Absolute Oral Bioavailability of Creatine Monohydrate in Rats: Debunking a Myth




Abstract
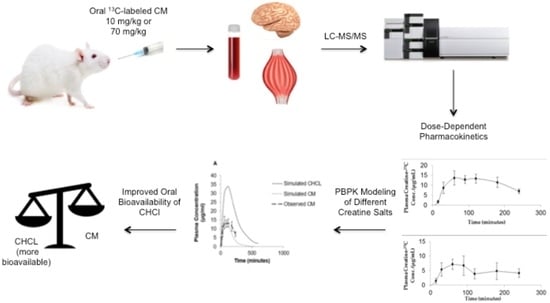
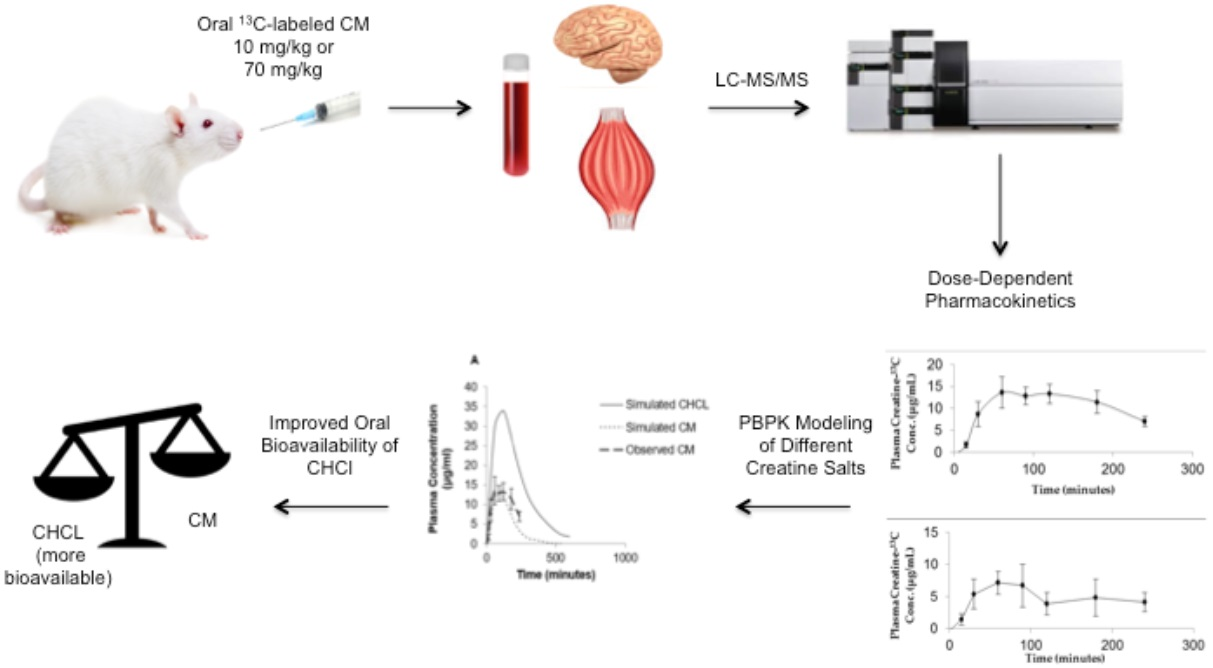
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. PK Study in Rats
2.3. Sample Preparation
2.3.1. Plasma and RBC Samples Preparation
2.3.2. Brain and Muscle Samples Preparation
2.4. LC-MS/MS Analysis
