Synergy of Phospholipid—Drug Formulations Significantly Deactivates Profibrogenic Human Hepatic Stellate Cells

 ,
, 
Abstract
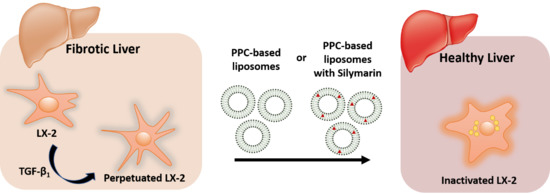
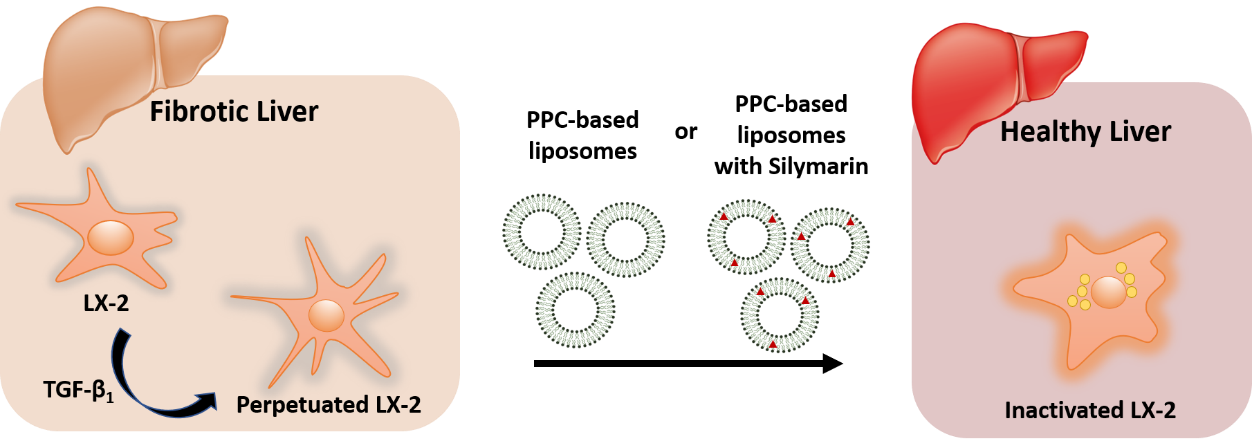{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Development of PPC-Based Formulations
2.3. Development of Silymarin PPC-Based Formulations
2.4. Characterization of Particle Size and Size Distribution
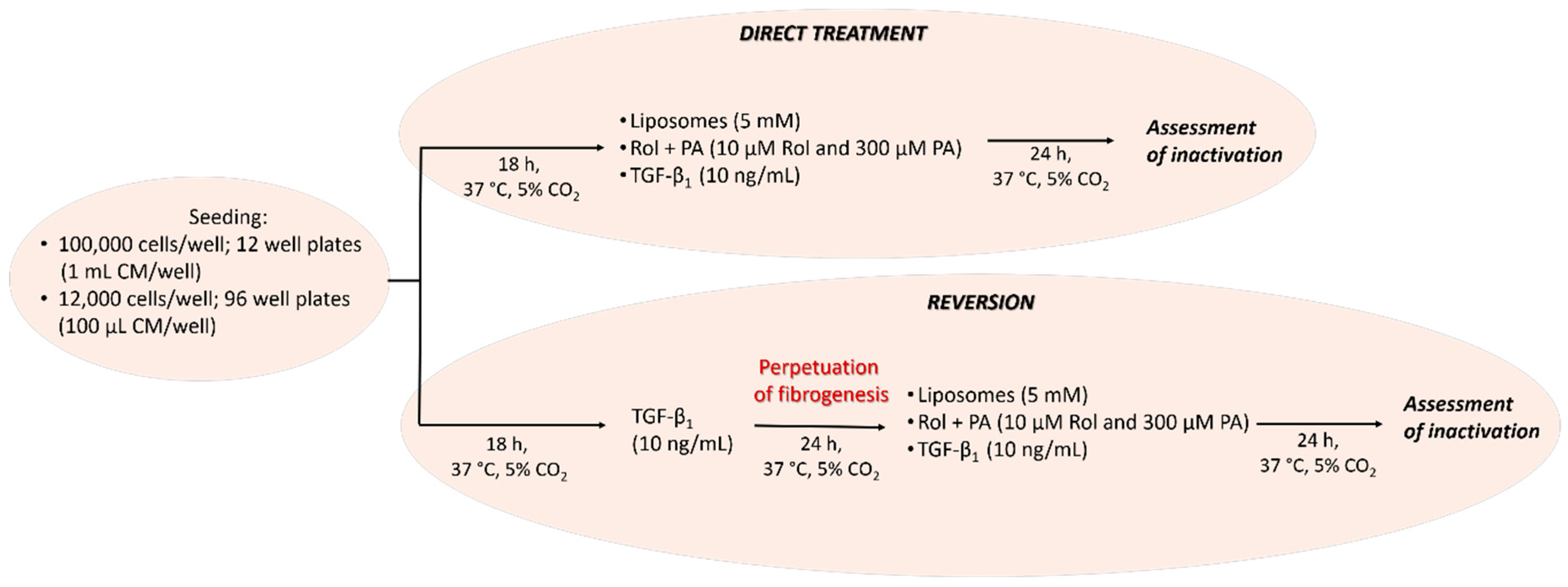
2.5. Cell Culture and General Design of Cell Experiments
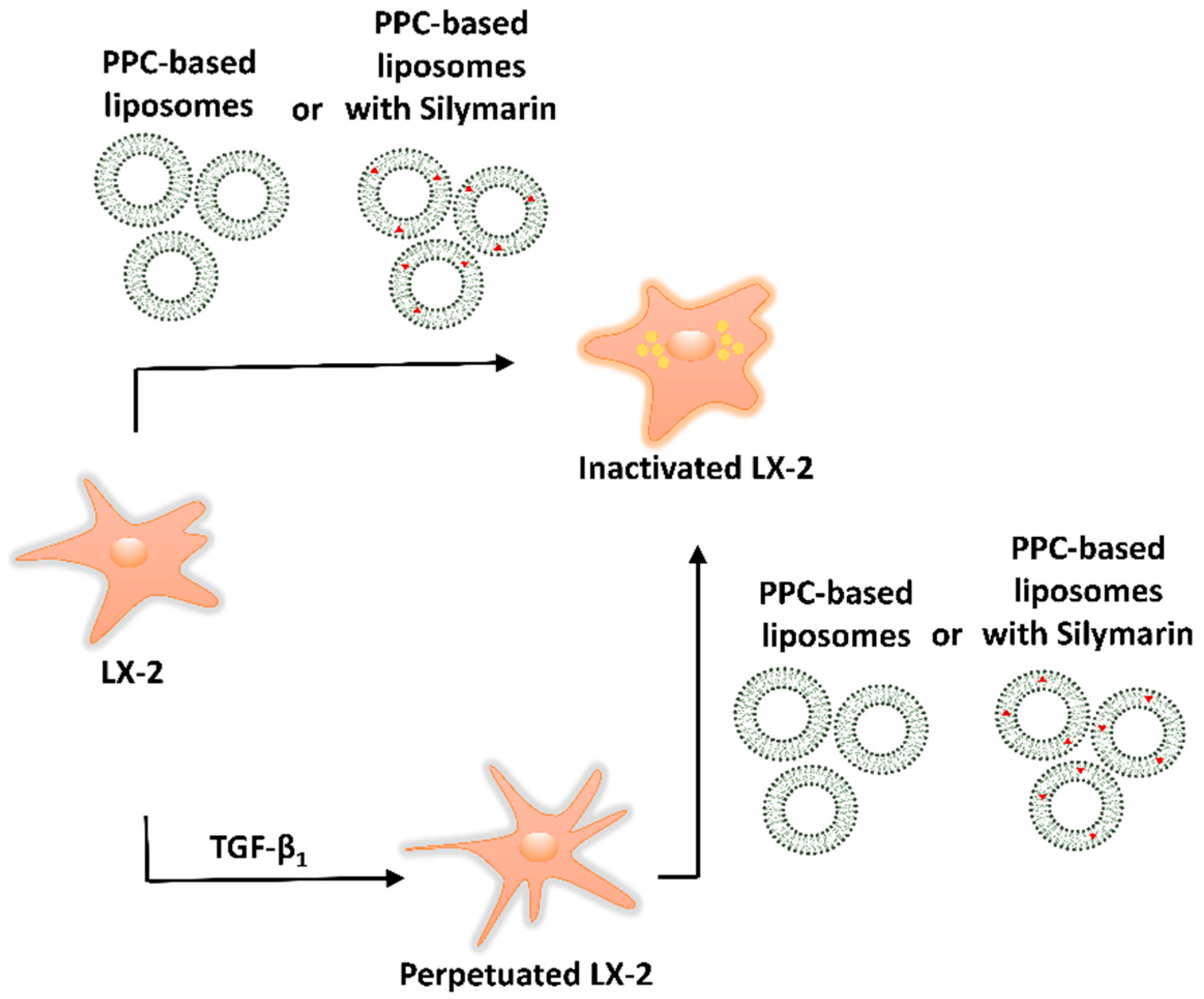
2.6. Experimental Approach: Direct Treament of LX-2 Cells and Treatment of Perpetuated LX-2 Cells (pLX-2 Cells)
2.7. Induction of Quiescent-Like LX-2 Cells with Retinol and Palmitic Acid (Rol + PA)
2.8. LX-2 Cells and pLX-2 Cells Treatment with PPC-Based Formulations
2.9. Analysis of Lipid Droplet Content
2.10. Analysis of Fibrillar Collagen
2.11. Analysis of α-SMA Expression
2.12. Cell Proliferation Assay
2.13. Motional Order of the Cell Membrane in Adherent LX-2 Cells
2.14. Statistical Analysis
3. Results and Discussion
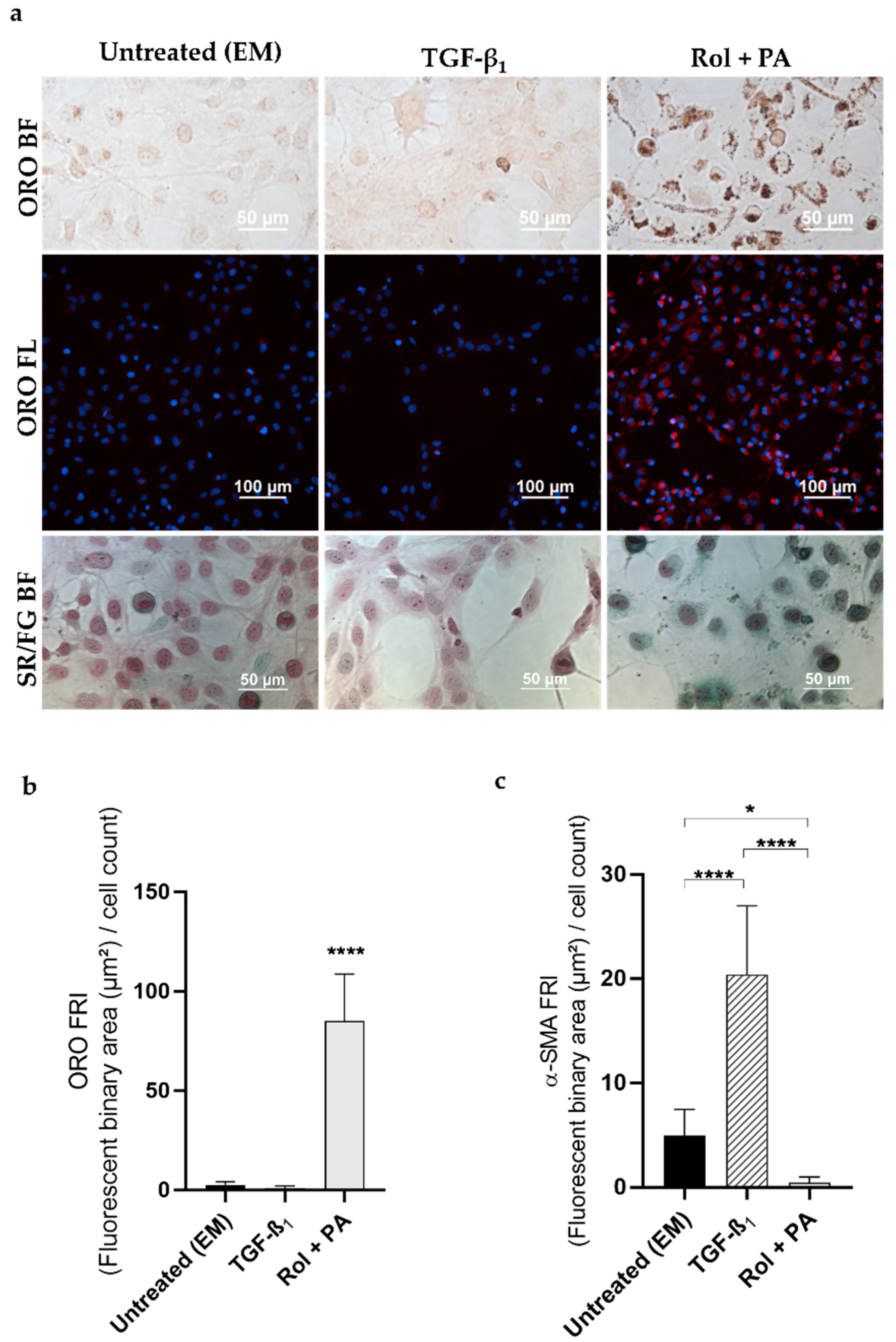
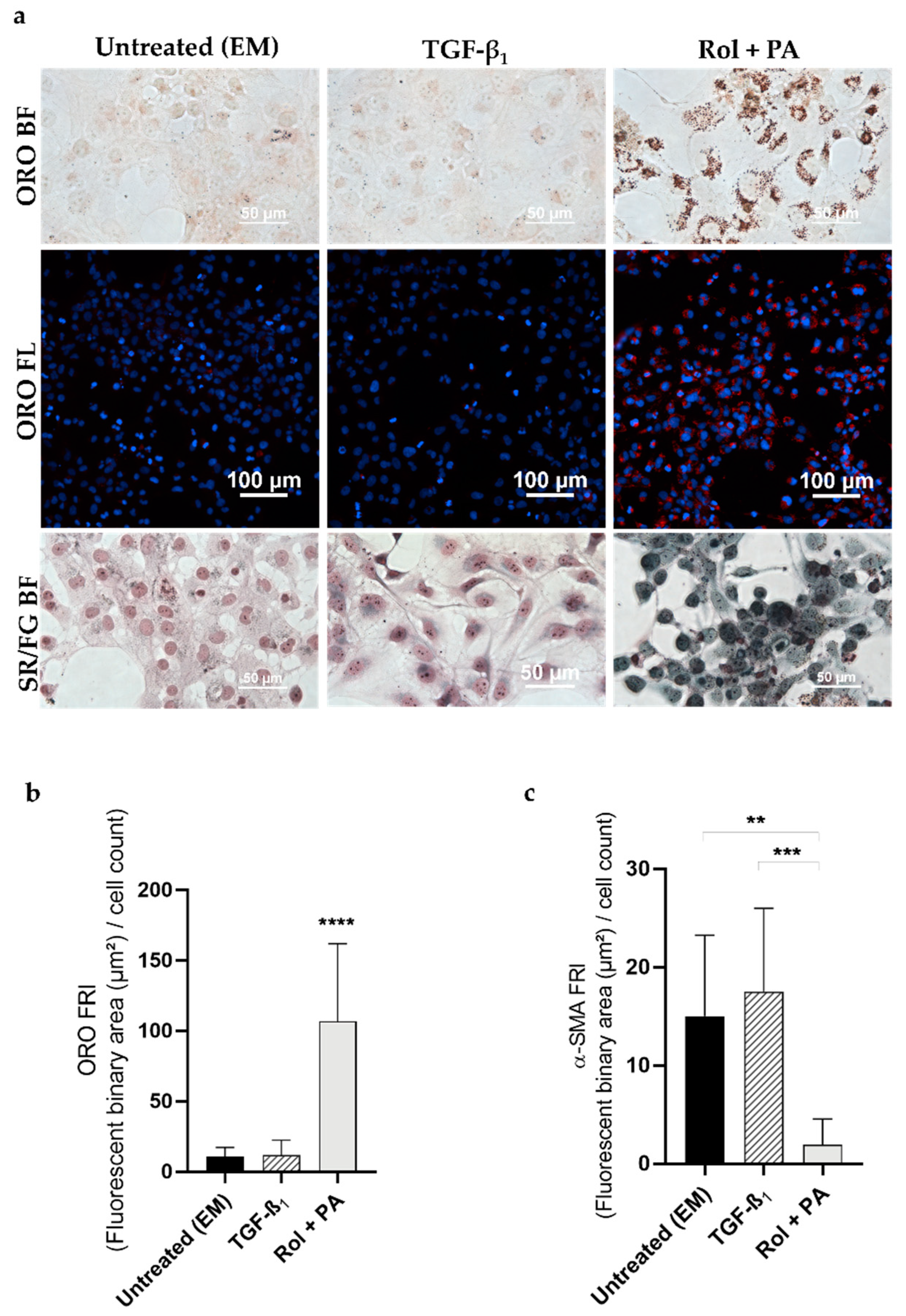
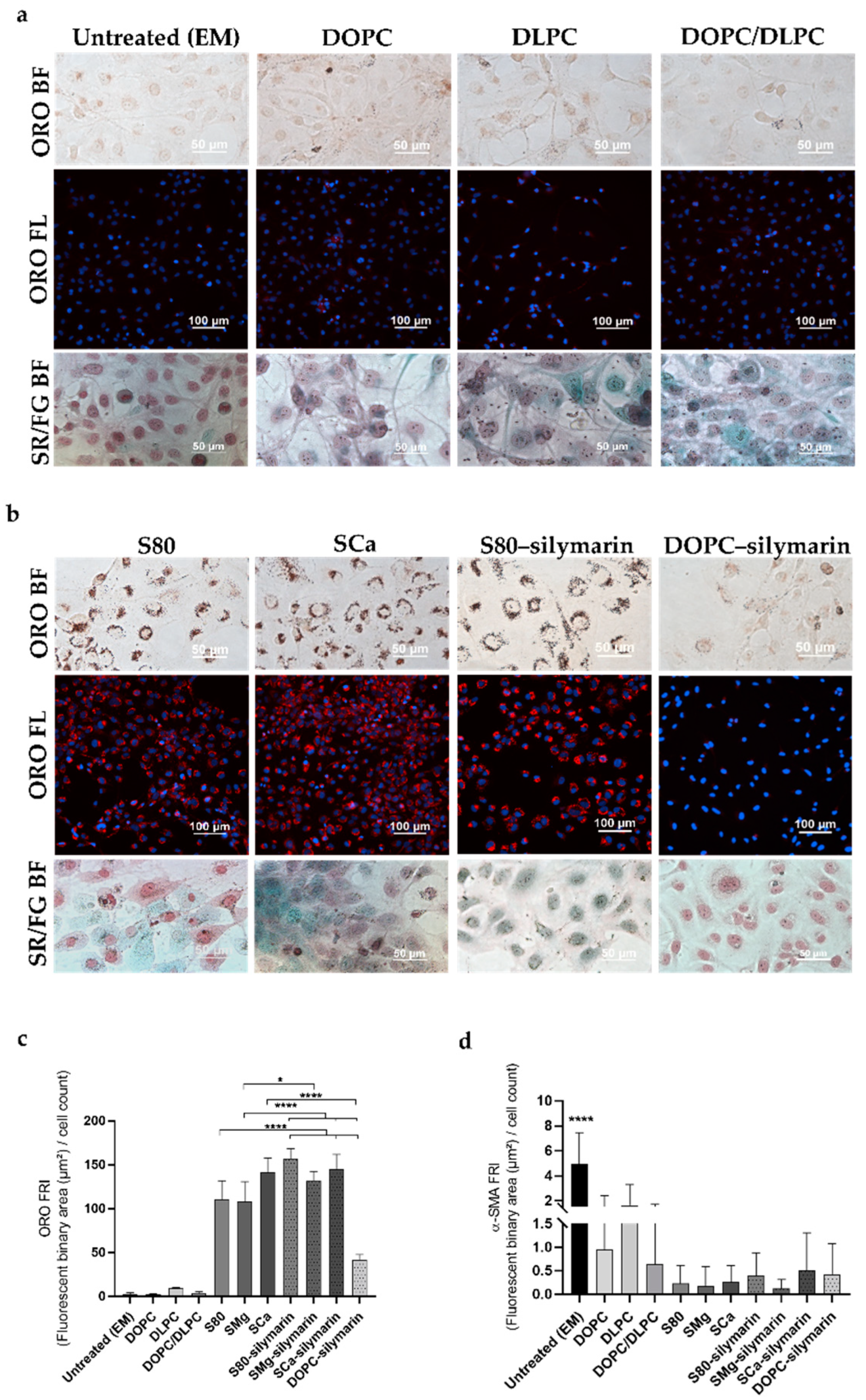
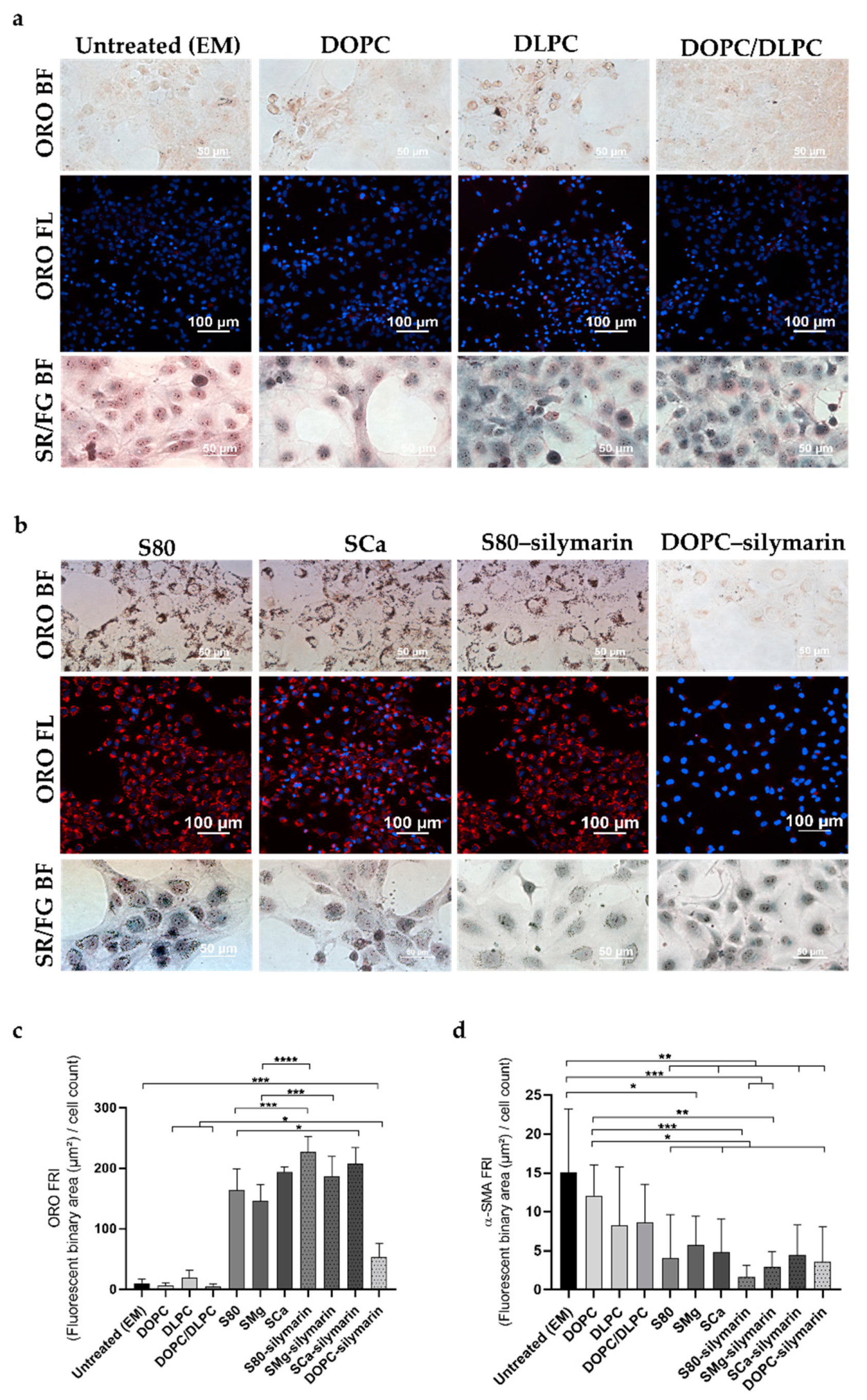
3.1. Progression and Regression of Fibrogenic Features in LX-2 Cells
3.2. In Vitro Antifibrotic Effect of Phospholipid-Based Formulations
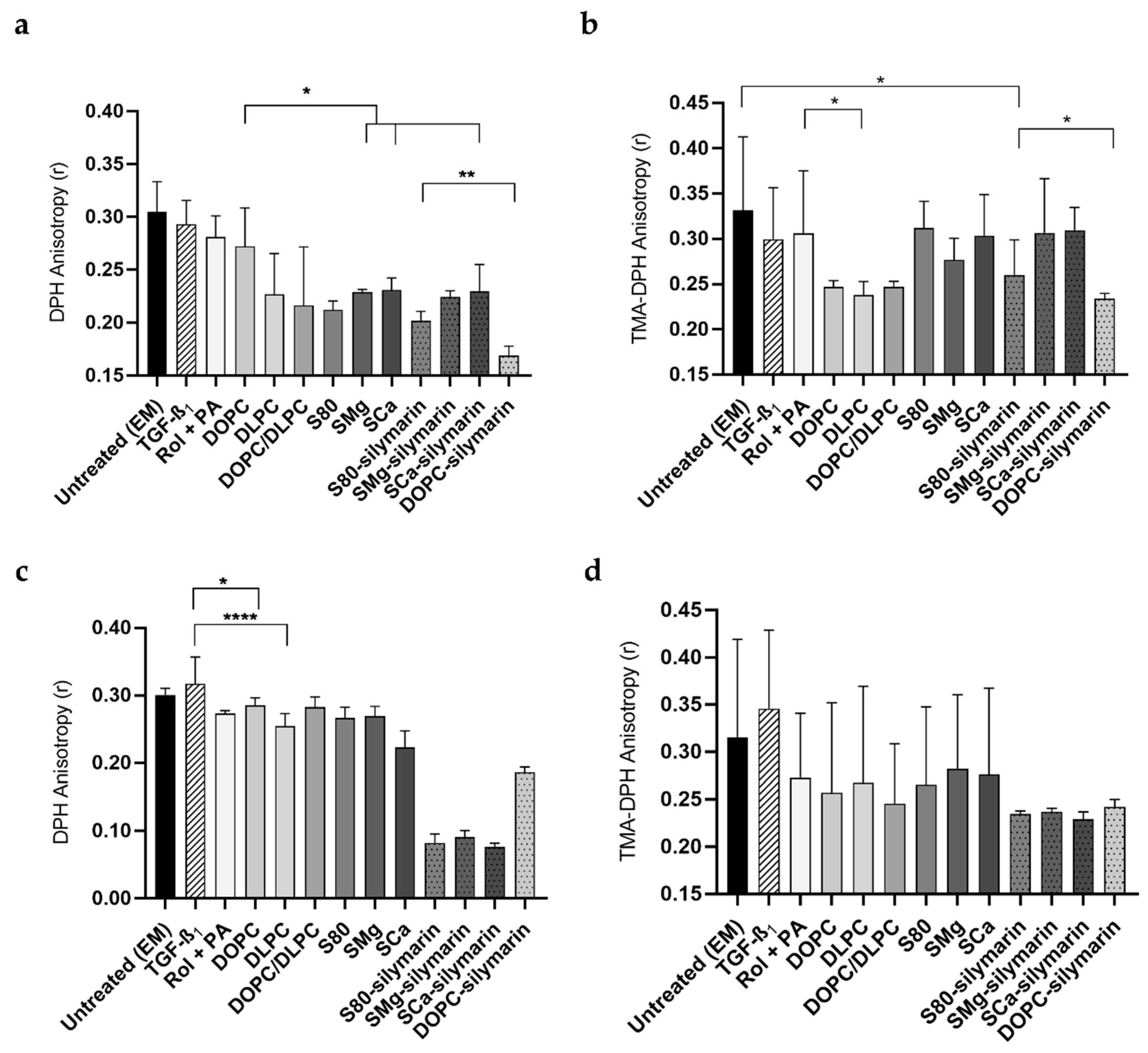
3.3. Cell Membrane Motional Order of Adherent LX-2 Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Friedman, S.L. Liver fibrosis—From bench to bedside. J. Hepatol. 2003, 38, 38–53. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Kutala, B.K. Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. 2018, 38 (Suppl. 1), 2–6. [Google Scholar] [CrossRef] [PubMed]
