Simultaneous Determination of Procainamide and N-acetylprocainamide in Rat Plasma by Ultra-High-Pressure Liquid Chromatography Coupled with a Diode Array Detector and Its Application to a Pharmacokinetic Study in Rats



Abstract
1. Introduction
2. Materials and Methods
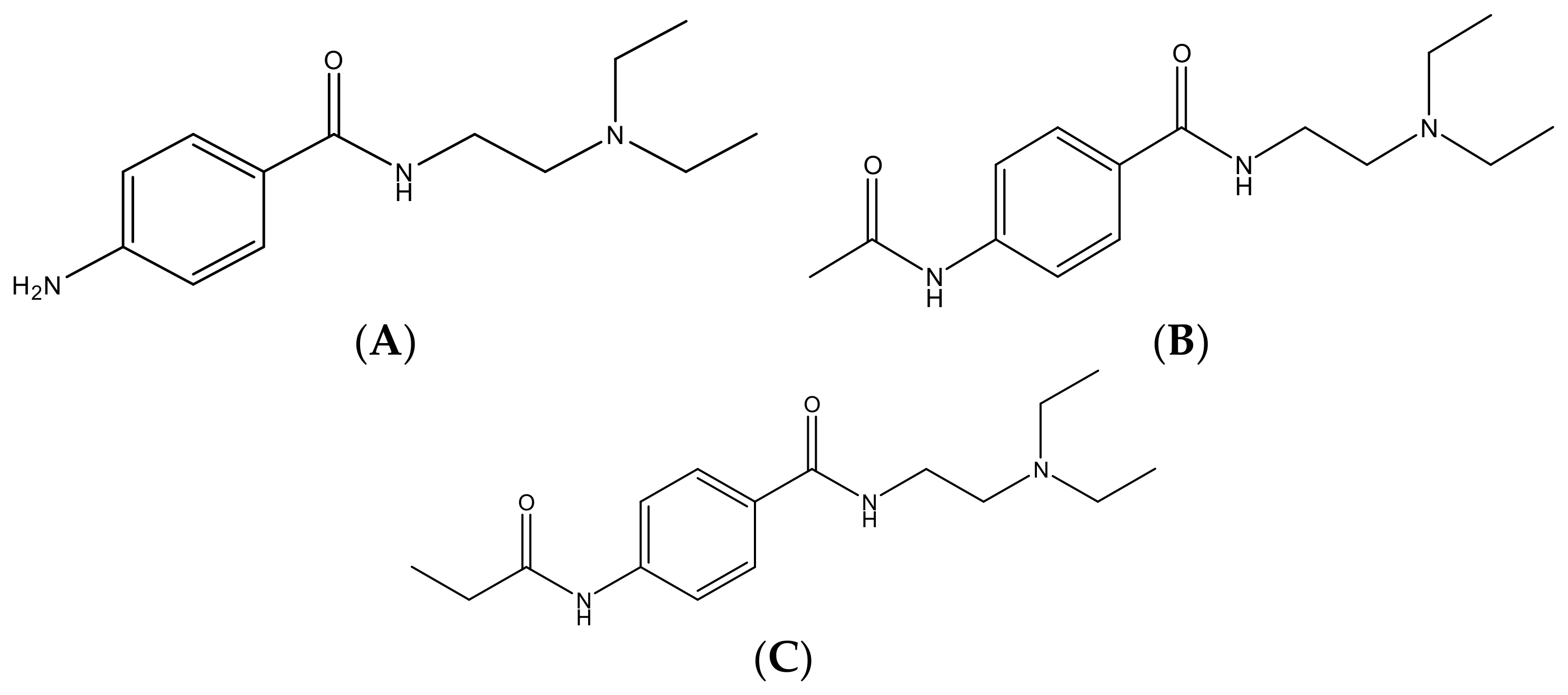
2.1. Chemicals and Reagents
2.2. UHPLC System and Chromatographic Condition
2.3. Preparation of Calibration Standards and Quality Control (QC) Samples
2.4. Sample Preparation
2.5. Method Validation
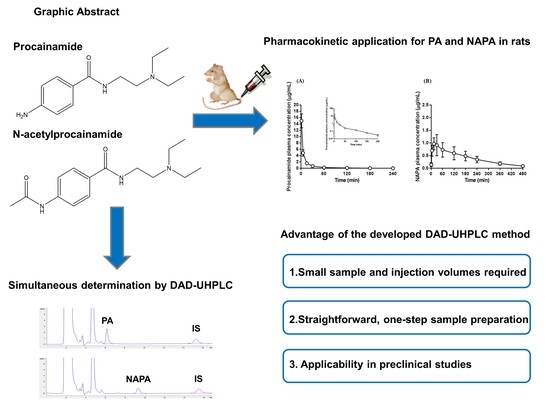
2.6. Application to Pharmacokinetic Studies of Procainamide HCl
3. Results and Discussion
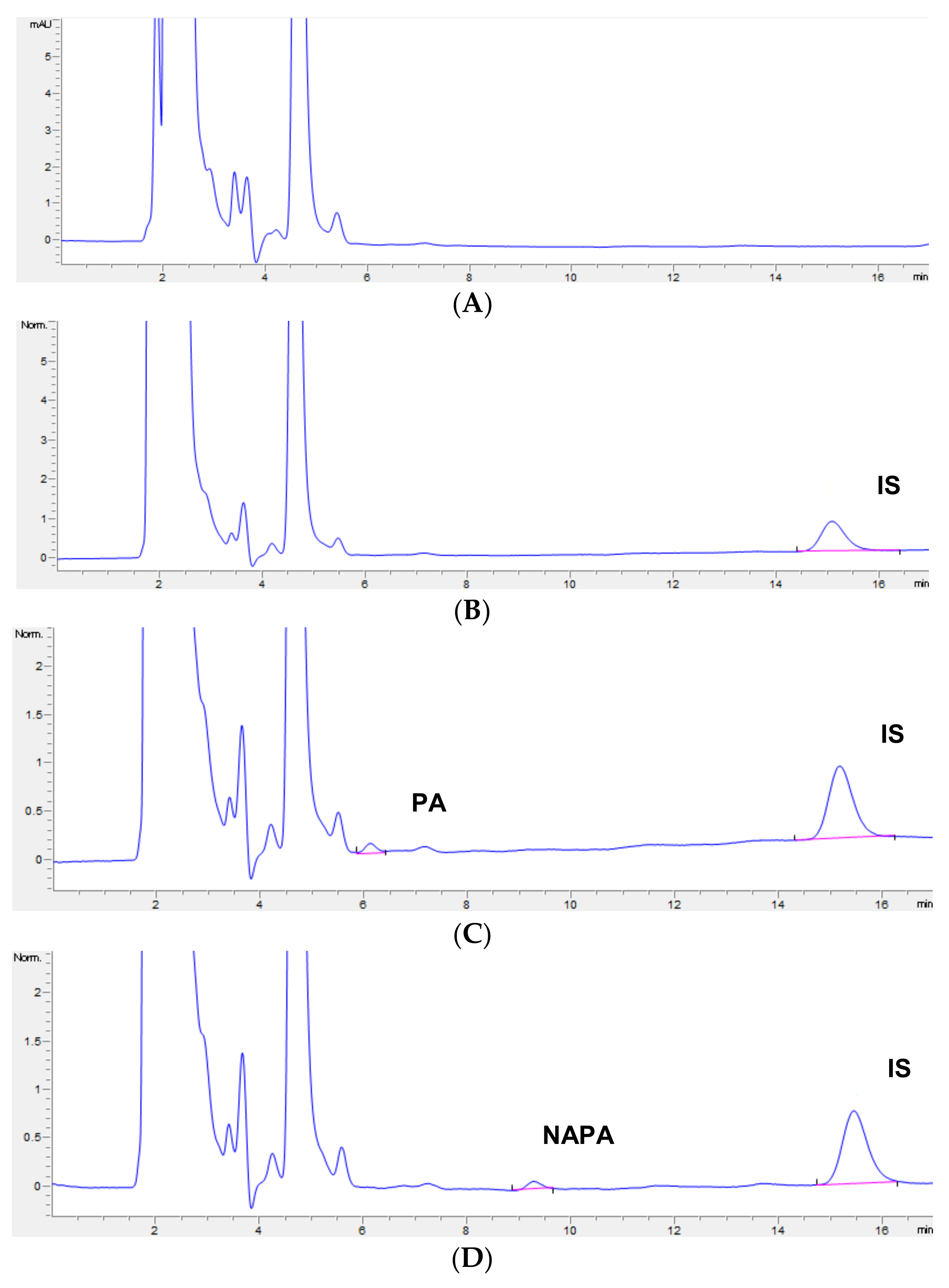
3.1. Optimization of Chromatographic Analysis
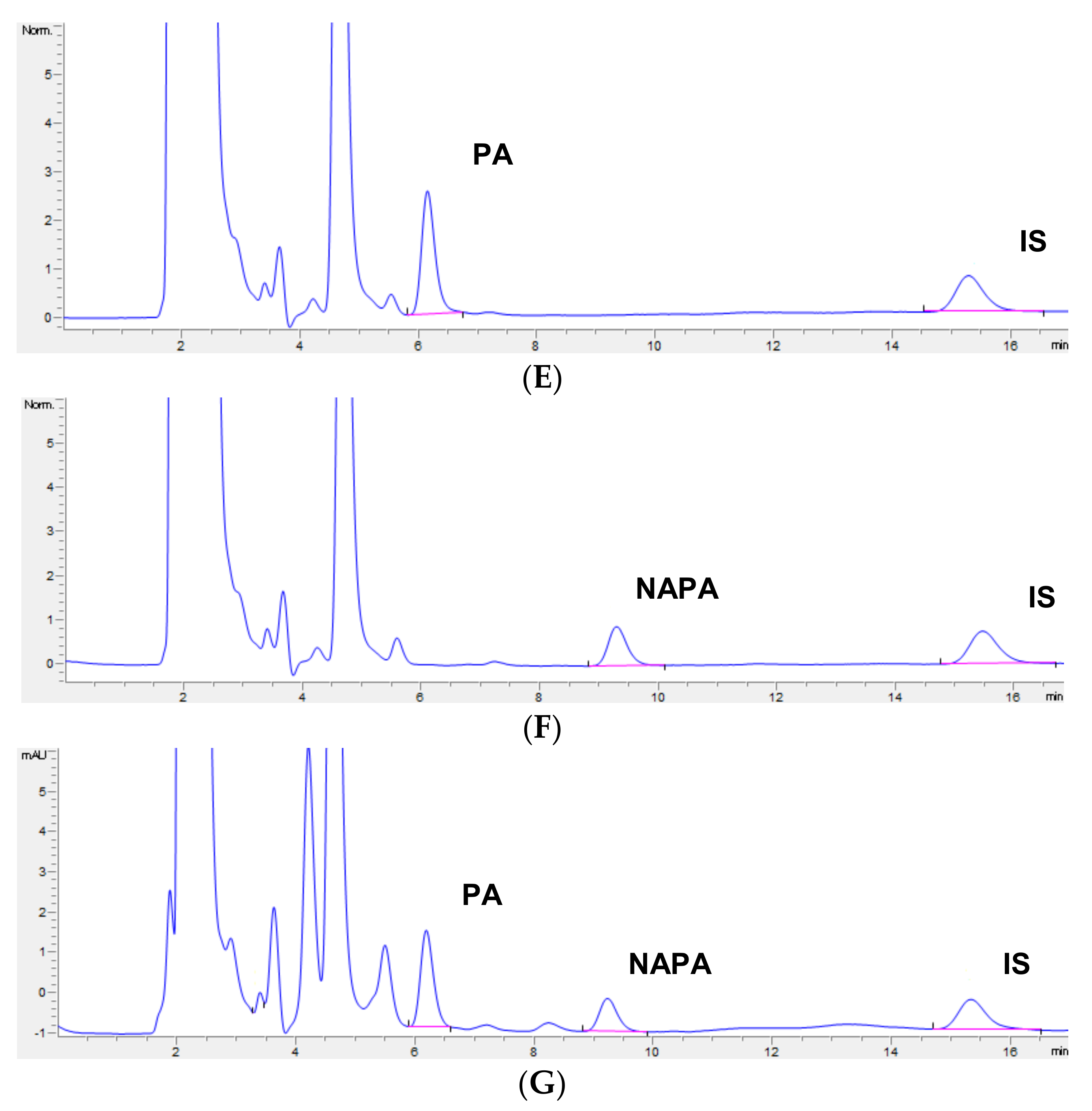
3.2. Selectivity
3.3. Linearity
3.4. Precision and Accuracy
3.5. Sensitivity
3.6. Recovery
3.7. Stability
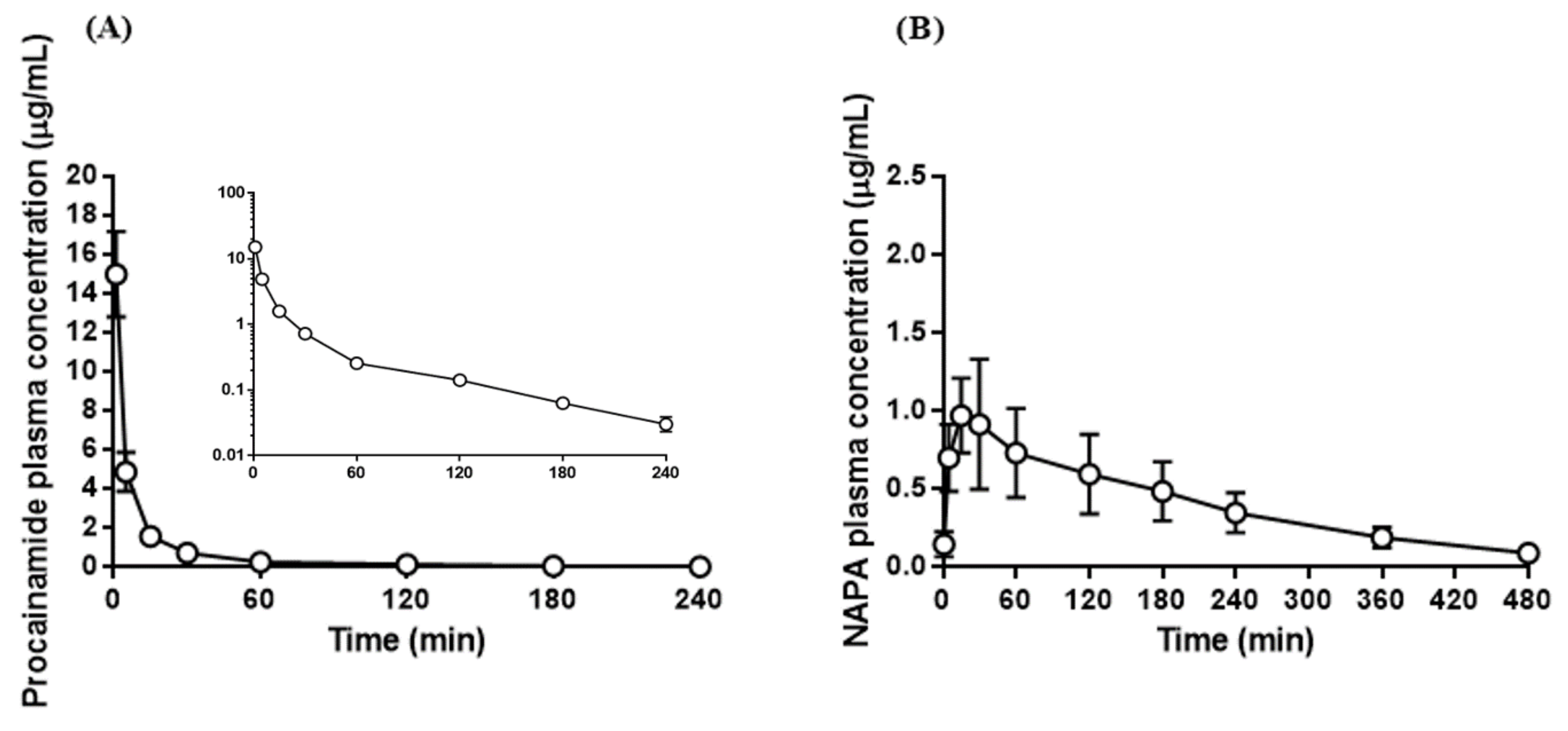
3.8. Application to Pharmacokinetic Studies of Procainamide HCl
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ellenbogen, K.A.; Wood, M.A.; Stambler, B.S. Procainamide: A perspective on its value and danger. Heart Dis. Stroke 1993, 2, 473–476. [Google Scholar] [PubMed]
- Giardina, E.V. Procainamide: Clinical pharmacology and efficacy against ventricular arrhythmias. Ann. N. Y. Acad. Sci. 1984, 432, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Bauer, L.A. Applied Clinical Pharmacokinetics, 2nd ed.; McGrow-Hill: New York, NY, USA, 2008. [Google Scholar]
