Application of Permeation Enhancers in Oral Delivery of Macromolecules: An Update





Abstract
1. Introduction
2. Permeation Enhancer (PE) Categories
3. Targets for Intestinal Permeation Enhancement: Beyond Insulin
4. Recent Highlights
4.1. Oral Semaglutide Reduces HBA1c in Type 2 Diabetics by over 1.5% in Phase II Trials
4.2. The Ionic Liquid Choline Geranate (CAGE) Has a Major Effect on Oral BA of Insulin
4.3. Mode of Action Studies on the PE, PIP 640
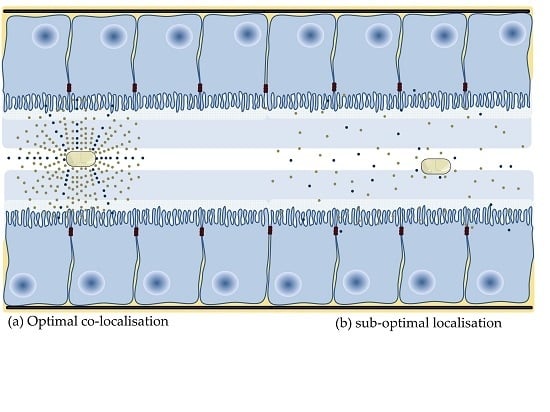
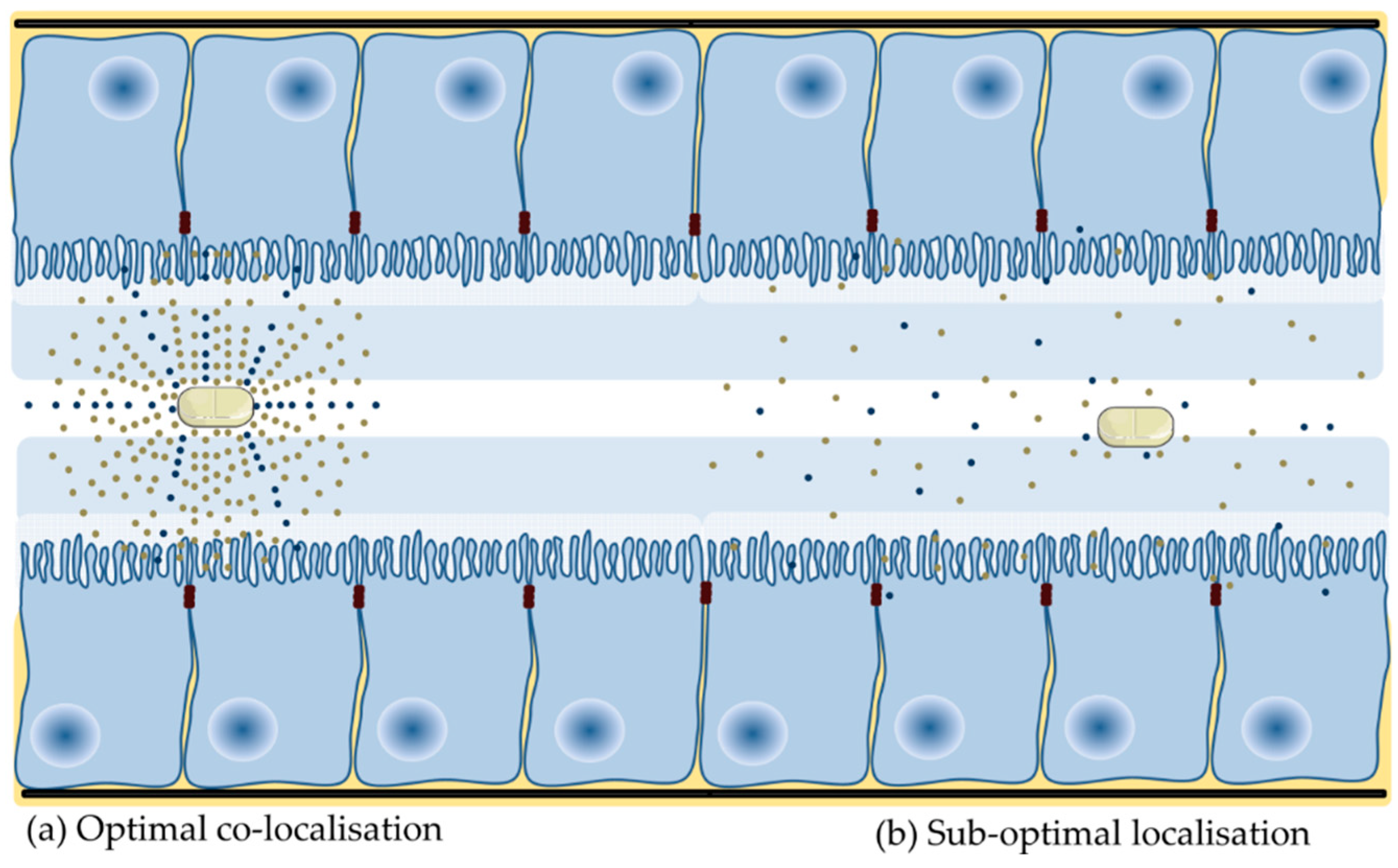
4.4. Application of Nanoparticles to Co-Localise Active and PE
4.5. Application of PEs in Delivery of Nutraceuticals
4.6. Can Non-Ionic Surfactants be More Effective than Ionizable Surfactants?
4.7. Can Physical Hydrophobization Improve Passive Intestinal Flux?
4.8. Mode of Action Studies are Required to Provide Evidence for a Paracellular Effect
4.9. Growing Need for Simulated Intestinal Fluid in PE Experiments
4.10. Improving PE Action in the Dynamic GI Tract
4.11. Intestinal Patches to Co-Localise PE and Active
4.12. Is Safety of PEs a Real Impediment to Translation?
4.13. Convergence between Delivery Concepts, Intestinal Physiology, and Formulation Science
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Barton, P.; Riley, R.J. A new paradigm for navigating compound property related drug attrition. Drug Discov. Today 2016, 21, 72–81. [Google Scholar] [CrossRef]
- Leeson, P.D. Molecular inflation, attrition and the rule of five. Adv. Drug Deliv. Rev. 2016, 101, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Crew, M.; Lipinski, C. Where to Invest? The Drug or the Delivery System? American Association of Pharmaceutical Scientists Blog. 2016. Available online: https://aapsblog.aaps.org/2016/11/05/where-to-invest-the-drug-or-the-delivery-system/#more-9226 (accessed on 17 January 2019).