- Byass, P. The global burden of liver disease: A challenge for methods and for public health. BMC Med. 2014, 12, 159. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Hagström, H.; Nasr, P.; Ekstedt, M.; Hammar, U.; Stål, P.; Hultcrantz, R.; Kechagias, S. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J. Hepatol. 2017, 67, 1265–1273. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Asp. Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 139–148. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, C.R. Hepatic stellate cell activation and pro-fibrogenic signals. J. Hepatol. 2017, 67, 1104–1105. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic Stellate Cells and Liver Fibrosis. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Volume 3, pp. 1473–1492. [Google Scholar]
- El Taghdouini, A.; Najimi, M.; Sancho-Bru, P.; Sokal, E.; van Grunsven, L.A. In vitro reversion of activated primary human hepatic stellate cells. Fibrogenes. Tissue Repair 2015, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Trautwein, C.; Friedman, S.L.; Schuppan, D.; Pinzani, M. Hepatic fibrosis: Concept to treatment. J. Hepatol. 2015, 62, S15–S24. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef]
- Wang, X.; Dong, F.; Zhang, S.; Yang, W.; Yu, W.; Wang, Z.; Zhang, S.; Wang, J.; Ma, S.; Wu, P.; et al. TGF-β1 Negatively Regulates the Number and Function of Hematopoietic Stem Cells. Stem Cell Rep. 2018, 11, 274–287. [Google Scholar] [CrossRef]
- Lotersztajn, S.; Mallat, A. Chapter 11—Hepatic Stellate Cells as Target for Reversal of Fibrosis/Cirrhosis; Elsevier Inc.: Amsterdam, The Netherlands, 2015; ISBN 9780128005446. [Google Scholar]