- Gorgels, A.P.M.; van den Dool, A.; Hofs, A.; Mulleneers, R.; Smeets, J.L.; Vos, M.A.; Wellens, H.J. Comparison of procainamide and lidocaine in terminating sustained monomorphic ventricular tachycardia. Am. J. Cardiol. 1996, 78, 43–46. [Google Scholar] [CrossRef]
- American Heart Association. Guidelines 2000 for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Part 6: Advanced cardiovascular life support: Section 5: Pharmacology I: Agents for arrhythmias. Circulation 2000, 102, I112–I128. [Google Scholar]
- Fenoglio, C.; Boncompagni, E.; Chiavarina, B.; Cafaggi, S.; Cilli, M.; Viale, M. Morphological and histochemical evidence of the protective effect of procainamide hydrochloride on tissue damage induced by repeated administration of low doses of cisplatin. Anticancer Res. 2005, 25, 4123–4128. [Google Scholar] [PubMed]
- Shih, C.C.; Liao, M.H.; Hsiao, T.S.; Hii, H.P.; Shen, C.H.; Chen, S.J.; Ka, S.M.; Chang, Y.L.; Wu, C.C. Procainamide inhibits DNA methylation and alleviates multiple organ dysfunction in rats with endotoxic shock. PLoS ONE 2016, 11, e0163690. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M.; Reele, S.B.; Higgins, S.B.; Wilkinson, G.R.; Smith, R.F.; Oates, J.A.; Woosley, R.L. Antiarrhythmic efficacy, pharmacokinetics and safety of N-acetylprocainamide in human subjects: Comparison with procainamide. Am. J. Cardiol. 1980, 46, 463–468. [Google Scholar] [CrossRef]