- Buckley, S. Oral Semaglutide: Delivering new possibilities in the treatment of diabetes. In Proceedings of the Annual Meeting and Exposition of the Controlled Release Society (Oral Peptide Workship), New York, NY, USA, 22–24 July 2018. [Google Scholar]
- Maher, S. An Outlook on Oral Peptide Delivery. AAPS Blog. 2017. Available online: https://aapsblog.aaps.org/2017/02/08/an-outlook-on-oral-peptide-delivery/#more-9531 (accessed on 17 January 2019).
- Kondoh, M.; Yoshida, T.; Kakutani, H.; Yagi, K. Targeting tight junction proteins-significance for drug development. Drug Discov. Today 2008, 13, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal Permeation Enhancers for Oral Peptide Delivery. Adv. Drug Deliv. Rev. 2016, 106, 277–319. [Google Scholar] [CrossRef] [PubMed]
- Eichner, M.; Protze, J.; Piontek, A.; Krause, G.; Piontek, J. Targeting and alteration of tight junctions by bacteria and their virulence factors such as Clostridium perfringens enterotoxin. Pflügers Arch. Eur. J. Physiol. 2017, 469, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Eldor, R.; Arbit, E.; Corcos, A.; Kidron, M. Glucose-reducing effect of the ORMD-0801 oral insulin preparation in patients with uncontrolled type 1 diabetes: A pilot study. PLoS ONE 2013, 8, e59524. [Google Scholar] [CrossRef]
- Zupančič, O.; Bernkop-Schnürch, A. Lipophilic peptide character—What oral barriers fear the most. J. Control. Release 2017, 255, 242–257. [Google Scholar] [CrossRef]
- Maher, S.; Heade, J.; McCartney, F.; Waters, S.; Bleiel, S.B.; Brayden, D.J. Effects of surfactant-based permeation enhancers on mannitol permeability, histology, and electrogenic ion transport responses in excised rat colonic mucosae. Int. J. Pharm. 2018, 539, 11–22. [Google Scholar] [CrossRef]
- Perinelli, D.R.; Cespi, M.; Casettari, L.; Vllasaliu, D.; Cangiotti, M.; Ottaviani, M.F.; Giorgioni, G.; Bonacucina, G.; Palmieri, G.F. Correlation among chemical structure, surface properties and cytotoxicity of N-acyl alanine and serine surfactants. Eur. J. Pharm. Biopharm. 2016, 109, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Sinko, P.J. Oral delivery of salmon calcitonin. Adv. Drug Deliv. Rev. 2000, 42, 225–238. [Google Scholar] [CrossRef]
- Leonard, T.W.; Lynch, J.; McKenna, M.J.; Brayden, D.J. Promoting absorption of drugs in humans using medium-chain fatty acid-based solid dosage forms: GIPET. Expert Opin. Drug Deliv. 2006, 3, 685–692. [Google Scholar] [CrossRef]
- Tuvia, S.; Pelled, D.; Marom, K.; Salama, P.; Levin-Arama, M.; Karmeli, I.; Idelson, G.H.; Landau, I.; Mamluk, R. A novel suspension formulation enhances intestinal absorption of macromolecules via transient and reversible transport mechanisms. Pharm. Res. 2014, 31, 2010–2021. [Google Scholar] [CrossRef] [PubMed]
- Khedkar, A.; Iyer, H.; Anand, A.; Verma, M.; Krishnamurthy, S.; Savale, S.; Atignal, A. A dose range finding study of novel oral insulin (IN-105) under fed conditions in type 2 diabetes mellitus subjects. Diabetes Obes. Metab. 2010, 12, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Cilek, A.; Celebi, N.; Tirnaksiz, F. Lecithin-based microemulsion of a peptide for oral administration: Preparation, characterization, and physical stability of the formulation. Drug Deliv. 2006, 13, 19–24. [Google Scholar] [CrossRef]
- Blikslager, A.T.; Moeser, A.J.; Gookin, J.L.; Jones, S.L.; Odle, J. Restoration of barrier function in injured intestinal mucosa. Physiol. Rev. 2007, 87, 545–564. [Google Scholar] [CrossRef]
- Brayden, D.J.; Gleeson, J.; Walsh, E.G. A head-to-head multi-parametric high content analysis of a series of medium chain fatty acid intestinal permeation enhancers in Caco-2 cells. Eur. J. Pharm. Biopharm. 2014, 88, 830–839. [Google Scholar] [CrossRef]
- Matsuura, J.; Powers, M.E.; Manning, M.C.; Shefter, E. Structure and stability of insulin dissolved in 1-octanol. J. Am. Chem. Soc. 1993, 115, 1261–1264. [Google Scholar] [CrossRef]
- Zupancic, O.; Leonaviciute, G.; Lam, H.T.; Partenhauser, A.; Podricnik, S.; Bernkop-Schnurch, A. Development and in vitro evaluation of an oral SEDDS for desmopressin. Drug Deliv. 2016, 23, 2074–2083. [Google Scholar] [CrossRef]
- Bonengel, S.; Jelkmann, M.; Abdulkarim, M.; Gumbleton, M.; Reinstadler, V.; Oberacher, H.; Prufert, F.; Bernkop-Schnurch, A. Impact of different hydrophobic ion pairs of octreotide on its oral bioavailability in pigs. J. Control. Release 2018, 273, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Menzel, C.; Holzeisen, T.; Laffleur, F.; Zaichik, S.; Abdulkarim, M.; Gumbleton, M.; Bernkop-Schnurch, A. In vivo evaluation of an oral self-emulsifying drug delivery system (SEDDS) for exenatide. J. Control. Release 2018, 277, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.; Gomez-Orellana, I. Challenges for the oral delivery of macromolecules. Nat. Rev. Drug Discov. 