- Bataller, R.; Brenner, D.A. Hepatic stellate cells as a target for the treatment of liver fibrosis. Semin. Liver Dis. 2001, 21, 437–451. [Google Scholar] [CrossRef]
- Gundermann, K.-J.; Gundermann, S.; Drozdzik, M.; Mohan Prasad, V.G. Essential phospholipids in fatty liver: A scientific update. Clin. Exp. Gastroenterol. 2016, 9, 105–117. [Google Scholar]
- Varganova, D.L.; Pavlov, C.S.; Casazza, G.; Nikolova, D.; Gluud, C. Essential phospholipids for people with non-alcoholic fatty liver disease (Protocol). Cochrane Database Syst. Rev. 2019. [Google Scholar] [CrossRef]
- van Hoogevest, P.; Wendel, A. The use of natural and synthetic phospholipids as pharmaceutical excipients. Eur. J. Lipid Sci. Technol. 2014, 116, 1088–1107. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Dallio, M.; Loguercio, C.; Federico, A.; Dallio, M.; Loguercio, C. Silymarin/Silybin and Chronic Liver Disease: A Marriage of Many Years. Molecules 2017, 22, 191. [Google Scholar] [CrossRef] [PubMed]
- Trappoliere, M.; Caligiuri, A.; Schmid, M.; Bertolani, C.; Failli, P.; Vizzutti, F.; Novo, E.; Manzano, C.; Marra, F.; Loguercio, C.; et al. Silybin, a component of sylimarin, exerts anti-inflammatory and anti-fibrogenic effects on human hepatic stellate cells. J. Hepatol. 2009, 50, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.Y.; Kalantar, K.; Shahin, K.; Ghayour, M.; Rajabi Bazl, M.; Fattahi, M.-R.; Moini, M.; Amirghofran, Z. Comparison of the In Vitro Antifibrogenic Effects of Silymarin, Silybin A and 18α-Glycyrrhizin on Activated Hepatic Stellate Cells. Jundishapur J. Nat. Pharm. Prod. 2016, 12, e40285. [Google Scholar] [CrossRef]
