1. Introduction
Vitamin K1 (VK1) is a type of oily drug that is given to treat or prevent excessive bleeding in people with insufficient VK1 or who have taken excessive coumarin anticoagulant medication [
1]. VK1 is mainly administered intramuscularly or intravenously; these routes have rapid effects, but they occasionally result in allergic reactions, anaphylactic shock, or hemolysis [
2]. To overcome these adverse reactions, a new formulation of VK1 which was called Konakion
® MM was launched in 1991; Konakion
® MM is based on mixed micelles of lecithin and glycocholic acid [
3]. Compared with classical products containing synthetic solubilizers such as Cremophor EL, tween, and Pluronics, Konakion
® MM is a safe and effective treatment with excellent biocompatibility for both oral and intravenous administration. However, for better treatment compliance and convenience of using, a solid formulation would be useful.
The low aqueous solubility of poorly water-soluble drugs often leads to poor oral bioavailability because of their slow and limited release of drug in gastrointestinal fluid [
4]. Different strategies can be used to address this problem, including the use of solid dispersions, micronization, complexation with cyclodextrins, and nanoparticles. Recently, lipid-based formulations, especially self-emulsifying drug delivery systems (SEDDS), have been widely accepted as a promising approach for delivering poorly water-soluble drugs. A SEDDS is an isotropic pre-concentrate consisting of oil, surfactant, cosurfactant, and drug. The SEDDS spontaneously forms thermodynamically stable oil-in-water emulsions by gentle agitation in the presence of an aqueous phase. When the droplet size of the emulsion ranges between 20 and 200 nm, the SEDDS is a self-nanoemulsifying drug delivery system (SNEDDS) [
5]. Poorly water-soluble drugs are dissolved in small oil droplets such that the dissolution process is no longer the rate-limiting step of in vivo absorption [
6]. Moreover, a SNEDDS enhances the oral bioavailability of hydrophobic drugs by avoiding the hepatic first-pass effect, reducing metabolism by the cytochrome-P450 family of enzymes, inhibiting P-glycoprotein (P-gp) efflux, and promoting drug transport via lymph circulation [
7]. As a liquid formulation, however, inherent defects, such as, migration of the components, potential drug leakage, low stability during manufacturing, have limited its practical industrial application [
8]. To overcome these difficulties, solid SNEDDS (S-SNEDDS) formulations have been investigated as an alternative approach.
S-SNEDDS formulations, which can be prepared by spray drying, spray congealing, liquisolid technique, melt granulation, freeze drying technique and extrusion/spheronization, transform lipid formulations into various solid counterparts with the added advantage of convenient process control [
9]. Among these methods, liquisolid technique is a convenient and economical way to transform liquid to solid powders without influencing the self-emulsifying properties [
10]. Liquisolid systems refer to formulations formed by conversion of liquid drugs, drug suspensions or drug solution in non-volatile solvents into dry, nonadherent, free-flowing, compressible and compactible powder mixtures by blending the suspension or solution with selected carriers and coating materials [
11]. Liquisolid systems have been reported to enhance drug release and improve bioavailability owing to increased surface area of the drug, increased aqueous solubility, and improved wettability of drug particles [
12,
13]. There is little reported literature about the application of the solid self-emulsifying technology in the tablet preparation of cyclosporine A [
9], nimodipine [
14], CoQ10 [
15], and valsartan [
16].
In the present study, to enhance solubility and bioavailability of VK1, the self-nanoemulsifying liquisolid (SNE-L) tablets were designed and prepared. The formulation of a liquid SNEDDS (L-SNEDDS) was optimized using a central composite design (CCD) and response surface methodology. The optimal liquid formulation was adsorbed onto suitable carriers and then converted into liquisolid powder. Finally, SNE-L tablets were prepared by direct compression. Uniform nano-emulsion suspension rapidly formed while the SNE-L tablets disintegrated in dissolution media. The dissolution rate in vitro and relative bioavailability in vivo were significantly enhanced by combined application of SNEDDS and liquisolid technology, which can be a useful approach for providing patient-approved dosage form of oily drug VK1.