- Strasberg, B.; Sclarovsky, S.; Erdberg, A.; Duffy, C.E.; Lam, W.; Swiryn, S.; Agmon, J.; Roden, K.M. Procainamide-induced polymorphous ventricular tachycardia. Am. J. Cardiol. 1981, 47, 1309–1314. [Google Scholar] [CrossRef]
- Karlsson, E. Clinical pharmacokinetics of procainamide. Clin. Pharmacokin. 1978, 3, 97–107. [Google Scholar] [CrossRef]
- Campbell, T.J.; Williams, K.M. Therapeutic drug monitoring: Antiarrhythmic drugs. Br. J. Clin. Pharmacol. 1998, 46, 307–319. [Google Scholar] [CrossRef]
- Okumura, K.; Kita, T.; Chikazawa, S.; Komada, F.; Iwakawa, S.; Tanigawara, Y. Genotyping of N-acetylation polymorphism and correlation with procainamide metabolism. Clin. Pharmacol. Ther. 1997, 61, 509–517. [Google Scholar] [CrossRef]
- Kark, B.; Sistovaris, N.; Keller, A. Thin-layer chromatographic determination of procainamide and N-acetylprocainamide in human serum and urine at single-dose levels. J. Chromatogr. 1983, 277, 261–272. [Google Scholar] [CrossRef]
- Wesley-Hadzija, B.; Mattocks, A.M. Quantitative thin-layer chromatographic method for the determination of procainamide and its major metabolite in plasma. J. Chromatogr. 1977, 143, 307–313. [Google Scholar] [CrossRef]
- Kessler, K.M.; Ho-Tung, P.; Steele, B.; Silver, J.; Pickoff, A.; Narayanan, S.; Myerburg, R. Simultaneous quantitation of quinidine, procainamide, and N-acetylprocainamide in serum by gas-liquid chromatography with a nitrogen-phosphorus selective detector. Clin. Chem. 1982, 28, 1187–1190. [Google Scholar] [PubMed]
