1. Introduction
The blood–brain barrier (BBB), formed by endothelial cells of the brain capillaries, astrocytes, and pericytes, is a regulatory interface between the central nervous system (CNS) and the circulatory bloodstream [
1]. Movement of endogenous and xenobiotic substances across the BBB is tightly regulated by various transporter proteins expressed in the endothelial cells. On the one hand, it is a unique challenge to develop drugs which can overcome the BBB and reach their pharmacological targets within the CNS. On the other hand, it can also be important to prevent drugs with peripheral targets from entering the CNS due to their potential undesired side effects.
Two membrane transporters have been identified as the major players regulating the transport of drugs across the BBB: multidrug resistance 1 (MDR1)/P-glycoprotein (P-gp)/ATP-binding cassette sub-family B member 1 (ABCB1) and breast cancer resistance protein (BCRP)/ATP-binding cassette super-family G member 2 (ABCG2) [
2,
3]. Both transporters belong to the ATP-binding cassette (ABC) superfamily of transporters and are localized in the luminal membrane of the brain capillary endothelial cells. Due to their very broad substrate spectrum both transporters can efficiently limit the entry of drugs into brain and thus are considered as major hurdles in the discovery of CNS drugs. There is, therefore, a strong interest to evaluate the impact of both transporters on the brain distribution of drug candidates early in the drug discovery process. In recent years, the partition coefficient of unbound drug between brain and plasma (K
p,uu,brain) has become more and more relevant for the description of the extent of brain penetration of a drug [
4,
5]. Under steady-state conditions, K
p,uu,brain defines the ratio between the unbound drug concentration in brain (C
u,brain), i.e., the interstitial fluid (ISF) and the unbound drug concentration in plasma(C
u,plasma) (Equation (1)).
Using microdialysis, the concentrations in brain ISF can be determined. Since the concentrations in the ISF are compared to unbound plasma concentrations to derive K
p,uu,brain, in addition to the ISF and plasma concentrations derived from the in vivo experiment, the plasma protein binding of the investigated compound has to be determined by an in vitro method. As method development for microdialysis is time consuming and recovery can be an issue, this method is not suitable for the investigation of large numbers of compounds as typically needed in drug discovery research. A commonly used method with higher throughput is the brain homogenate method where drug concentrations in brain and plasma are determined and converted to the unbound concentrations using fraction unbound in plasma (f
u,plasma) and fraction unbound in brain homogenate (f
u,brain), both being determined in vitro (Equation (2)).
where C
plasma and C
brain are the plasma and brain concentration, respectively. K
p is the partition coefficient of total drug in brain and plasma:
The assumption behind Equation (2) is that the in vitro fraction unbound in brain homogenate (f
u,brain) equals the in vivo fraction unbound in brain. This assumption is, however, no longer valid in case of strong bases which partition strongly into acidic organelles (e.g., lysosomes). The pH partitioning is destroyed by the disruption of organelles during the preparation of brain homogenate, which leads to an overestimation of f
u,brain [
6,
7]. Measurement of volume of distribution of unbound drug (V
u,brain) in brain slices overcomes this partitioning issue [
8]; however, it is not feasible for higher throughput either. In the past, the partition coefficient of total drug, K
p, was frequently used to describe the extent of brain distribution. Since the binding of a drug to plasma protein could differ strongly to the binding to brain tissue, use of K
p for this purpose can be misleading [
8] and is no longer considered a state of the art approach. Instead of measuring the unbound drug concentration in ISF, drug concentration in the cerebrospinal fluid (C
CSF) is often used as a surrogate. Because of the different physiological origin of CSF and ISF and the different expression of drug transporters in the BBB and blood–CSF barrier, the value of C
CSF is limited, especially for strong substrates of drug transporters [
9,
10,
11]. Since all these methods include at least one in vivo experiment, their throughput is rather limited. For compound screening in the early drug discovery phase, several in vitro methods have been established in the pharmaceutical industry. P-gp and BCRP-transfected Madin-Darby canine kidney (MDCK) cells were shown to be valuable tools [
12,
13]. In such assays, the bidirectional transport of a drug across a cell monolayer grown on a Transwell
® insert is measured. The transport activities of P-gp or BCRP in these cells for a certain compound are expressed by the efflux ratio (ER):
with P
appAB and P
appBA being apparent permeability coefficients in the apical to basolateral and basolateral to apical directions, respectively, J
efflux being the rate of transporter-mediated efflux, and PS
Diff the rate of passive diffusion. By setting a certain threshold for the ER, compounds can be easily filtered with regard to potential limitation in brain distribution by P-gp and/or BCRP. These in vitro data, however, have to be used with caution. Firstly, the expression levels of the transport proteins in the transfectants may differ to those in the BBB. ER measured in the in vitro assays may therefore over- or underestimate the transporter activities in vivo. Secondly, the plasma exposure of the compounds can differ strongly to the standard concentration used in the in vitro assays and over- or underestimation of transporter activities using in vitro data may occur in case of nonlinearity of the transporter activities.