2003, 2, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.C.; Friedman, K.; Sherry, J.; Brazzillo, K.; Genoble, L.; Bhargava, P.; Riley, M.G.I. Comparing the Efficacy and Tolerability of a New Daily Oral Vitamin B12 Formulation and Intermittent Intramuscular Vitamin B12 in Normalizing Low Cobalamin Levels: A Randomized, Open-Label, Parallel-Group Study. Clin. Ther. 2011, 33, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Pieber, T.R.; Hartoft-Nielsen, M.L.; Hansen, O.K.H.; Jabbour, S.; Rosenstock, J. Effect of Oral Semaglutide Compared With Placebo and Subcutaneous Semaglutide on Glycemic Control in Patients With Type 2 Diabetes: A Randomized Clinical Trial. JAMA 2017, 318, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Byrjalsen, I.; Alexandersen, P.; Bihlet, A.; Andersen, J.R.; Riis, B.J.; Bay-Jensen, A.C.; Christiansen, C. Treatment of symptomatic knee osteoarthritis with oral salmon calcitonin: Results from two phase 3 trials. Osteoarthr. Cartil. 2015, 23, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, M.E.; Hallbrink, M.M.; Elmquist, A.M.; Langel, U. Passage of cell-penetrating peptides across a human epithelial cell layer in vitro. Biochem. J. 2004, 377, 69–76. [Google Scholar] [CrossRef]
- Kamei, N.; Shigei, C.; Hasegawa, R.; Takeda-Morishita, M. Exploration of the Key Factors for Optimizing the in Vivo Oral Delivery of Insulin by Using a Noncovalent Strategy with Cell-Penetrating Peptides. Biol. Pharm. Bull. 2018, 41, 239–246. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Rehmani, S.; Dixon, J.E. Oral delivery of anti-diabetes therapeutics using cell penetrating and transcytosing peptide strategies. Peptides 2018, 100, 24–35. [Google Scholar] [CrossRef]
- Lewis, A.L.; Richard, J. Challenges in the delivery of peptide drugs: An industry perspective. Ther. Deliv. 2015, 6, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Semaglutide: First Global Approval. Drugs 2018, 78, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Feeney, O.M.; Crum, M.F.; McEvoy, C.L.; Trevaskis, N.L.; Williams, H.D.; Pouton, C.W.; Charman, W.N.; Bergström, C.A.S.; Porter, C.J.H. 50 years of oral lipid-based formulations: Provenance, progress and future perspectives. Adv. Drug Deliv. Rev. 2016, 101, 167–194. [Google Scholar] [CrossRef] [PubMed]
- Benet, L.Z.; Cummins, C.L. The drug efflux-metabolism alliance: Biochemical aspects. Adv. Drug Deliv. Rev. 2001, 50 (Suppl. 1), S3–S11. [Google Scholar] [CrossRef]
- Maher, S.; Ryan, B.; Duffy, A.; Brayden, D.J. Formulation strategies to improve oral peptide delivery. Pharm. Pat. Anal. 2014, 3, 313–336. [Google Scholar] [CrossRef]
- Mahmood, A.; Bernkop-Schnurch, A. SEDDS: A game changing approach for the oral administration of hydrophilic macromolecular drugs. Adv. Drug Deliv. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of Octanol−Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Buckley, S.T.; Bækdal, T.A.; Vegge, A.; Maarbjerg, S.J.; Pyke, C.; Ahnfelt-Rønne, J.; Madsen, K.G.; Schéele, S.G.; Alanentalo, T.; Kirk, R.K.; et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci. Transl. Med. 2018, 10, eaar7047. [Google Scholar] [CrossRef]
- Novo Nordisk AS. Tablet Formulation Comprising Semaglutide and a Delivery Agent. U.S. Patent US9993430B2, 12 June 2018. [Google Scholar]
- Hjerpsted, J.B.; Flint, A.; Brooks, A.; Axelsen, M.B.; Kvist, T.; Blundell, J. Semaglutide improves postprandial glucose and lipid metabolism, and delays first-hour gastric emptying in subjects with obesity. Diabetes Obes. Metab. 2018, 20, 610–619. [Google Scholar] [CrossRef]
- Banerjee, A.; Ibsen, K.; Brown, T.; Chen, R.; Agatemor, C.; Mitragotri, S. Ionic liquids for oral insulin delivery. Proc. Natl. Acad. Sci. USA 2018, 115, 7296–7301. [Google Scholar] [CrossRef]
- Frade, R.F.M.; Matias, A.; Branco, L.C.; Afonso, C.A.M.; Duarte, C.M.M. Effect of ionic liquids on human colon carcinoma HT-29 and CaCo-2 cell lines. Green Chem. 2007, 9, 873–877. [Google Scholar] [CrossRef]
- Novo Nordisk AS. Stable Non-Aqueous Pharmaceutical Compositions. U.S. Patent US20100190706, 29 July 2010. [Google Scholar]
- Taverner, A.; Dondi, R.; Almansour, K.; Laurent, F.; Owens, S.E.; Eggleston, I.M.; Fotaki, N.; Mrsny, R.J. Enhanced paracellular transport of insulin can be achieved via transient induction of myosin light chain phosphorylation. J. Control. Release 2015, 210, 189–197. [Google Scholar] [CrossRef]
- Almansour, K.; Taverner, A.; Eggleston, I.M.; Mrsny, R.J. Mechanistic studies of a cell-permeant peptide designed to enhance myosin light chain phosphorylation in polarized intestinal epithelia. J. Control. Release 2018, 279, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Almansour, K.; Taverner, A.; Turner, J.R.; Eggleston, I.M.; Mrsny, R.J. An intestinal paracellular pathway biased toward positively-charged macromolecules. J. Control. Release 2018, 288, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Date, A.A.; Hanes, J.; Ensign, L.M. Nanoparticles for oral delivery: Design, evaluation and state-of-the-art. J. Control. Release 2016, 240, 504–526. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Ryan, K.B.; Ahmad, T.; O’Driscoll, C.M.; Brayden, D.J. Chapter 2.1 Nanostructures Overcoming the Intestinal Barrier: Physiological Considerations and Mechanistic Issues. In Nanostructured Biomaterials for Overcoming Biological Barriers; The Royal Society of Chemistry: London, UK, 2012; pp. 39–62. [Google Scholar] [CrossRef]
