–163. [Google Scholar] [CrossRef]
- Ebube, N.K.; Jones, A.B. Sustained release of acetaminophen from a heterogeneous mixture of two hydrophilic non-ionic cellulose ether polymers. Int. J. Pharm. 2004, 272, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, I. Factors affecting drug release from hydroxypropyl methylcellulose matrix systems in the light of classical and percolation theories. Expert Opin. Drug Deliv. 2010, 7, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Moosa, R.M.; Choonara, Y.E.; Du Toit, L.C.; Tomar, L.K.; Tyagi, C.; Kumar, P.; Carmichael, T.R.; Pillay, V. In vivo evaluation and in-depth pharmaceutical characterization of a rapidly dissolving solid ocular matrix for the topical delivery of timolol maleate in the rabbit eye model. Int. J. Pharm. 2014, 466, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.M.; Braga, C.R.; Fook, M.V.; Raposo, C.M.; Carvalho, L.H.; Canedo, E.L. Application of Infrared Spectroscopy to Analysis of Chitosan/Clay Nanocomposites. Infrared Spectrosc. Mater. Sci. Eng. Technol. 2012. [Google Scholar] [CrossRef]
- Dimzon, I.K.D.; Knepper, T.P.; Dimzon, I.K. Degree of deacetylation of chitosan by infrared spectroscopy and partial least squares. Int. J. Boil. Macromol. 2015, 72, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Tranquilan-Aranilla, C.; Nagasawa, N.; Bayquen, A.; Rosa, A.D. Synthesis and characterization of carboxymethyl derivatives of kappa-carrageenan. Carbohydr. Polym. 2012, 87, 1810–1816. [Google Scholar] [CrossRef]
- Tekade, B.W.; Thakare, V.M.; Jadhao, U.T.; Kazi, F. Optimization and in vitro evaluation of verapamil hydrochloride floating bilayer tablet. Pharma. Innov. 2014, 3, 48–56. [Google Scholar]
- Kunta, J.R.; Lee, S.-H.; Perry, B.A.; Sinko, P.J. Differentiation of gut and hepatic first-pass loss of verapamil in intestinal and vascular access-ported (IVAP) rabbits. Drug Metab. Dispos. 2004, 32, 1293–1298. [Google Scholar] [CrossRef]
- Marvola, M.; Kannikoski, A.; Taskinen, J.; Ottoila, P. Assessment of bioavailabilitv of experimental single-unit sustained release tablets 6f verapamil hydrochloride using the stable isotope technique. J. Pharm. Pharmacol. 1985, 37, 766–770. [Google Scholar] [CrossRef]
- Ravi, P.R.; Vats, R.; Balija, J.; Adapa, S.P.N.; Aditya, N. Modified pullulan nanoparticles for oral delivery of lopinavir: Formulation and pharmacokinetic evaluation. Carbohydr. Polym. 2014, 110, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Rao, B.S.; Seshasayana, A.; Saradhi, S.P.; Kumar, N.R.; Narayan, C.P.; Murthy, K.R. Correlation of ‘in vitro’ release and ‘in vivo’ absorption characteristics of rifampicin from ethylcellulose coated nonpareil beads. Int. J. Pharm. 2001, 230, 1–9. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System # | Ch MW | Ch % (w/v) | EE % | t90% (h) |
|---|---|---|---|---|
| F1 | Low | 1 | 97.00 ± 0.97 | 2.75 ± 0.03 |
| F2 | 1.5 | 94.19 ± 4.19 | 4.31 ± 1.56 | |
| F3 | 2 | 96.49 ± 2.53 | 4.09 ± 1.53 | |
| F4 | Medium | 1 | 95.82 ± 3.08 | 2.33 ± 0.29 |