- Lieber, C.S. New concepts of the pathogenesis of alcoholic liver disease lead to novel treatments. Curr. Gastroenterol. Rep. 2004, 6, 60–65. [Google Scholar] [CrossRef]
- Lieber, C.S.; Robins, S.J.; Li, J.; DeCarli, L.M.; Mak, K.M.; Fasulo, J.M.; Leo, M.A. Phosphatidylcholine protects against fibrosis and cirrhosis in the baboon. Gastroenterology 1994, 106, 152–159. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, J.; Lieber, C.S. Polyenylphosphatidylcholine attenuates non-alcoholic hepatic fibrosis and accelerates its regression. J. Hepatol. 1996, 24, 604–613. [Google Scholar] [CrossRef]
- Aleynik, S.I.; Leo, M.A.; Ma, X.; Aleynik, M.K.; Lieber, C.S. Polyenylphosphatidylcholine prevents carbon tetrachloride-induced lipid peroxidation while it attenuates liver fibrosis. J. Hepatol. 1997, 27, 554–561. [Google Scholar] [CrossRef]
- Buang, Y.; Wang, Y.-M.; Cha, J.-Y.; Nagao, K.; Yanagita, T. Dietary phosphatidylcholine alleviates fatty liver induced by orotic acid. Nutrition 2005, 21, 867–873. [Google Scholar] [CrossRef]
- Li, J.; Kim, C.I.; Leo, M.A.; Mak, K.M.; Rojkind, M.; Lieber, C.S. Polyunsaturated lecithin prevents acetaldehyde-mediated hepatic collagen accumulation by stimulating collagenase activity in cultured lipocytes. Hepatology 1992, 15, 373–381. [Google Scholar] [CrossRef]
- Poniachik, J.; Baraona, E.; Zhao, J.; Lieber, C.S. Dilinoleoylphosphatidylcholine decreases hepatic stellate cell activation. J. Lab. Clin. Med. 1999, 133, 342–348. [Google Scholar] [CrossRef]
- Cao, Q.; Mak, K.M.; Lieber, C.S. DLPC and SAMe prevent a 1(I) collagen mRNA up-regulation in human hepatic stellate cells, whether caused by leptin or menadione. Biochem. Biophys. Res. Commun. 