- Brayden, S. Oral Peptide Delivery: The Potential of Combining Nanoparticle Constructs with Permeation Enhancers. In Proceedings of the Annual Meeting and Exposition of the Controlled Release Society, New York City, NY, USA, 22–24 July 2018. [Google Scholar]
- Alonso, J.M. Learning from the EU TRANS-INT consortium: Oral peptide formulations using nanotechnologies. In Proceedings of the Annual Meeting and Exposition of the Controlled Release Society, New York City, NY, USA, 22–24 July 2018. [Google Scholar]
- Gonçalves, R.F.S.; Martins, J.T.; Duarte, C.M.M.; Vicente, A.A.; Pinheiro, A.C. Advances in nutraceutical delivery systems: From formulation design for bioavailability enhancement to efficacy and safety evaluation. Trends Food Sci. Technol. 2018, 78, 270–291. [Google Scholar] [CrossRef]
- Thanou, M.; Verhoef, J.C.; Junginger, H.E. Chitosan and its derivatives as intestinal absorption enhancers. Adv. Drug Deliv. Rev. 2001, 50 (Suppl. 1), S91–S101. [Google Scholar] [CrossRef]
- Onishi, H.; Imura, Y.; Uchida, M.; Machida, Y. Enhancement potential of sucrose laurate (L-1695) on intestinal absorption of water-soluble high molecular weight compounds. Curr. Drug Deliv. 2012, 9, 487–494. [Google Scholar] [CrossRef]
- Lucarini, S.; Fagioli, L.; Campana, R.; Cole, H.; Duranti, A.; Baffone, W.; Vllasaliu, D.; Casettari, L. Unsaturated fatty acids lactose esters: Cytotoxicity, permeability enhancement and antimicrobial activity. Eur. J. Pharm. Biopharm. 2016, 107, 88–96. [Google Scholar] [CrossRef]
- Dimitrijevic, D.; Shaw, A.J.; Florence, A.T. Effects of some non-ionic surfactants on transepithelial permeability in Caco-2 cells. J. Pharm. Pharmacol. 2000, 52, 157–162. [Google Scholar] [CrossRef]
- Gleeson, J.P.; Frías, J.M.; Ryan, S.M.; Brayden, D.J. Sodium caprate enables the blood pressure-lowering effect of Ile-Pro-Pro and Leu-Lys-Pro in spontaneously hypertensive rats by indirectly overcoming PepT1 inhibition. Eur. J. Pharm. Biopharm. 2018, 128, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Moghimipour, E.; Ameri, A.; Handali, S. Absorption-Enhancing Effects of Bile Salts. Molecules 2015, 20, 14451–14473. [Google Scholar] [CrossRef] [PubMed]
- Shoba, G.; Joy, D.; Joseph, T.; Majeed, M.; Rajendran, R.; Srinivas, P.S. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998, 64, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.D.; Kwon, S.J.; Ju, J.; Bose, M.; Lee, M.J.; Hong, J.; Hao, X.; Yang, C.S. Effect of genistein on the bioavailability and intestinal cancer chemopreventive activity of (−)-epigallocatechin-3-gallate. Carcinogenesis 2008, 29, 2019–2024. [Google Scholar] [CrossRef] [PubMed]
- McCartney, F.; Rosa, M.; Coulter, I.; Brayden, D.J. A sucrose ester is a novel permeation enhancer using isolated rat colonic mucosae mounted in Ussing chambers. In Proceedings of the Annual Meeting and Exposition of the American Association of Pharmaceutical Scientists (Poster Presentation), San Antonio, TX, USA, 10–14 November 2013. [Google Scholar]
- McCartney, F.; Rosa, M.; Coulter, I.; Brayden, D.J. Sucrose laurate is an effective permeation enhancer for insulin: Rat intestinal instillations. In Proceedings of the Annual meeting and Exposition of the Controlled Release Society (Oral Presentation), Boston, MA, USA, 16–19 July 2017. [Google Scholar]
- Casettari, L. Ex-vivo evaluation of intestinal permeability-enhancing effects of mono-esterified sugar based surfactants. In Proceedings of the 11th World Meeting on Pharmaceutics, Biopharmaceutics, and Pharmaceutical Technology (Oral Presentation), Granada, Spain, 19–22 March 2017. [Google Scholar]
- Maher, S.; Medani, M.; Carballeira, N.N.; Winter, D.C.; Baird, A.W.; Brayden, D.J. Development of a Non-Aqueous Dispersion to Improve Intestinal Epithelial Flux of Poorly Permeable Macromolecules. AAPS J. 2017, 19, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Gradauer, K.; Nishiumi, A.; Unrinin, K.; Higashino, H.; Kataoka, M.; Pedersen, B.L.; Buckley, S.T.; Yamashita, S. Interaction with Mixed Micelles in the Intestine Attenuates the Permeation Enhancing Potential of Alkyl-Maltosides. Mol. Pharm. 2015, 12, 2245–2253. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Jarvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Liang, N.; Kawashima, Y.; Xia, D.; Cui, F. Hydrophobic ion pairing of an insulin-sodium deoxycholate complex for oral delivery of insulin. Int. J. Nanomed. 2011, 6, 3049–3056. [Google Scholar] [CrossRef]
- Griesser, J.; Hetenyi, G.; Moser, M.; Demarne, F.; Jannin, V.; Bernkop-Schnurch, A. Hydrophobic ion pairing: Key to highly payloaded self-emulsifying peptide drug delivery systems. Int. J. Pharm. 2017, 520, 267–274. [Google Scholar] [CrossRef]
- Li, P.; Nielsen, H.M.; Fano, M.; Mullertz, A. Preparation and characterization of insulin-surfactant complexes for loading into lipid-based drug delivery systems. J. Pharm. Sci 2013, 102, 2689–2698. [Google Scholar] [CrossRef]
- Hintzen, F.; Laffleur, F.; Sarti, F.; Müller, C.; Bernkop-Schnürch, A. In vitro and ex vivo evaluation of an intestinal permeation enhancing self-microemulsifying drug delivery system (SMEDDS). J. Drug Deliv. Sci. Technol. 2013, 23, 261–267. [Google Scholar] [CrossRef]