In the present paper, we describe a new in vivo method for the fast estimation of in vivo Kp,uu,brain. We validated this method with compounds from our running discovery programs by comparing it to literature known in vivo and in vitro methods. We describe the derived screening strategy applied in Boehringer-Ingelheim’s drug discovery research which helps to select compounds more efficiently and more reliably with regard to BBB penetration.
4. Discussion
We describe in this publication a new method for the rapid estimation of the in vivo BBB efflux of a compound by using the partition coefficient K
p,br/mu. The underlying assumptions are: (1) that binding within brain and muscle tissue, which is based upon pH partitioning and binding to neutral lipids, neutral phospoholipids and acidic phospholipids [
19,
20] is comparable due to similar intracellular pH and similar amounts of the different phospholipids in both tissues; and (2) that the efflux transporters which limit distribution into brain, especially P-gp and BCRP, are not expressed in muscle tissue in relevant amounts.
Since the pH of the intracellular water of both brain and muscle tissue is reported to be identical, pH 7.0 [
19], pH partitioning should not contribute to differences in distribution of drugs between both tissues. The fractional tissue volume of neutral lipids is approximately four-fold higher in brain than in muscle tissue, whereas the tissue concentration of acidic phospholipids is approximately fourfold higher in muscle than in brain tissue [
19,
20]. According to the models of Rodgers and Rowland, the dominating factor for moderate to strong bases is binding to acidic phospholipids, whereas for weak bases, acids, and neutral compounds it is binding to neutral lipids [
19,
20]. Calculation of the distribution of unbound drug between plasma and both tissues (K
p,u,brain and K
p,u,muscle) for the compounds described in both publications shows that both values typically are within a threefold range, what leads to comparable values for the tissue partitioning coefficients K
p,u,brain and K
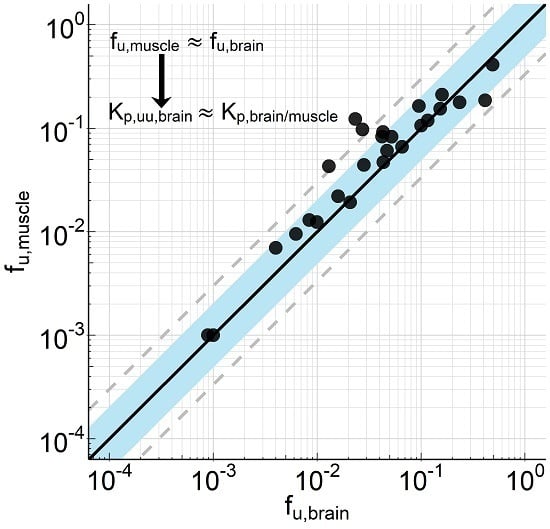
p,u,muscle. Consistent with these considerations, we could show for 26 compounds (for which calculated or measured pKa values ranged between 2 and 11) very similar binding in homogenates from brain and skeletal muscle of rats (
Figure 1). Assuming that transporters do not relevantly affect the distribution into muscle, differences in the tissue partitioning coefficients can therefore be attributed to transporters at the blood–brain barrier. That allows us to estimate efflux at the blood–brain barrier (K
p,uu,br) using the ratio of the tissue partitioning coefficients K
p,u,brain/K
p,u,muscle and with the further assumption of identical binding in tissues (f
u,brain = f
u,muscle) to use the ratio K
p,br/mu. However, for weak bases, acids, and neutral compounds, K
p,u,brain is typically slightly greater than K
p,u,muscle and for moderate to strong bases K
p,u,muscle is typically slightly greater than K
p,u,brain, what would lead to a small overestimation of free brain concentration and underestimation of efflux for weak bases, acids and neutral compounds and a small underestimation of free brain concentration and overestimation of efflux for moderate to strong bases (data not shown). It should be mentioned that we have not done a systematic comparison of partitioning within a broad set of different tissues. Although we see muscle as an appropriate reference tissue, there is a possibility that other tissues could also be suitable or even better for this purpose.