- Butterfield, A.G.; Cooper, J.K.; Midha, K.K. Simultaneous determination of procainamide and N-acetylprocainamide in plasma by high-performance liquid chromatography. J. Pharm. Sci. 1978, 67, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Carr, K.; Woosley, R.L.; Oates, J.A. Simultaneous quantification of procainamide and N-acetylprocainamide with high-performance liquid chromatography. J. Chromatogr. 1976, 129, 363–368. [Google Scholar] [CrossRef]
- Coyle, J.D.; Mackichan, J.J.; Boudoulas, H.; Lima, J.J. Reversed-phase liquid chromatography method for measurement of procainamide and three metabolites in serum and urine; percent of dose excreted as deemethyl metabolites. J. Pharm. Sci. 1987, 76, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.M.; Kamath, B.L.; Look, Z.M.; Yacobi, A. Determination of procainamide and N-Acetylprocainamide in biological fluids by high pressure liquid chromatography. J. Pharm. Sci. 1980, 69, 982–984. [Google Scholar] [CrossRef] [PubMed]
- Lessard, E.; Fortin, A.; Coquet, A.; Bélanger, P.M.; Hamelin, B.A.; Turgeon, J. Improved high-performance liquid chromatographic assay for the determination of procainamide and its N-acetylated metabolite in plasma: Application to a single-dose pharmacokinetic study. J. Chromatogr. Sci. 1998, 36, 49–54. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Patel, C.P. Improved liquid chromatographic determination of procainamide and N-acetylprocainamide in serum. Ther. Drug. Monit. 