- Meyer, J.D.; Manning, M.C. Hydrophobic Ion Pairing: Altering the Solubility Properties of Biomolecules. Pharm. Res. 1998, 15, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Günday Türeli, N.; Türeli, A.E.; Schneider, M. Counter-ion complexes for enhanced drug loading in nanocarriers: Proof-of-concept and beyond. Int. J. Pharm. 2016, 511, 994–1001. [Google Scholar] [CrossRef] [PubMed]
- Krug, S.M.; Amasheh, M.; Dittmann, I.; Christoffel, I.; Fromm, M.; Amasheh, S. Sodium caprate as an enhancer of macromolecule permeation across tricellular tight junctions of intestinal cells. Biomaterials 2013, 34, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Krug, S.M.; Fromm, M.; Günzel, D. Two-Path Impedance Spectroscopy for Measuring Paracellular and Transcellular Epithelial Resistance. Biophys. J. 2009, 97, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Koeplinger, K.A. Oral peptide-protein interaction inhibitors (PPI): Excited about cycling. In Proceedings of the Annual meeting and Exposition of the Controlled Release Society (Oral Presentation, Oral Peptide Workshop), New York, NY, USA, 22–24 July 2018. [Google Scholar]
- Wang, X.; Maher, S.; Brayden, D.J. Restoration of rat colonic epithelium after in situ intestinal instillation of the absorption promoter, sodium caprate. Ther. Deliv. 2010, 1, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A.; Nataro, J.P. Intestinal epithelial tight junctions as targets for enteric bacteria-derived toxins. Adv. Drug Deliv. Rev. 2004, 56, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, S.; Jannin, V.; Rodier, J.D.; Ritter, N.; Mahler, B.; Carriere, F. Comparative study on digestive lipase activities on the self emulsifying excipient Labrasol, medium chain glycerides and PEG esters. Biochim. Biophys. Acta 2007, 1771, 633–640. [Google Scholar] [CrossRef]
- Sadhukha, T.; Layek, B.; Prabha, S. Incorporation of lipolysis in monolayer permeability studies of lipid-based oral drug delivery systems. Drug Deliv. Transl. Res. 2018, 8, 375–386. [Google Scholar] [CrossRef]
- Shaw, D.J. Introduction to Colloid and Surface Chemistry; Butterworth-Heinemann: Oxford, UK, 1992. [Google Scholar]
- Dahlgren, D.; Roos, C.; Lundqvist, A.; Tannergren, C.; Sjoblom, M.; Sjogren, E.; Lennernas, H. Effect of absorption-modifying excipients, hypotonicity, and enteric neural activity in an in vivo model for small intestinal transport. Int. J. Pharm. 2018, 549, 239–248. [Google Scholar] [CrossRef]
- Anderberg, E.K.; Lindmark, T.; Artursson, P. Sodium Caprate Elicits Dilatations in Human Intestinal Tight Junctions and Enhances Drug Absorption by the Paracellular Route. Pharm. Res. 1993, 10, 857–864. [Google Scholar] [CrossRef]
- Tippin, T.K.; Thakker, D.R. Biorelevant refinement of the Caco-2 cell culture model to assess efficacy of paracellular permeability enhancers. J. Pharm. Sci. 2008, 97, 1977–1992. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.; Karr, N.; Mitragotri, S. Discovery of synergistic permeation enhancers for oral drug delivery. J. Control. Release 2008, 128, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Heade, J.; Maher, S.; Bleiel, S.B.; Brayden, D.J. Labrasol((R)) and Salts of Medium-Chain Fatty Acids Can Be Combined in Low Concentrations to Increase the Permeability of a Macromolecule Marker Across Isolated Rat Intestinal Mucosae. J. Pharm. Sci. 2018, 107, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.-H.; Li, H.; Levons, J.; Lentz, K.; Gandhi, R.B.; Raghavan, K.; Smith, R.L. Predicting Effect of Food on Extent of Drug Absorption Based on Physicochemical Properties. Pharm. Res. 2007, 24, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Stappaerts, J.; Wuyts, B.; Tack, J.; Annaert, P.; Augustijns, P. Human and simulated intestinal fluids as solvent systems to explore food effects on intestinal solubility and permeability. Eur. J. Pharm. Sci. 2014, 63, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Riethorst, D.; Mols, R.; Duchateau, G.; Tack, J.; Brouwers, J.; Augustijns, P. Characterization of Human Duodenal Fluids in Fasted and Fed State Conditions. J. Pharm. Sci. 2016, 105, 673–681. [Google Scholar] [CrossRef]
- Jantratid, E.; Janssen, N.; Reppas, C.; Dressman, J.B. Dissolution media simulating conditions in the proximal human gastrointestinal tract: An update. Pharm. Res. 2008, 25, 1663–1676. [Google Scholar] [CrossRef]
- Markopoulos, C.; Thoenen, F.; Preisig, D.; Symillides, M.; Vertzoni, M.; Parrott, N.; Reppas, C.; Imanidis, G. Biorelevant media for transport experiments in the Caco-2 model to evaluate drug absorption in the fasted and the fed state and their usefulness. Eur. J. Pharm. Biopharm. 2014, 86, 438–448. [Google Scholar] [CrossRef]
- Birch, D.; Diedrichsen, R.G.; Christophersen, P.C.; Mu, H.; Nielsen, H.M. Evaluation of drug permeation under fed state conditions using mucus-covered Caco-2 cell epithelium. Eur. J. Pharm. Sci. 2018, 118, 144–153. [Google Scholar] [CrossRef]