2.4.1. Stock and Working Standard Solutions
2.4.2. Sample Preparation for Standards
2.5. Physiologically Based Pharmacokinetic Modeling (PBPK)
2.6. Statistics
3. Results
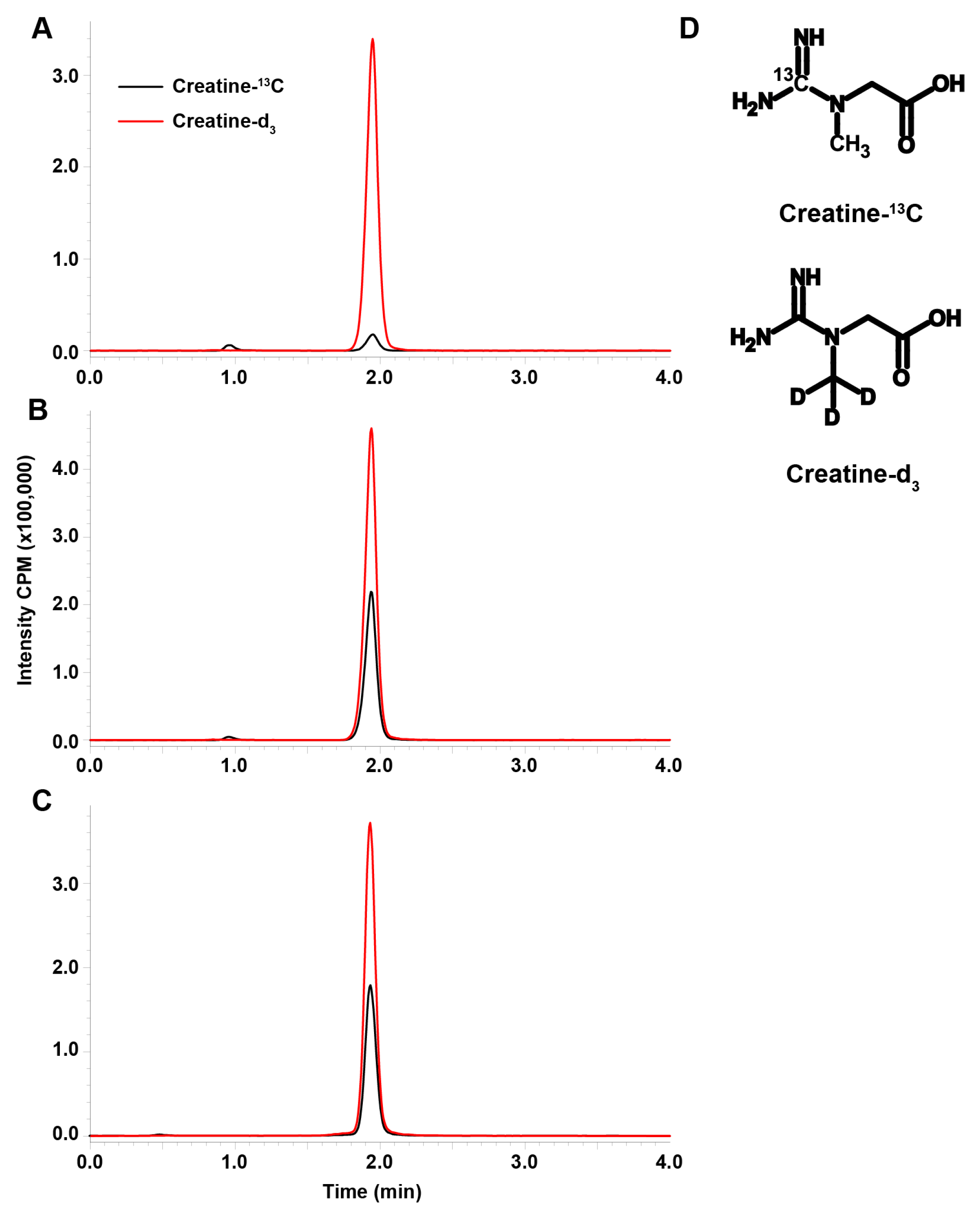
3.1. LC-MS/MS Assay
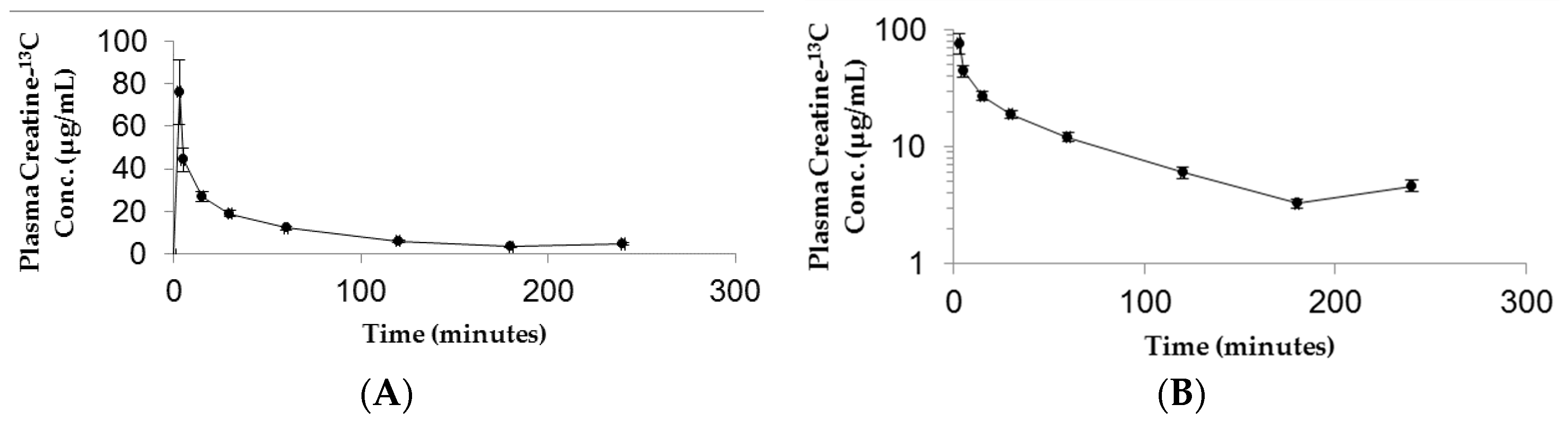
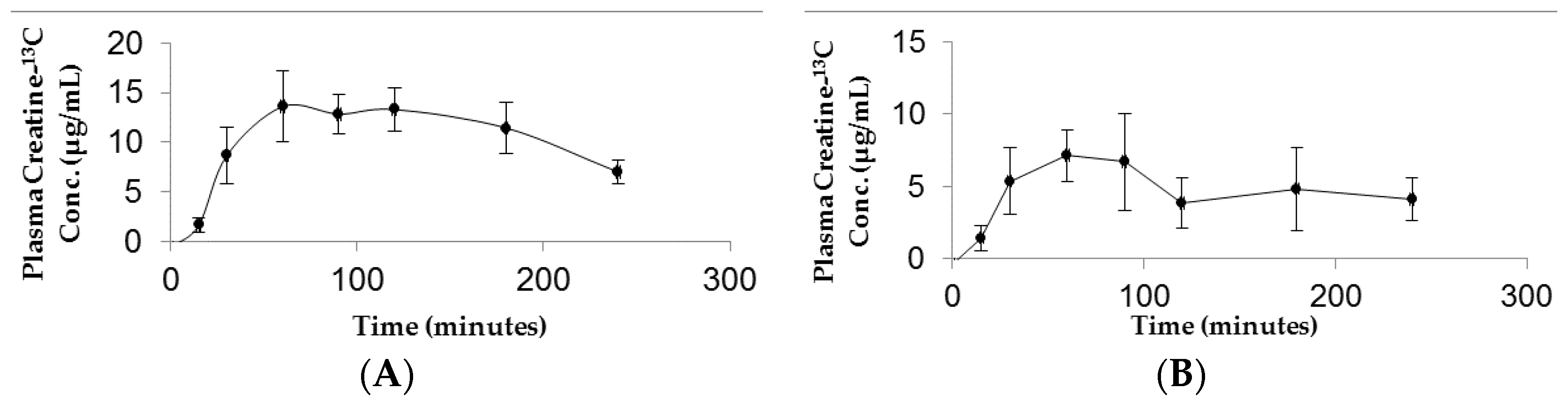
3.2. Plasma Kinetics and Oral Bioavailability of Low Dose and High Dose CM