1983, 5, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Raphanaud, D.; Borensztejn, M.; Dupeyron, J.P.; Guyon, F. High performance liquid chromatography of procainamid and N-scetylprocainamide in human blood plasma. Ther. Drug. Monit. 1986, 8, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Stearns, F.M. Determination of procainamide and N-acetylprocainamide by “high-performance” liquid chromatography. Clin. Chem. 1981, 27, 2064–2067. [Google Scholar] [PubMed]
- Su, S.C.; Au, W.Y.W. An improved microanalytical procedure for the quantitationof procainamide and N-acetyl procainamide in serum by high-performance liquid chromatography. J. Liq. Chromatogr. 1978, 1, 783–794. [Google Scholar] [CrossRef]
- Weddle, O.H.; Mason, W.D. Rapid determination of procainamide and its N-acetyl derivative in human plasma by high-pressure liquid chromatography. J. Pharm. Sci. 1977, 66, 874–875. [Google Scholar] [CrossRef] [PubMed]
- Wesley, J.F.; Lasky, F.D. High performance liquid chromatographic analysis of the antiarrhythmic drugs procainamide, disopyramide, quinidine, propranolol and metabolites from serum extracts. Clin. Biochem. 1981, 14, 113–118. [Google Scholar] [CrossRef]
- Nováková, L.; Matysová, L.; Solich, P. Advantages of application of UPLC in pharmaceutical analysis. Talanta 2006, 68, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.W.; Zhang, K. Ultra-high-pressure liquid chromatography (UHPLC) in method development. Trends Analyt. Chem. 2014, 63, 21–30. [Google Scholar] [CrossRef]