- Wuyts, B.; Riethorst, D.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Evaluation of fasted state human intestinal fluid as apical solvent system in the Caco-2 absorption model and comparison with FaSSIF. Eur. J. Pharm. Sci. 2015, 67, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Roos, C.; Lundqvist, A.; Tannergren, C.; Sjoblom, M.; Sjogren, E.; Lennernas, H. Time-dependent effects on small intestinal transport by absorption-modifying excipients. Eur. J. Pharm. Biopharm. 2018, 132, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Roos, C.; Dahlgren, D.; Sjogren, E.; Tannergren, C.; Abrahamsson, B.; Lennernas, H. Regional Intestinal Permeability in Rats: A Comparison of Methods. Mol. Pharm. 2017, 14, 4252–4261. [Google Scholar] [CrossRef] [PubMed]
- Thanou, M.; Verhoef, J.C.; Verheijden, J.H.M.; Junginger, H.E. Intestinal Absorption of Octreotide Using Trimethyl Chitosan Chloride: Studies in Pigs. Pharm. Res. 2001, 18, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Baluom, M.; Friedman, M.; Assaf, P.; Haj-Yehia, A.I.; Rubinstein, A. Synchronized release of sulpiride and sodium decanoate from HPMC matrices: A rational approach to enhance sulpiride absorption in the rat intestine. Pharm. Res. 2000, 17, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Sutton, S.C.; LeCluyse, E.L.; Engle, K.; Pipkin, J.D.; Fix, J.A. Enhanced Bioavailability of Cefoxitin Using Palmitoylcarnitine. II. Use of Directly Compressed Tablet Formulations in the Rat and Dog. Pharm. Res. 1993, 10, 1516–1520. [Google Scholar] [CrossRef] [PubMed]
- Schiller, C.; Fröhlich, C.-P.; Giessmann, T.; Siegmund, W.; Mönnikes, H.; Hosten, N.; Weitschies, W. Intestinal fluid volumes and transit of dosage forms as assessed by magnetic resonance imaging. Aliment. Pharmacol. Ther. 2005, 22, 971–979. [Google Scholar] [CrossRef]
- Dorkoosh, F.A.; Verhoef, J.C.; Verheijden, J.H.M.; Rafiee-Tehrani, M.; Borchard, G.; Junginger, H.E. Peroral Absorption of Octreotide in Pigs Formulated in Delivery Systems on the Basis of Superporous Hydrogel Polymers. Pharm. Res. 2002, 19, 1532–1536. [Google Scholar] [CrossRef]
- Koziolek, M.; Grimm, M.; Becker, D.; Iordanov, V.; Zou, H.; Shimizu, J.; Wanke, C.; Garbacz, G.; Weitschies, W. Investigation of pH and Temperature Profiles in the GI Tract of Fasted Human Subjects Using the Intellicap® System. J. Pharm. Sci. 2015, 104, 2855–2863. [Google Scholar] [CrossRef]
- Baluom, M.; Friedman, M.; Rubinstein, A. The importance of intestinal residence time of absorption enhancer on drug absorption and implication on formulative considerations. Int. J. Pharm. 1998, 176, 21–30. [Google Scholar] [CrossRef]
- Tillman, L.G.; Geary, R.S.; Hardee, G.E. Oral delivery of antisense oligonucleotides in man. J. Pharm. Sci. 2008, 97, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Junginger, H. Excipients as Absorption Enhancers. In Biopharmaceutics Applications in Drug Development; Krishna, R., Yu, L., Eds.; Springer: New York, NY, USA, 2008; pp. 139–174. [Google Scholar] [CrossRef]
- Brayden, D.J.; Walsh, E. Efficacious intestinal permeation enhancement induced by the sodium salt of 10-undecylenic acid, a medium chain fatty acid derivative. AAPS J. 2014, 16, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.; Shen, Z.; Mitragotri, S. Oral delivery of macromolecules using intestinal patches: Applications for insulin delivery. J. Control. Release 2004, 98, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Hwang, B.H.; Doshi, N.; Banerjee, A.; Anselmo, A.C.; Mitragotri, S. Delivery of Exenatide and Insulin Using Mucoadhesive Intestinal Devices. Ann. Biomed. Eng. 2016, 44, 1993–2007. [Google Scholar] [CrossRef] [PubMed]
- Eiamtrakarn, S.; Itoh, Y.; Kishimoto, J.; Yoshikawa, Y.; Shibata, N.; Murakami, M.; Takada, K. Gastrointestinal mucoadhesive patch system (GI-MAPS) for oral administration of G-CSF, a model protein. Biomaterials 2002, 23, 145–152. [Google Scholar] [CrossRef]
- Grabovac, V.; Foger, F.; Bernkop-Schnurch, A. Design and in vivo evaluation of a patch delivery system for insulin based on thiolated polymers. Int. J. Pharm. 2008, 348, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, N.; Uchino, K.; Amagase, K.; Ito, Y.; Shibata, N.; Takada, K. Gastro-intestinal patch system for the delivery of erythropoietin. J. Control. Release 2006, 111, 19–26. [Google Scholar] [CrossRef]
- Banerjee, A.; Wong, J.; Gogoi, R.; Brown, T.; Mitragotri, S. Intestinal micropatches for oral insulin delivery. J. Drug Target. 2017, 25, 608–615. [Google Scholar] [CrossRef]
- McCartney, F.; Gleeson, J.P.; Brayden, D.J. Safety concerns over the use of intestinal permeation enhancers: A mini-review. Tissue Barriers 2016, 4, e1176822. [Google Scholar] [CrossRef]
- Fasano, A.; Fiorentini, C.; Donelli, G.; Uzzau, S.; Kaper, J.B.; Margaretten, K.; Ding, X.; Guandalini, S.; Comstock, L.; Goldblum, S.E. Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. J. Clin. Investig. 1995, 96, 710–720. [Google Scholar] [CrossRef]
- Bogman, K.; Zysset, Y.; Degen, L.; Hopfgartner, G.; Gutmann, H.; Alsenz, J.; Drewe, J. P-glycoprotein and surfactants: Effect on intestinal talinolol absorption. Clin. Pharmacol. Ther. 2005, 77, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, A.; Takeda-Morishita, M.; Maeda, K.; Banba, H.; Takayama, K.; Kumagai, Y.; Kusuhara, H.; Sugiyama, Y. Effects of Cremophor EL on the absorption of orally administered saquinavir and fexofenadine in healthy subjects. Drug Metab. Pharmacokinet. 2015, 30, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Lindmark, T.; Soderholm, J.D.; Olaison, G.; Alvan, G.; Ocklind, G.; Artursson, P. Mechanism of absorption enhancement in humans after rectal administration of ampicillin in suppositories containing sodium caprate. Pharm. Res. 1997, 14, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Leonard, T.W.; Jacobsen, J.; Brayden, D.J. Safety and efficacy of sodium caprate in promoting oral drug absorption: From in vitro to the clinic. Adv. Drug Deliv. Rev. 2009, 61, 1427–1449. [Google Scholar] [CrossRef]