2. Materials and Methods
2.1. Materials and Animals
VK1 was purchased from Anhui Wanhe Pharmaceutical Co., Ltd. (Anhui, China). Soy lecithin was purchased from Lipoid GmbH (Ludwigshafen, Germany). Glycocholic acid was purchased from Ark Pharm (Libertyville, IL, USA). Transcutol HP was a kind gift from Gattefosse SAS (Saint Priest Cedex, France). Polyethylene glycol 400 (PEG 400) and propylene glycol were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Microcrystalline cellulose (Vivapur® PH-102) was purchased from JRS Pharma (New York, NY, USA). Lactose (Cellatose® 80) was purchased from Molkerei Meggle Wasserburg GmbH & Co., KG (Wasserburg, Germany). Anhydrous dibasic calcium phosphate (Fujicalin®) and magnesium aluminometasilicate (Neusilin® US2) were generous gifts from Fuji Chemical Industries Co., Ltd. (Tokyo, Japan). Colloidal silicon dioxide (CAB-O-SIL® M-5P) was purchased from Cabot Rheinfelden GmbH (Rheinfelden, Germany). Granulated silicon dioxide (Aeroperl® 300) was obtained as a gift sample from Evonik (Shanghai, China). Sodium carboxymethyl starch, crosslinked sodium carboxymethyl cellulose, crosslinked polyvinylpyrrolidone, and magnesium stearate were purchased from Beijing Fengli Jingqiu Commerce and Trade Co., Ltd. (Beijing, China). High-performance liquid chromatography (HPLC)-grade methanol and acetonitrile were purchased from Sigma Aldrich Trading Co., Ltd. (Shanghai, China). All other reagents were analytical grade.
Beagle dogs were purchased from the Laboratory Animals Center of Beijing Institute of Pharmacology and Toxicology (Beijing, China). All animals were handled according to the code of ethics in research, training, and testing of drugs established by the Animal Care and Use Ethics Committee of the Beijing Institute of Pharmacology and Toxicology (Certificate number: 2017041).
2.2. Determination of Drug Concentration
The drug concentration was determined by HPLC analysis with a Waters Alliance HPLC system (UV detector 2487, Separations module 2695, Waters, Milford, MA, USA). Chromatographic separation was accomplished using a reversed phase C18 analytical column (Capcell core C18, 2.1 × 100 mm, 2.7 μm, Shiseido Co., Ltd., Tokyo, Japan). The concentration of VK1 was measured by absorbance at 254 nm with a column heater set to 25 °C. The mobile phase consisted of a mixture of methanol, acetonitrile, and water in the ratio 88:10:2. The flow rate of the mobile phase was 0.3 mL/min, and a 10-μL sample was injected.
2.3. Preparation and Characterization of L-SNEDDS
2.3.1. Formulation Screening of L-SNEDDS
Ternary phase diagrams were used for the formulation screening of L-SNEDDS which obtained the optimal oil phase, surfactant, and cosurfactant for self-emulsification. In this study, water-insoluble VK1 was used as the oil phase. Soybean lecithin and glycocholic acid were used together as surfactants, and Transcutol HP, polyethylene glycol 400 (PEG 400), and propylene glycol were selected as candidates for cosurfactants. The oil phase, surfactant, and cosurfactant were set as three vertices of the ternary phase diagram. All materials were weighed and mixed in accordance with specific points representing different component proportions in the ternary phase diagram. Each formulation was magnetically stirred in a water bath at 60 °C until a yellow transparent liquid self-emulsifying drug solution formed. Then, 1 mL of the L-SNEDDS was added to 100 mL distilled water (37 °C) under constant magnetic stirring to evaluate the self-emulsifying peformance. The self-emulsifying results and the appearance of the generated emulsion (clear, light blue or opalescent) were recorded and plotted in the phase diagrams. Then, the effective self-emulsifying region was plotted to select the type of cosurfactant and the optimal ratio of the two surfactants.