- Kim, Y.C.; Kim, I.B.; Noh, C.K.; Quach, H.P.; Yoon, I.S.; Chow, E.C.Y.; Kim, M.; Jin, H.E.; Cho, K.H.; Chung, S.J.; et al. Effects of 1α,25-dihydroxyvitamin D3, the natural vitamin D receptor ligand, on the pharmacokinetics of cefdinir and cefadroxil, organic anion transporter substrates, in rat. J. Pharm. Sci. 2014, 103, 3793–3805. [Google Scholar] [CrossRef] [PubMed]
- US Food Drug Administration. Guidance for Industry, Bioanalytical Methods Validation. 2013. Available online: https://www.fda.gov/downloads/Drugs/Guidances/ucm368107.pdf (accessed on 5 March 2018).
- Basseches, P.J.; DiGregorio, G.J. Pharmacokinetics of procainamide in rats with extrahepatic biliary obstruction. J. Pharm. Sci. 1982, 71, 1256–1259. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Knowlton, P.W.; Svensson, C.K. Effect of amiodarone on the disposition of procaiamide in the rat. J. Pharm. Sci. 1988, 77, 662–665. [Google Scholar] [CrossRef] [PubMed]
- Schneck, D.W.; Grove, K.; Dewitt, F.O.; Shiroff, R.A.; Hayes, A.H., Jr. The quantitative disposition of procainamide and N-acetylprocainamide in rat. J. Pharm. Exp. Ther. 1978, 204, 219–225. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Nominal Concentration (ng/mL) | Intra-day (n = 6) | Inter-day (n = 30) | ||||
|---|---|---|---|---|---|---|---|
| Measured Concentration (ng/mL) | Precision (RSD %) | Accuracy (%) | Measured Concentration (ng/mL) | Precision (RSD %) | Accuracy (%) | ||
| PA | 20 | 22.1 | 2.0 | 110.3 | 19.5 | 10.5 | 97.7 |
| 60 | 60.5 | 7.5 | 110.9 | 59.9 | 7.1 | 99.8 | |
| 8000 | 7890 | 4.2 | 98.6 | 8011 | 5.2 | 100.1 | |