Typical drug transporters like organic anion transporting polypetide (OATP) 1B1, OATP1B3, organic anion transporter (OAT) 1, OAT3, P-gp, BCRP, and multidrug resistance protein (MRP) 2 are not detected in skeletal muscle [
23]. However, OATP2B1, MRP1, MRP4, and MRP5, which are relevant for the uptake (OATP2B1) and efflux (MRPs) of statins (e.g., atorvastatin and rosuvastatin) in skeletal muscle, were detected in skeletal muscle by mRNA expression, western blot, and immunolocalization [
23]. Thus, if other transporters than P-gp or BCRP are involved in the brain and/or muscle distribution of a drug the potential role of these transporters has therefore to be carefully assessed before applying this method. On the other hand, discrepancies between in vitro efflux data for P-gp/BCRP and K
p,uu,brain determined using K
p,br/m or between K
p,uu,brain determined using K
p,br/m and other methods can be an indication that other transporters or mechanisms are potentially involved.
Our results demonstrate that the new method described here is fast, robust, and versatile and provides sufficient comparability to the standard method for the estimation of BBB efflux (K
p,uu,brain), the brain homogenate method (K
p,uu,brain(hom)). It is certainly debatable whether the K
p,uu,brain calculation using f
u,brain determined from brain homogenate is appropriate for all compounds, e.g., the unbound fraction of basic compounds with strong tendency of pH partitioning could be overestimated [
6,
7,
8]. In this regard, the K
p,br/mu method could be even superior to the K
p,uu,brain calculation using f
u,brain determined from brain homogenate method since the pH partitioning into brain and skeletal muscle seems to be similar [
24]. A more accurate method to determine the unbound concentration of basic compounds in brain is the determination of V
u,brain with the brain slice method [
6,
7,
8]. Due to the limited throughput of this method, however, it is not suitable for large-scale compound screening. Moreover, most of our compounds with CNS targets are neutral or weakly basic. For those compounds, the effect of pH partitioning on tissue distribution is considered minor. Indeed, we measured the V
u,brain of a small set of our compounds and could confirm the minor effect of pH partitioning on the K
p,uu,brain calculation using the brain homogenate method (data not shown). For the brain homogenate method three separate experiments are necessary: one in vivo tissue distribution study and two in vitro studies to determine f
u,plasma and f
u,brain. For K
p,br/mu determination, one single in vivo tissue distribution study is sufficient. In addition to the lower experimental effort needed, K
p,br/mu determination is considered less prone to experimental variability than the brain homogenate method: Since experimental variability is intrinsic in all data, the more data points are used in a calculation, the stronger the amplification of the variability. Especially for highly bound compounds, the unbound fraction measurement in the screening assays is often hampered by the limit of quantification in the analytical assay, despite the use of LC-MS/MS. In those cases the K
p,uu,brain calculation using the brain homogenate method can become unreliable. Here, K
p,br/mu measurement provides a more reliable estimation of in vivo efflux at BBB as it does not need those very high sensitivity of the analytical methods. As discussed earlier [
9,
11], CSF is a less reliable surrogate for C
u,brain compared to other methods when the compounds are substrates of efflux transporters. The poorer correlation between K
p,uu,CSF and K
p,br/mu compared to K
p,uu,brain and K
p,br/mu in our experiments (
Figure 2) confirm this.
A few compounds tested are reference compounds with published brain partitioning data. As shown in
Table 4, K
p,uu,brain values of the P-gp substrate quinidine and the non-efflux compound carbamazapine reported by Kodaira et al. fit well to our K
p,br/mu values [
11]. For the BCRP substrate dantrolene, however, there is a strong discrepancy between the value reported by Kodaira et al. and the K
p,br/mu in this study. Interestingly, our K
p,uu,brain value determined using the brain homogenate method is only 2-fold higher than that reported by Kodaira et al., suggesting that the discrepancy might not result from lab-to-lab variability, but rather from the difference in the methods. Moreover, brain distribution studies with dantrolene in Bcrp knockout rats demonstrated that dantrolene is rather a low efflux substrate with a K
p,uu,brain of 0.27 [
22], very close to our K
p,br/mu value. Since K
p,uu,brain calculation using brain slice method or brain homogenate method includes an in vitro f
u,brain determination, whereas distribution studies using knockout animals and the K
p,br/mu method include only in vivo measurements, the discrepancy might represent a underestimation of f
u,brain by the in vitro measurements. Consistent to this consideration, we observed a 2-fold bias of K
p,br/mu towards higher values compared to K
p,uu,brain values (
Figure 2A). Underestimation of K
p,uu,brain might result from the underestimation of f
u,brain by the in vitro method.