2.3.2. Formulation Optimization of L-SNEDDS
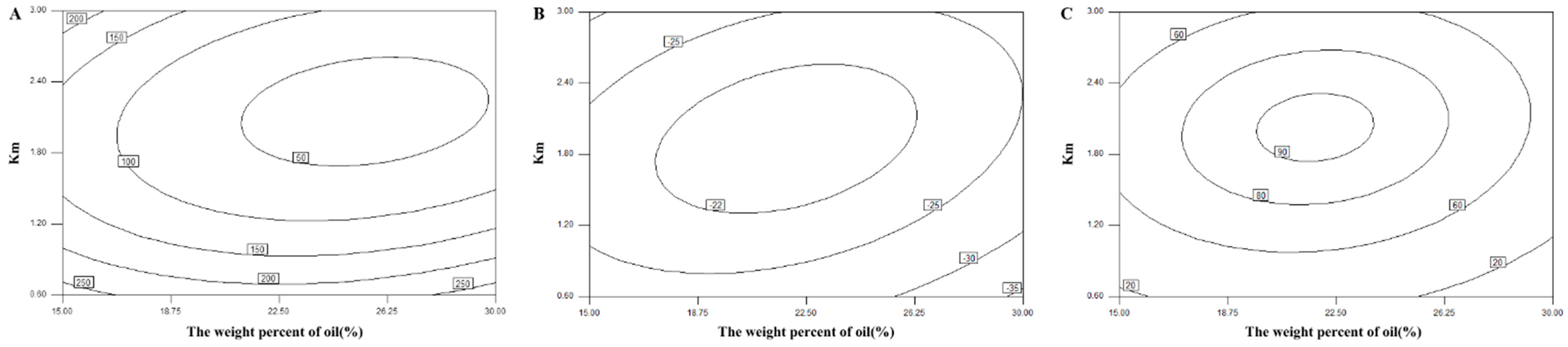
The central composite design and response surface methodology were adopted to optimize the formulation of the L-SNEDDS. Based on the results of ternary phase diagram, the weight percent of oil (X
1, ranging from 15% to 30%) and the weight ratio of surfactant to cosurfactant (K
m; X
2, ranging from 0.6 to 3) were set as two factors. Thus, a central composite design with two-factor and five-level (−a, −1, 0, 1, a) (
Table 1) was selected. The droplet size (DS), zeta potential, and drug dissolution efficiency after 20 min (DE
20min) were selected as three responses.
2.3.3. Characterization of L-SNEDDS
• Dispersibility Test
The dispersibility test was performed to observe the phase clarity of the emulsions and self-emulsification time. One milliliter of the optimized L-SNEDDS was added to 100 mL distilled water (37 °C) with gentle agitation to assess emulsion formation and the appearance of the formed nanoemulsion.
• Robustness to Dilution
Different dilution ratios and diluents might significantly affect the physical stability of the spontaneously emulsifying systems. Accordingly, nanoemulsions diluted to different proportions (10-, 100-, and 1000-fold) with different diluents (purified water, 0.1 N HCl, phosphate buffer pH 6.8, and 0.5% SDS aqueous solution), and then stored for 24 h at room temperature for physical stability study.
• Droplet Size, Zeta Potential and Medium Viscosity
The optimized L-SNEDDS was diluted with distilled water (1:100) under constant magnetic stirring. The droplet size (DS), polydispersibility index (PDI), and zeta potential of the droplets were then determined by dynamic light scattering using a Malvern Zetasizer (Nano ZS-90, Malvern instruments Ltd., Malvern, UK). The medium viscosity of the solutions were achieved with a viscometer (Fungilab, VISCOLEAD, Co., Ltd., Barcelona, Spain). All values recorded were the average of three measurements.
• In Vitro Dissolution Studies
The in vitro dissolution profile of L-SNEDDS was assessed using the USP paddle method with a USP dissolution apparatus (SR8PLUS dissolution tester, Hanson Research Corporation, Chatsworth, CA, USA) in 500 mL water at 100 rpm and 37 ± 0.5 °C. Hard capsules were each filled with an L-SNEDDS containing 10 mg VK1 and placed in dissolution media. Aliquot samples of 5 mL were withdrawn at certain time intervals (5, 10, 20, 30 min) and filtered using a 0.45 μm glass microfiber filter. The filtered samples were diluted with ethanol and the drug concentration was measured spectrophotometrically using a Shimadzu-1750 UV-visible spectrophotometer (Shimadzu, Japan) and the absorbance had a λmax of 273 nm.
• Transmission Electron Microscopy (TEM)
TEM was used to evaluate the morphology of the L-SNEDDS emulsion droplets. The optimized L-SNEDDS was diluted using distilled water (1:50) with constant magnetic stirring. A drop of nanoemulsions was placed on a copper grid coated with carbon film and then stained with 3% (w/v) phosphotungstic acid solution for 2 min, and then observed under a transmission electron microscope (Hitachi H-7650, Tokyo, Japan) while the grid was air-dried at room temperature.