| 80,000 | 79,079 | 4.0 | 98.9 | 78,994 | 6.0 | 98.7 | |
| NAPA | 20 | 20.0 | 6.1 | 99.7 | 20.9 | 10.5 | 104.3 |
| 60 | 62.1 | 3.3 | 103.4 | 60.5 | 9.1 | 100.8 | |
| 8000 | 873.2 | 1.0 | 109.2 | 850.1 | 4.6 | 106.3 | |
| 80,000 | 8031 | 5.1 | 100.4 | 8238 | 6.1 | 103.0 | |
| Concentration (ng/mL) | Stability (%) | |
|---|---|---|
| PA | NAPA | |
| Freeze-thaw stability (3 cycles) | ||
| 20 | 103.6 ± 7.9 | 97.3 ± 6.5 |
| 60 | 98.9 ± 1.5 | 96.3 ± 2.0 |
| 8000 | 99.8 ± 1.0 | 102.7 ± 1.8 |
| 80,000 | 95.6 ± 0.8 | 100.3 ± 2.2 |
| Auto-sampler stability (24 h at 4 °C) | ||
| 20 | 104.6 ± 6.0 | 102.7 ± 0.9 |
| 60 | 103.1 ± 2.2 | 98.3 ± 1.2 |
| 8000 | 103.4 ± 2.2 | 101.1 ± 3.0 |
| 80,000 | 94.4 ± 1.5 | 100.2 ± 1.7 |
| Short-term stability (4 h at room temperature) | ||
| 20 | 108.1 ± 6.7 | 97.2 ± 9.4 |
| 60 | 103.5 ± 11.1 | 99.0 ± 1.6 |
| 8000 | 104.0 ± 2.3 | 102.0 ± 2.7 |
| 80,000 | 91.6 ± 4.6 | 95.3 ± 2.4 |
| Long-term stability (4 week at −20 °C) | ||
| 20 | 107.8 ± 1.1 | 92.4 ± 2.5 |
| 60 | 106.1 ± 12.1 | 102.6 ± 2.5 |
| 8000 | 101.5 ± 2.4 | 96.4 ± 1.1 |
| 80,000 | 91.6 ± 4.6 | 97.3 ± 3.2 |
| Parameter | Procainamide | N-acetylprocainamide |
|---|---|---|
| AUClast (µg·min/mL) | 134 ± 12.5 | 156 ± 36.8 |
| AUCinf (µg·min/mL) | 136 ± 12.1 | 177 ± 29.6 |
| t1/2 (min) | 52.4 ± 2.20 | 131 ± 31.7 |
| MRT (min) | 28.5 ± 2.99 | 181 ± 38.3 |
| Vss (mL/kg) | 2070 ± 316 | - |
| CL (mL/min/kg) | 73.8 ± 6.51 | - |
| Cmax (µg/mL) | - | 0.949 ± 0.124 |
| Tmax (min) | - | 21.0 ± 8.22 |