Bidirectional permeability measurement using transfected MDCK cells is a well-accepted in vitro screening assay in the pharmaceutical industry to address the blood–brain barrier efflux [
12,
13,
21]. We describe in this work the calibration of our in-house transfected cell lines with the in vivo K
p,br/mu data. The IVIVC is in general very good with regard to P-gp mediated efflux, although the substrate concentration in the permeability assay has to be reduced for certain compound classes. Our standard substrate concentration of 10 µM was a balance between assay efficiency and relevance of the results. The unbound plasma concentration of a test compound in a pharmacokinetic (PK) or a pharmacodynamics (PD) study is in most cases lower than 10 µM, therefore a lower substrate concentration in the in vitro assay would be more appropriate. However, especially for compounds with lower permeability or those with bad ionization behavior, sensitivity issues of the LC-MS/MS methods prevent investigations at lower substrate concentration. A retrospective analysis showed that for ~20% of our research compounds the limit of quantification would be insufficient for a substrate concentration of 1 µM (data not shown). Using such a concentration as standard substrate concentration would lead to too many repeats of the experiments at higher concentrations. Our screening strategy was thus to get an IVIVC using standard substrate concentration of 10 µM at an early stage of a research project. In case of insufficient correlation, the substrate concentration can be reduced to 1 µM accompanied by investment in improving the analytical methods for these specific compounds/compound classes. Otherwise, IVIVC needs to be spot-checked regularly.
For some of compounds, insufficient IVIVC with MDCK-MDR1 cells was not resolved by applying a lower substrate concentration, but rather caused by the involvement of other transporters at the BBB. The MDCK-BCRP assay was a valuable tool to identify such compounds. Due to the very high expression level of recombinant BCRP in the transfected cells, a correction factor has to be applied for translation of the in vitro efflux to in vivo efflux. Using the reference BCRP substrate dantrolene, we determined a correction factor of about 6. A similar approach was already described by Liu et al. [
13]. Since some compounds are substrates of both P-gp and BCRP, estimation of in vivo efflux needs to combine data from both in vitro assays. Although a synergism between P-gp and BCRP at the BBB was hypothesized [
25], the total efflux of both transporters is considered rather additive [
26]. Based on these considerations, we developed Equation (17) to calculate a total in vitro efflux using data from both in vitro assays. The validity of this equation was confirmed by the regression analysis of data from a set of 133 compounds (
Table 3). As the expression of recombinant transporters differs from laboratory to laboratory, our equation is essentially laboratory-specific and needs an individual calibration for the respective transfected cell lines in the different laboratories. It is important to note that all in vitro data were obtained with cells expressing recombinant human transporters and the in vivo data were from rats. The reported similar activities of rodent and human P-gp in vitro is consistent with the good IVIVC we observed here [
12]. Quantitative proteomics indicates a three to four-fold higher P-gp protein concentration in the BBB of rat than in human BBB, while BCRP protein concentrations are comparable in rats and humans [
27,
28]. Consistent with these data, positron emission tomography studies with [
11C]verapamil indicated a roughly three-fold higher BBB efflux of Verapamil in rat than in human [
29]. However, Braun et al. demonstrated in dogs that up to 5-fold expression differences of P-gp as well as BCRP between cerebrum, cerebellum, and brain stem of dogs did not affect the K
p,brain of quinidine, apafant, dantrolene, and daidzeine, two P-gp and two BCRP substrates in these brain regions of the same dogs [
9]. Thus it is currently unclear whether differences in transporter expression really affect the drug concentrations in brain. Due to the proposed, at maximum three fold difference between rats and humans, it is thus reasonable to assume that the efflux measured in rats is close to that in humans. The good correlation between in vivo data in rat and data from our in vitro assays therefore qualifies these assays for compound screening and selection of development candidates.