2006, 350, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, R.; Ishii, K.; Hoshikawa, Y.; Azumi, J.; Arakaki, Y.; Yasui, T.; Matsuura, S.; Matsumi, Y.; Kono, Y.; Mizuta, Y.; et al. Reactive oxygen species and NADPH oxidase 4 induced by transforming growth factor β1 are the therapeutic targets of polyenylphosphatidylcholine in the suppression of human hepatic stellate cell activation. Inflamm. Res. 2011, 60, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.L.; Ash, M.E. Membrane Lipid Replacement for chronic illnesses, aging and cancer using oral glycerolphospholipid formulations with fructooligosaccharides to restore phospholipid function in cellular membranes, organelles, cells and tissues. Biochim. Biophys. Acta 2017, 1859, 1704–1724. [Google Scholar] [CrossRef]
- Lieber, C.S.; Robins, S.J.; Leo, M.A. Hepatic phosphatidylethanolamine methyltransferase activity is decreased by ethanol and increased by phosphatidylcholine. Alcohol. Clin. Exp. Res. 1994, 18, 592–595. [Google Scholar] [CrossRef]
- Rahnfeld, L.; Thamm, J.; Steiniger, F.; van Hoogevest, P.; Luciani, P. Study on the in situ aggregation of liposomes with negatively charged phospholipids for use as injectable depot formulation. Colloids Surf. B Biointerfaces 2018, 168, 10–17. [Google Scholar] [CrossRef]
- Castilla, A.; Prieto, J.; Fausto, N. Transforming Growth Factors β1 and α in Chronic Liver Disease. N. Engl. J. Med. 1991, 324, 933–940. [Google Scholar] [CrossRef]
- Dewidar, B.; Soukupova, J.; Fabregat, I.; Dooley, S. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis: Updated. Curr. Pathobiol. Rep. 2015, 3, 291–305. [Google Scholar] [CrossRef]
- Yu, J.; Hu, Y.; Gao, Y.; Li, Q.; Zeng, Z.; Li, Y.; Chen, H. Kindlin-2 regulates hepatic stellate cells activation and liver fibrogenesis. Cell Death Discov. 2018, 4, 35. [Google Scholar] [CrossRef]