2.4. Preparation and Characterization of S-SNEDDS
2.4.1. Screening of the Adsorbing Agents
The liquisolid technology was used to solidify the L-SNEDDS to S-SNEDDS. Different porous adsorbents including Vivapur® PH102, Cellatose® 80, Fujicalin®, Neusilin® US2, Cab-O-Sil® M5P, and Aeroperl® 300 were used to absorb the L-SNEDDS.
Firstly, Scanning electron microscopy (SEM) was utilized in order to assess the morphological characteristics of the adsorbents using a Hitachi (S4800, Tokyo, Japan) scanning electron microscope. The samples were coated with a thin gold layer and examined in the microscope using an accelerating voltage of 15 kV.
Secondly, a compatibility test was performed to investigate the influence of solid adsorbents on the stability of the drug. Accurately weighed samples of the L-SNEDDS (100 mg) and each of selected adsorbents (200 mg) were mixed by co-grinding them manually in an agate mortar to obtain non-sticky solid powder. Then, the liquid-solid mixture was placed in an oven at 60 °C/92% RH protected from light for 10 days. The total impurity contents of the samples were analyzed by HPLC after 1, 5, and 10 days to evaluate the compatibility of the drug and adsorbents.
Finially, the liquid adsorption capacity of each type of adsorbent was evaluated. However, preparation of a liquisolid with desired flow rate and compressibility was possible when the amount of the carrier was appropriate. The maximum amount of liquid was termed as liquid load factor (
Lf). The value of
Lf was calculated as follows:
where Φ
ca and Φ
co were carrier flowable liquid-retention potential (Φ-value) and coating Φ-value, respectively. R represented the ratio between the carrier materials and the coating materials. In order to get the optimal flowable liquid-retention potential for adsorbents, an experiment was designed to measure angle of slide. Use 10 g adsorbent to adsorb L-SNEDDS, and put the admixture on a metal plate which was polished. Thentilte the plategradually, the angle of the plate was the angle of slide (θ) when the admixture slid. The Φ-value was calculated based on the following equation. The Φ-value at 33° was reported to represent the optimal Φ-value of the adsorbent in L-SNEDDS [
17].
2.4.2. Preparation of S-SNEDDS
Then the liquisolid powder was prepared using L-SNEDDS as liquid medication and selected adsorbent as both the carrier and coating material. Determine the amount of L-SNEDDS(W) required based on the drug concentration in L-SNEDDS to ensure that each SNE-L tablet contains 10 mg VK1. According to the Equations (3) and (4) [
18], the amounts of the carrier (Q) and the coating (q) can be obtained. For all liquid solid systems, the carrier: coating ratio (R) was chosen to be 20.
Then was incorporated the calculated quantitiesad sorbent with the appropriate amount of L-SNEDDS using the way of Spireas et al. [
19]. The prepared liquisolid powder was yellow uniform granules with good flowability.
2.4.3. Characterization of Angle of Repose
The angle of repose was measured by the fixed height funnel method. Powder with a lower angle of repose showed better flow properties due to decreased adhesion. The height (
h) and radius (
r) of the powder cone were recorded after the mixture passed through the funnel, and the angle of repose (𝜃) was calculated using the following equation:
• Carr’s Index
The flowability of all liquisolid powders was assessed based on Carr’s compressibility index (
CI), which is derived from the tapped density (
Pt) and poured bulk density (
Pb) of the liquisolid powders. The prepared liquisolid powders were weighed and poured into a graduated cylinder and the Pb and Pt can be obtained from poured bulk volume (
Vb) and the tapped volume (
Vt). Then the
CI % was calculated as follows [
20]:
• Hausner’s Ratio
The Hausner ratio was calculated as the quotient of tapped density (
Pt) and bulk density (
Pb).
2.5. Preparation and Characterization of Self-Nanoemulsifying Liquisolid (SNE-L) Tablets
2.5.1. Preparation of SNE-L Tablets
The liquisolid powder was blended with fillers, disintegrants and lubricants and then compressed into a tablet in a single-punch tablet press (Shanghai Tianhe Pharmaceutical Device Co., Ltd., Shanghai, China) using a 10-mm diameter punch. Conventional VK1 tablets were prepared by the same procedure of SNE-L tablets except VK1 replaced the L-SNEDDS. Each tablet weighed 400 mg and contained 10 mg VK1.