| AUCNAPA/AUCPA ratio | 1.30 ± 0.191 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balla, A.; Cho, K.H.; Kim, Y.C.; Maeng, H.-J. Simultaneous Determination of Procainamide and N-acetylprocainamide in Rat Plasma by Ultra-High-Pressure Liquid Chromatography Coupled with a Diode Array Detector and Its Application to a Pharmacokinetic Study in Rats. Pharmaceutics 2018, 10, 41. https://doi.org/10.3390/pharmaceutics10020041
Balla A, Cho KH, Kim YC, Maeng H-J. Simultaneous Determination of Procainamide and N-acetylprocainamide in Rat Plasma by Ultra-High-Pressure Liquid Chromatography Coupled with a Diode Array Detector and Its Application to a Pharmacokinetic Study in Rats. Pharmaceutics. 2018; 10(2):41. https://doi.org/10.3390/pharmaceutics10020041
Chicago/Turabian StyleBalla, Anusha, Kwan Hyung Cho, Yu Chul Kim, and Han-Joo Maeng. 2018. "Simultaneous Determination of Procainamide and N-acetylprocainamide in Rat Plasma by Ultra-High-Pressure Liquid Chromatography Coupled with a Diode Array Detector and Its Application to a Pharmacokinetic Study in Rats" Pharmaceutics 10, no. 2: 41. https://doi.org/10.3390/pharmaceutics10020041
APA StyleBalla, A., Cho, K. H., Kim, Y. C., & Maeng, H.-J. (2018). Simultaneous Determination of Procainamide and N-acetylprocainamide in Rat Plasma by Ultra-High-Pressure Liquid Chromatography Coupled with a Diode Array Detector and Its Application to a Pharmacokinetic Study in Rats. Pharmaceutics, 10(2), 41. https://doi.org/10.3390/pharmaceutics10020041