- Kiss, L.; Walter, F.R.; Bocsik, A.; Veszelka, S.; Ozsvari, B.; Puskas, L.G.; Szabo-Revesz, P.; Deli, M.A. Kinetic analysis of the toxicity of pharmaceutical excipients Cremophor EL and RH40 on endothelial and epithelial cells. J. Pharm. Sci. 2013, 102, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Ribet, D.; Cossart, P. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect. 2015, 17, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Gunzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed]
- Drewe, J.; Beglinger, C.; Fricker, G. Effect of ischemia on intestinal permeability of lipopolysaccharides. Eur. J. Clin. Investig. 2001, 31, 138–144. [Google Scholar] [CrossRef]
- Tomlinson, J.E.; Blikslager, A.T. Interactions between lipopolysaccharide and the intestinal epithelium. J. Am. Vet. Med Assoc. 2004, 224, 1446–1452. [Google Scholar] [CrossRef]
- Barmeyer, C.; Schulzke, J.D.; Fromm, M. Claudin-related intestinal diseases. Semin. Cell Dev. Boil. 2015, 42, 30–38. [Google Scholar] [CrossRef]
- Tippin, T.K.; Thakker, D.R. Novel Approaches to Assess the Efficacy and Toxicity of Intestinal Absorption Enhancers. Ph.D. Thesis, ProQuest, Ann Arbor, MI, USA, 2006. [Google Scholar]
- Nielsen, E.J.; Kamei, N.; Takeda-Morishita, M. Safety of the cell-penetrating peptide penetratin as an oral absorption enhancer. Boil. Pharm. Bull. 2015, 38, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, A.C.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015, 519, 92–96. [Google Scholar] [CrossRef]
- Kararli, T.T. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm. Drug Dispos. 1995, 16, 351–380. [Google Scholar] [CrossRef] [PubMed]
- Dressman, J.B. Comparison of canine and human gastrointestinal physiology. Pharm. Res. 1986, 3, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, P.; Pechenov, S.; Anand Subramony, J. Oral peptide delivery: Translational challenges due to physiological effects. J. Control. Release 2018, 287, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Lui, C.Y.; Amidon, G.L.; Berardi, R.R.; Fleisher, D.; Youngberg, C.; Dressman, J.B. Comparison of gastrointestinal pH in dogs and humans: Implications on the use of the beagle dog as a model for oral absorption in humans. J. Pharm. Sci. 1986, 75, 271–274. [Google Scholar] [CrossRef]
- Polentarutti, B.; Albery, T.; Dressman, J.; Abrahamsson, B. Modification of gastric pH in the fasted dog. J. Pharm. Pharmacol. 2010, 62, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Henze, L.J.; Koehl, N.J.; O’Shea, J.P.; Kostewicz, E.S.; Holm, R.; Griffin, B.T. The pig as a preclinical model for predicting oral bioavailability and in vivo performance of pharmaceutical oral dosage forms: A PEARRL review. J. Pharm. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Sjogren, E.; Abrahamsson, B.; Augustijns, P.; Becker, D.; Bolger, M.B.; Brewster, M.; Brouwers, J.; Flanagan, T.; Harwood, M.; Heinen, C.; et al. In vivo methods for drug absorption—Comparative physiologies, model selection, correlations with in vitro methods (IVIVC), and applications for formulation/API/excipient characterization including food effects. Eur. J. Pharm. Sci. 2014, 57, 99–151. [Google Scholar] [CrossRef]
- Hatton, G.B.; Madla, C.M.; Rabbie, S.C.; Basit, A.W. Gut reaction: Impact of systemic diseases on gastrointestinal physiology and drug absorption. Drug Discov. Today 2018. [Google Scholar] [CrossRef]
- Davis, S.S.; Wilding, I.R. Oral drug absorption studies: The best model for man is man! Drug Discov. Today 2001, 6, 127–130. [Google Scholar] [CrossRef]
- Hatton, G.B.; Yadav, V.; Basit, A.W.; Merchant, H.A. Animal Farm: Considerations in Animal Gastrointestinal Physiology and Relevance to Drug Delivery in Humans. J. Pharm. Sci. 2015, 104, 2747–2776. [Google Scholar] [CrossRef] [PubMed]
- McConnell, E.L.; Basit, A.W.; Murdan, S. Measurements of rat and mouse gastrointestinal pH, fluid and lymphoid tissue, and implications for in-vivo experiments. J. Pharm. Pharmacol. 2008, 60, 63–70. [Google Scholar] [CrossRef]
- Bak, A.; Ashford, M.; Brayden, D.J. Local delivery of macromolecules to treat diseases associated with the colon. Adv. Drug Deliv. Rev. 2018, 136–137, 2–27. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Henriksen, K.; Bay-Jensen, A.C.; Molloy, B.; Arnold, M.; John, M.R.; Byrjalsen, I.; Azria, M.; Riis, B.J.; Qvist, P.; et al. Lessons learned from the development of oral calcitonin: The first tablet formulation of a protein in phase III clinical trials. J. Clin. Pharmacol. 2011, 51, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Riis, B.J.; Mehta, N.; Stern, W.; Arbit, E.; Christiansen, C.; Henriksen, K. Lessons learned from the clinical development of oral peptides. Br. J. Clin. Pharmacol. 2015, 79, 720–732. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Active | Mw | Dose | Frequency | Route | t½ | LogP † | BCS | Oral BA |