2.5.2. Characterization of SNE-L Tablets
• In vitro Dissolution Studies
The in vitro dissolution profile of SNE-L tablets was assessed as described in the section of Liquefaction Time. The distilled water, pH 1.0, hydrochloric acid solution, pH 6.8 phosphate buffer solutions and 0.5% sodium dodecyl sulfate (SDS) solution were used as dissolution media separately.
• Liquefaction Time
One tablet of the optimum formulation was wrapped in a transparent polyethylene film and tied to the bulb of a thermometer with thread. The thermometer with attached tablet was placed in a dissolution vessel containing 500 mL dissolution media and maintained at 37 ± 0.5 °C. The tablet was observed carefully, and the melt time was recorded. An average of 10 measurements were performed for each aqueous medium.
• Physical Characterization Tests
SNE-L tablets were evaluated for hardness and friability. Hardness was measured using a manual hardness tester (Eyweka). The friability (Fr) was determined (
n = 3) using a friabilator (TA, Erweka, Heusenstamm, Germany). Twenty accurately weighed tablets (
M1) were placed in the drum, and the sample was subjected to falling shocks for 4 min at a rotational speed of 25 rpm. Then, the weight of the tablets after the test (
M2) was accurately measured and used to calculate the friability by the following equation:
• Reconstitution Study
A SNE-L tablet (400 mg) was dispersed in a volumetric flask containing 25 mL of distilled water by shaking gently. The resulting suspension was incubated for 30 min at room temperature to completely precipitate excipients, and the suspension was then filtered through a 0.45-μm filter. The DS and PDI of the emulsion were then determined.
2.6. In Vivo Pharmacokinetic Study
2.6.1. Drug Administration
A single-dose, randomized, two-period crossover design for pharmacokinetic study with a washout period of a week was carried out. Six healthy beagle dogs (9.9 ± 0.8 kg) were randomly divided into two groups (SNE-L tablets and conventional VK1 tablets). The dogs were fasted but had free access to water overnight. Each dog received either a single 10 mg SNE-L tablet or a single 10 mg conventional tablet. Dogs were giver free access to water and food 6 h after drug administration.
2.6.2. Blood Sampling
Serial blood samples (2 mL) were collected in heparinized tubes using an indwelling cannula before dosing and at 0.25, 0.5, 0.75, 1, 2, 3, 4, 5, 6, 8, 10, 12, and 24 h after dosing. Blood samples were cooled in an ice bath and centrifuged at 3000 rpm for at least 10 min at 4 °C. The harvested plasma was stored at −70 °C until analysis. The isolation process was performed with protection from light.
2.6.3. Analytical Methods
The VK1 concentration in dog plasma was quantified using a 1260 Infinity high-performance liquid chromatographic (HPLC) and G6460A triple-quad mass spectrometer (Agilent Technologies, Santa Clara, CA, USA). Fenofibrate was used as the internal standard. The HPLC-MS/MS analysis conditions were as follows: Agilent Zorbax Eclipse XDB C8 analytical column (2.1 × 100 mm, 3.5 μm, Agilent Technologies, Santa Clara, CA, USA); mobile phase, methanol containing 5 mM ammonium formate; flow rate, 0.3 mL/min; multiple-reaction monitoring (MRM) in negative ionization mode; transitions m/z, 451.3 → 187.0 for VK1 and 361.1 → 233.0 for fenofibrate; fragment voltage, 140 V and 135 V for VK1 and fenofibrate, respectively; capillary voltage, 4000 kV; gas temperature, 300 °C; gas flow, 11 L/min; nebulizer pressure, 25 psi.
2.6.4. Pharmacokinetic and Statistical Analysis
The pharmacokinetic analysis of both VK1 tablets was performed using a non-compartmental model. Cmax (the highest observed plasma concentration) and Tmax (the time required to reach Cmax) were observed from the individual plasma concentration-time curves. The area under the curve (AUC) was calculated using the linear trapezoidal rule. The half-life (t1/2) was calculated from the slope of the logarithm of concentration vs. time profile. The relative availability (Frel) was calculated as (AUCT/AUCR) × 100%.
A t-test was employed to compare the pharmacokinetic parameters between two formulatios. ANOVA was performed on un-transformed and log-transformed data for Cmax and AUC. All testings were performed by SPSS 16.0 (SPSS Inc., Somers, New York, NY, USA).

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}