- Shen, H.; Huang, G.J.; Gong, Y.W. Effect of transforming growth factor beta and bone morphogenetic proteins on rat hepatic stellate cell proliferation and trans-differentiation. World J. Gastroenterol. 2003, 9, 784–787. [Google Scholar] [CrossRef]
- Lee, T.F.; Mak, K.M.; Rackovsky, O.; Lin, Y.-L.; Kwong, A.J.; Loke, J.C.; Friedman, S.L. Downregulation of hepatic stellate cell activation by retinol and palmitate mediated by adipose differentiation-related protein (ADRP). J. Cell. Physiol. 2010, 223, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, S.; Fujimoto, T. Deformation of lipid droplets in fixed samples. Histochem. Cell Biol. 2002, 118, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.H.; Vucic, A. The effect of retinol on ito cell proliferation in vitro. Hepatology 1988, 8, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Pinzani, M.; Gentilini, P.; Hanna, E.A. Phenotypical modulation of liver fat-storing cells by retinoids: Influence on unstimulated and growth factor-induced cell proliferation. J. Hepatol. 1992, 14, 211–220. [Google Scholar] [CrossRef]
- Fabregat, I.; Caballero-Díaz, D. Transforming growth factor-β-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [PubMed]
- Duan, N.N.; Liu, X.J.; Wu, J. Palmitic acid elicits hepatic stellate cell activation through inflammasomes and hedgehog signaling. Life Sci. 2017, 176, 42–53. [Google Scholar] [CrossRef]
- Anfuso, B.; Giraudi, P.J.; Tiribelli, C.; Rosso, N. Silybin modulates collagen turnover in an in vitro model of NASH. Molecules 2019, 24, 1280. [Google Scholar] [CrossRef]
- Hong, Y.; Li, S.; Wang, J.; Li, Y. In vitro inhibition of hepatic stellate cell activation by the autophagy-related lipid droplet protein ATG2A. Sci. Rep. 2018, 8, 1–10. [Google Scholar]