|---|---|---|---|---|---|---|---|---|
| Desmopressin | 1069 Da | 1–4 mcg | Daily | sc | ~2.8 h | −4 | III | 0.17% |
| Octreotide | 1019 Da | 200 mcg | Thrice daily | sc | ~1.7 h | −1.4 | — | Phase 3 |
| Cyclosporin | 1203 Da | 280 mg | Daily | iv inf. | ~8.4 h | 7.5 | II | 27% |
| Vancomycin | 1449 Da | 1500 mg | Twice daily | iv inf. | ~7.2 h | −2.6 | III | Local |
| Salmon calcitonin | 3432 Da | 16.7 mcg | Daily | sc | ~1.3 h | −16.6 | — | Phase 3 |
| Semaglutide | 4114 Da | 500 mcg * | Weekly | sc | ~168 h | −5.8 | — | Phase 3 |
| Exenatide | 4186 Da | 10 mcg | Daily | sc | ~2.4 h | −21 | — | Phase 1 |
| Insulin degludec | 6108 Da | 350 mcg | Daily | sc | ~25 h | −4.9 | — | — |
| Insulin aspart | 5832 Da | 1.8 mg ** | — | sc | ~1.4 h | — | — | — |
| Formulation Additives | Disintegrant (% w/w) | Tableting Pressure (psi) | Disintegration Time (min) | Break Strength (N) |
|---|---|---|---|---|
| Labrasol and Neusilin® US2 (1:1) | 0 | 1000 | >60 | 29.1 ± 2.9 |
| Labrasol and Neusilin® US2 (1:1) | 0 | 2000 | >60 | 68.8 ± 3.2 |
| Labrasol and Neusilin® US2 (1:1) | 5 | 1000 | 5.5 ± 0.2 | 49.8 ± 6.2 |
| Labrasol and Neusilin® US2 (1:1) | 5 | 2000 | 4.9 ± 0.3 | 72.4 ± 2.7 |
| Anatomical/Physiological Property | Species | Influence on PE Action |
|---|---|---|
| Gastric emptying time (h) | Human: 1 h [130] Rat: 0.7–2.1 h [130] Dog: 3.9–5.3 h [130] Pig: 1.5–6 h [130] | For immediate release dosage forms, slower gastric emptying in pig and dog than in humans may increase gastric residence time of PE and payload, thus overestimating enhancement. For enteric dosage forms, slower gastric emptying, may delay dissolution in the GI tract and ultimately increase Tmax in these species versus humans. |
| Gastric fluid volume (mL) | Human: 118 mL [137] Rat: 2.29 [137] Dog: 500–1000 mL [137] Pig: 278 mL [137] | For immediate release dosage forms, the larger volume in dogs may result in greater dilution of PE to below a threshold for enhancement action, thereby underestimating enhancement. |
| Stomach pH | Human: 1.7 [128] Rat: 3.9 [138] Dog: 1.5 [128] Pig: 1.7 [128] | As many PEs that have progressed to clinical testing in oral formulations are weak acid surfactants, differences in solubility can be observed if there is variation in gastric pH. This gives rise to differences in enhancement as acidic surfactants are more effective in their ionizable form at high pH. |
| Small intestine transit time (Fasted state) (time (h) and length (m)) | Human: 3–4 h [139] Human: 6.25 m [137] Rat: 4–5 h [128] Rat: 0.34 m [137] Dog: 1.5 h [137] Dog: 2.48 m [137] Pig: 3–4 h [137] Pig: 14.2 m [137] | Faster transit may reduce the exposure of PE and payload at the epithelium, thereby reducing enhancement, and potentially underestimating the effects of the PE. A short transit time does not strictly mean faster movement, as length of the small intestine is different in different species. |
| Small intestine fluid volume (total and g/cm) | Human: 212 mL [137] Human: 0.6 g/cm [130] Rat: 3.9 mL [137] Rat: 0.06 g/cm [130] Dog: 300 mL [137] Dog: 0.9 [130] Pig: 476 mL [137] Pig: 0.62 [130] | Differences in fluid volume, or more specifically the volume and number of intestinal fluid pockets in the small intestine could lead to differences in the regional concentration of PE and payload, as well as differences in dissolution rate. This could lead to under- or overestimation of enhancement. |
| Duodenal mucus thickness (µm) | Human: 15.5 µm [137] Rat: 30.6 µm [137] Dog: — Pig: 25.6 µm [137] | Difference in the thickness of the protective mucus gel layer overlying the epithelium has potential to modulate enhancement. |
| Small intestine diameter | Human: 5 cm [137] Rat: 2.5–3 mm [137] Dog: — Pig: — | The diameter of the intestinal lumen may impact the proximity of enteric formulations to the epithelium and ultimately impact co-localization of PE and payload. |
| Plasma membrane phospholipid composition of intestinal epithelium | Human: — Rat: — Dog: — Pig: — | There are differences in phospholipid composition in different species [128], which may impact sensitivity to perturbation by surfactant PEs |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maher, S.; Brayden, D.J.; Casettari, L.; Illum, L. Application of Permeation Enhancers in Oral Delivery of Macromolecules: An Update. Pharmaceutics 2019, 11, 41. https://doi.org/10.3390/pharmaceutics11010041
Maher S, Brayden DJ, Casettari L, Illum L. Application of Permeation Enhancers in Oral Delivery of Macromolecules: An Update. Pharmaceutics. 2019; 11(1):41. https://doi.org/10.3390/pharmaceutics11010041
Chicago/Turabian StyleMaher, Sam, David J. Brayden, Luca Casettari, and Lisbeth Illum. 2019. "Application of Permeation Enhancers in Oral Delivery of Macromolecules: An Update" Pharmaceutics 11, no. 1: 41. https://doi.org/10.3390/pharmaceutics11010041
APA StyleMaher, S., Brayden, D. J., Casettari, L., & Illum, L. (2019). Application of Permeation Enhancers in Oral Delivery of Macromolecules: An Update. Pharmaceutics, 11(1), 41. https://doi.org/10.3390/pharmaceutics11010041