| F5 | 1.5 | 96.21 ± 0.79 | 3.25 ± 0.00 | |
| F6 | 2 | 96.25 ± 1.06 | 3.26 ± 0.25 | |
| F7 | High | 1 | 98.49 ± 0.48 | 3.02 ± 0.18 |
| F8 | 1.5 | 98.54 ± 0.78 | 4.04 ± 0.92 | |
| F9 | 2 | 98.82 ± 0.31 | 3.52 ± 0.59 |
| System # | High MW Ch % (w/v) | Kc % (w/v) | EE % (w/w) | t90% (h) |
|---|---|---|---|---|
| F8/1.50 | 1.5 | 1.5 | 95.16 ± 1.41 | 2.87 ± 0.24 |
| F8/2.00 | 1.5 | 2 | 97.10 ± 0.85 | 3.59 ± 0.01 |
| F8/2.50 | 1.5 | 2.5 | 97.85 ± 0.07 | 5.19 ± 0.84 |
| F8/3.00 | 1.5 | 3 | 97.1 ± 0.57 | 5.25 ± 0.58 |
| F8/3.50 | 1.5 | 3.5 | 97.05 ± 0.49 | 5.32 ± 0.65 |
| F8/4.00 | 15 | 4 | 97.3 ± 1.41 | 5.39 ± 0.69 |
| F8/4.50 | 1.5 | 4.5 | 96.84 ± 0.34 | 5.40 ± 0.24 |
| *F8/3.58 | 1.5 | 3.58 | 91.99 ± 0.24 | 5.36 ± 0.40 |
| System | Untreated Surface | NeverWet®-Treated Surface | ||||
|---|---|---|---|---|---|---|
| CA (°) | CA (°) | SA (°) | f | 1-f | % Trapped Air | |
| Water | 46.7 ± 0.85 | 151.03 ± 1.08 | 1.00 ± 0.00 | 0.0740 | 0.9258 | 92.58 |
| Olive Oil | 29.70 ± 4.19 | 23.62 ± 1.02 | - | 1.0250 | - | - |
| F8 | 56.40 ± 6.78 | 146.57 ± 2.45 | 11.67 ± 1.53 | 0.1065 | 0.8934 | 89.34 |
| F8/3.58 | 49.00 ± 3.39 | 151.40 ± 2.62 | 4.33 ± 0.58 | 0.0736 | 0.9263 | 92.63 |
| Parameter | Isoptin | F8/3.58 | p-Value |
|---|---|---|---|
| Cmax (ng/mL) | 41.78 ± 21.49 | 10.41 ± 6.97 b | 0.03 |
| AUC0-∞ (ng h/mL) | 165.39 ± 84.16 | 148.43 ± 76.92 | 0.81 |
| AUC0-24 (ng h/mL) | 138.08 ± 69.10 | 107.25 ± 44.46 | 0.24 |
| k (h−1) | 0.06 ± 0.02 | 0.05 ± 0.02 | 0.14 |
| t1/2 (h) | 11.50 ± 3.26 | 16.38 ± 6.85 | 0.13 |
| MRT (h) | 11.38 ± 2.29 | 25.19 ± 8.85 b | 0.03 |
| HVD (h) | 1.37± 0.12 | 7.18 ± 3.39 b | 0.02 |
| Tmax (h) | 1 (1–1.5) | 1 (1–6) | 0.66 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousry, C.; Ahmed, I.S.; Amin, M.M.; El Gazayerly, O.N. Superhydrophobic Substrates for Ultrahigh Encapsulation of Hydrophilic Drug into Controlled-Release Polyelectrolyte Complex Beads: Statistical Optimization and In Vivo Evaluation. Pharmaceutics 2019, 11, 257. https://doi.org/10.3390/pharmaceutics11060257
Yousry C, Ahmed IS, Amin MM, El Gazayerly ON. Superhydrophobic Substrates for Ultrahigh Encapsulation of Hydrophilic Drug into Controlled-Release Polyelectrolyte Complex Beads: Statistical Optimization and In Vivo Evaluation. Pharmaceutics. 2019; 11(6):257. https://doi.org/10.3390/pharmaceutics11060257
Chicago/Turabian StyleYousry, Carol, Iman S. Ahmed, Maha M. Amin, and Omaima N. El Gazayerly. 2019. "Superhydrophobic Substrates for Ultrahigh Encapsulation of Hydrophilic Drug into Controlled-Release Polyelectrolyte Complex Beads: Statistical Optimization and In Vivo Evaluation" Pharmaceutics 11, no. 6: 257. https://doi.org/10.3390/pharmaceutics11060257
APA StyleYousry, C., Ahmed, I. S., Amin, M. M., & El Gazayerly, O. N. (2019). Superhydrophobic Substrates for Ultrahigh Encapsulation of Hydrophilic Drug into Controlled-Release Polyelectrolyte Complex Beads: Statistical Optimization and In Vivo Evaluation. Pharmaceutics, 11(6), 257. https://doi.org/10.3390/pharmaceutics11060257