- Moriwaki, H.; Blaner, W.S.; Piantedosi, R.; Goodman, D.S. Effects of dietary retinoid and triglyceride on the lipid composition of rat liver stellate cells and stellate cell lipid droplets. J. Lipid Res. 1988, 29, 1523–1534. [Google Scholar]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.C.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta 2009, 1791, 467–473. [Google Scholar] [CrossRef]
- Mezaki, Y.; Morii, M.; Hebiguchi, T.; Yoshikawa, K.; Yamaguchi, N.; Yoshino, H.; Senoo, H. The role of retinoic acid receptors in activated hepatic stellate cells. Med. Hypotheses 2013, 81, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Jophlin, L.L.; Koutalos, Y.; Chen, C.; Shah, X.V.; Rockey, D.C. Hepatic stellate cells retain retinoid-laden lipid droplets after cellular transdifferentiation into activated myofibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 315, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Carpino, G.; Morini, S.; Ginannicorradini, S.; Franchitto, A.; Merlo, M.; Siciliano, M.; Gentili, F.; Onettimuda, A.; Berloco, P.; Rossi, M. Alpha-SMA expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig. Liver Dis. 2005, 37, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Pradere, J.P.; Friedman, R.A.; Schwabe, R.F. Deactivation of Hepatic Stellate Cells During Liver Fibrosis Resolution in Mice. Gastroenterology 2012, 143, 1073–1083. [Google Scholar]
- Ducharme, N.A.; Bickel, P.E. Minireview: Lipid Droplets in Lipogenesis and Lipolysis. Endocrinology 2008, 149, 942–949. [Google Scholar] [CrossRef]
- Kisseleva, T.; Cong, M.; Paik, Y.H.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef]