The rat is our primary species for pharmacokinetic optimization and thus also used in screening for optimization of BBB penetration since the available plasma pharmacokinetic profiles allow to select appropriate doses and time points for the brain distribution experiments. Since the mouse is often used for pharmacology studies there is also an interest in assessing the BBB efflux in this species. For a set of 135 compounds we investigated the correlation between in vitro efflux derived from MDCK-MDR1 cells with the mouse K
p,br/mu. We found, as for the rat, there was a good correlation, as depicted in
Supplement Figure S2 (data in
Table S7). Thus, the method is in principle also working in mice, which can be rationalized by the comparable expression of P-gp and BCRP in the BBB of rat and mouse (BCRP mouse/rat: 4.41 ± 0.69 fmol/µg protein/4.95 ± 0.32 fmol/µg protein; P-gp mouse/rat: 14.1 ± 2.1 fmol/µg protein/19.1 ± 1.0 fmol/µg protein) [
27,
28].
Our CNS screening strategies have changed over the last decade(s), driven by better understanding of the BBB as well as new assays being available for screening and in depth investigations. While considering CSF as major determinant of unbound brain concentrations in the past, we moved to determination of K
p,uu,br and now K
p,br/mu as efficient in vivo approach. Even more influence on our screening cascades came from the availability of the efflux transporter expressing MDCK cells. The improved predictivity of the in vitro assays generated the necessary trust in this data, thus ultimately reduced the number of compounds tested in vivo and sped up the decision process.
Scheme 1 depicts the CNS-specific ADME (absorption, distribution, metabolism, excretion) assays of a generic screening cascade applied at Boehringer Ingelheim. It applies to compounds with a reasonable potency, selectivity, and metabolic stability. In the CNS screening part of the cascade, in vitro efflux and f
u,plasma are measured in tier one. Usually, the MDCK-MDR1 assay is used as first line screening assay. As discussed above, spot-checking of the IVIVC by pharmacokinetic (PK) studies with the measurement of K
p,br/mu is performed in order to determine the optimal substrate concentration in the MDCK-MDR1 assay or the necessity of including the MDCK-BCRP assay. During regular compound screening, Equation (17) is used to calculate the total in vitro efflux.
There are two ways to rank the compounds with regard to efflux. If high systemic exposure is undesired, e.g., due to target-related side effects in peripheral tissues, a general cutoff can be used to filter out high-efflux compounds. Taking all experimental variabilities into account, a K
p,br/mu greater than 0.3 indicates a rather low or negligible efflux at BBB. A lower cutoff is conceivable depending on the nature of the off-targets and the associated side effects. If the question is whether or not a compound could achieve efficacious exposure in brain with a realistic plasma exposure at all, in vitro potency, in vitro efflux, and f
u,plasma can be used in combination to estimate the total plasma exposure (C
plasma,total) to be achieved in a pharmacodynamic (PD) study based on a simple equation:
Together with the in vitro data of metabolic stability, compounds can be rank-ordered for further profiling. Both approaches helped to reduce the number of compounds to be tested in PK and/or PD studies. In 2007, when the MDCK assay had not yet been established, brain exposure of nearly 500 compounds was investigated in rats through routine CSF sampling. In 2018, after implementation of our strategy, around 2000 Boehringer Ingelheim compounds were tested in at least one of the two in vitro efflux assays. Only about 100 compounds were further tested in a PK study with the measurement of K
p,br/mu. For development candidates, we perform in depth CNS profiling to further improve understanding of the compound(s). This includes determining K
p,uu,brain with the brain homogenate method and investigation of the time-dependence of brain exposure. Microdialysis and measurement of V
u,brain in brain slices (both considered to generate the most accurate data for estimation of unbound brain concentration) and in vivo studies with efflux transporter knock out animals are considered when we observe discrepancies in the available BBB data or an exact (less than 2-fold error) prediction of the human free brain concentrations is essential for the project. In case CSF sampling is considered for clinical development, we investigate this also in animals to provide the necessary translational data. Besides the CNS profiling, a battery of candidate profiling studies are done, such as in vitro drug-drug interaction (DDI) assessment and in vitro as well as in vivo metabolite identification (MetID) studies, via non-rodent PK studies enabling human PK prediction to perform selection and general pharmacology investigations.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}