- do Canto, A.M.T.M.; Robalo, J.R.; Santos, P.D.; Carvalho, A.J.P.; Ramalho, J.P.P.; Loura, L.M.S. Diphenylhexatriene membrane probes DPH and TMA-DPH: A comparative molecular dynamics simulation study. Biochim. Biophys. Acta 2016, 1858, 2647–2661. [Google Scholar] [CrossRef]
- Sauvant, P.; Abergel, A.; Partier, A.; Alexandre-Gouabau, M.-C.; Rock, E.; Sion, B.; Motta, C.; Sapin, V.; Azaïs-Bresco, V. Treatment of the rat hepatic stellate cell line, PAV-1, by retinol and palmitic acid leads to a convenient model to study retinoids metabolism. Biol. Cell 2002, 94, 401–408. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valentino, G.; Zivko, C.; Weber, F.; Brülisauer, L.; Luciani, P. Synergy of Phospholipid—Drug Formulations Significantly Deactivates Profibrogenic Human Hepatic Stellate Cells. Pharmaceutics 2019, 11, 676. https://doi.org/10.3390/pharmaceutics11120676
Valentino G, Zivko C, Weber F, Brülisauer L, Luciani P. Synergy of Phospholipid—Drug Formulations Significantly Deactivates Profibrogenic Human Hepatic Stellate Cells. Pharmaceutics. 2019; 11(12):676. https://doi.org/10.3390/pharmaceutics11120676
Chicago/Turabian StyleValentino, Gina, Cristina Zivko, Florian Weber, Lorine Brülisauer, and Paola Luciani. 2019. "Synergy of Phospholipid—Drug Formulations Significantly Deactivates Profibrogenic Human Hepatic Stellate Cells" Pharmaceutics 11, no. 12: 676. https://doi.org/10.3390/pharmaceutics11120676
APA StyleValentino, G., Zivko, C., Weber, F., Brülisauer, L., & Luciani, P. (2019). Synergy of Phospholipid—Drug Formulations Significantly Deactivates Profibrogenic Human Hepatic Stellate Cells. Pharmaceutics, 11(12), 676. https://doi.org/10.3390/pharmaceutics11120676