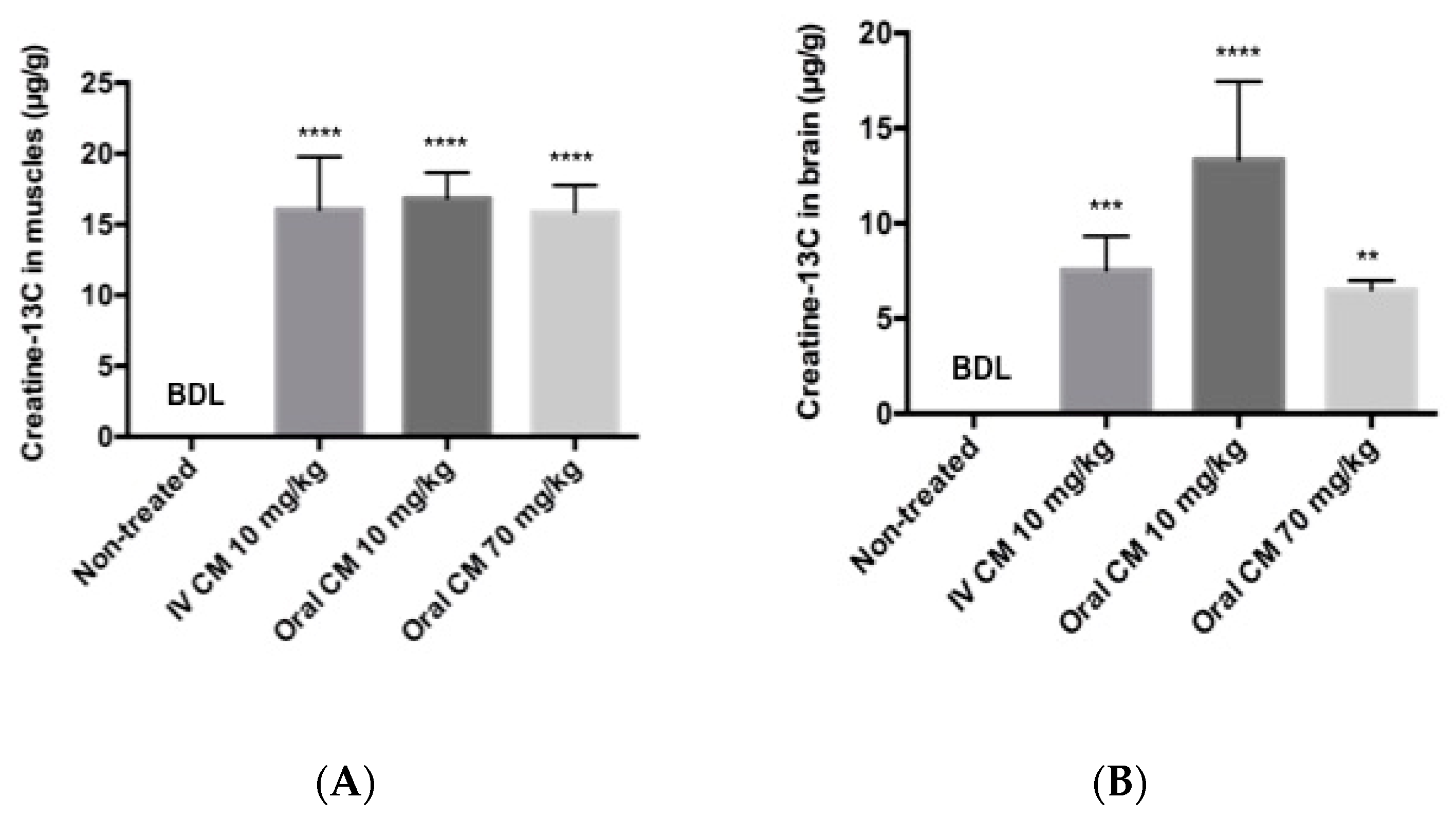
3.3. Tissue Distribution Following Low Dose and High Dose CM
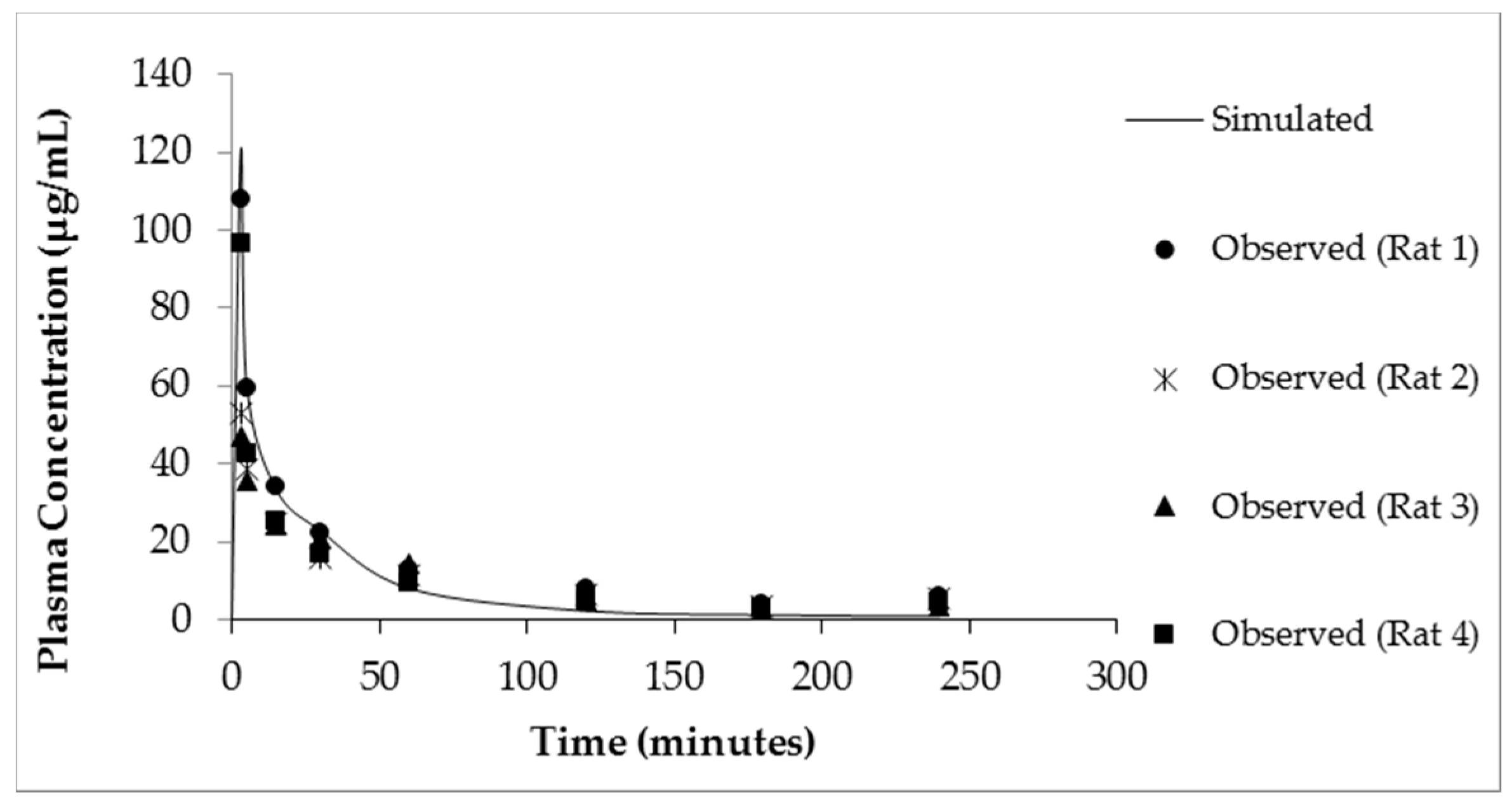
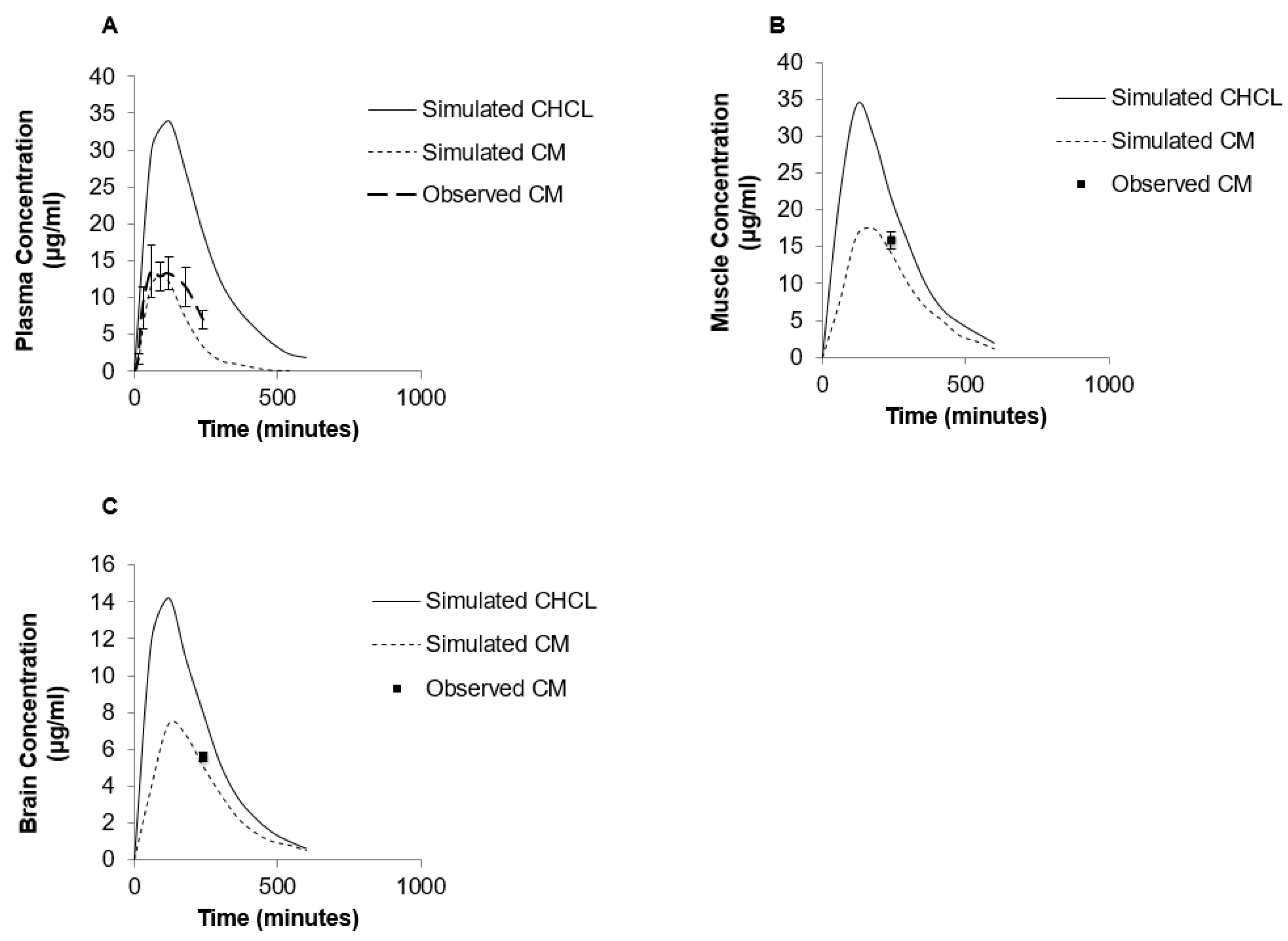
3.4. Modeling of Creatine Pharmacokinetics in Rats
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hall, M.; Trojian, T.H. Creatine supplementation. Curr. Sports Med. Rep. 2013, 12, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Persky, A.M.; Brazeau, G.A. Clinical pharmacology of the dietary supplement creatine monohydrate. Pharmacol. Rev. 2001, 53, 161–176. [Google Scholar] [PubMed]
- Wallimann, T.; Wyss, M.; Brdiczka, D.; Nicolay, K.; Eppenberger, H.M. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: The ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem. J. 1992, 281, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.H.; Branch, J.D. Creatine supplementation and exercise performance: An update. J. Am. Coll. Nutr. 1998, 17, 216–234. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Neuroprotective effects of creatine. Amino Acids 2011, 40, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Rosas, H.D.; Doros, G.; Gevorkian, S.; Malarick, K.; Reuter, M.; Coutu, J.P.; Triggs, T.D.; Wilkens, P.J.; Matson, W.; Salat, D.H.; et al. PRECREST: A phase II prevention and biomarker trial of creatine in at-risk Huntington disease. Neurology 2014, 82, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Wallimann, T.W. Protective effect of the energy precursor creatine against toxicity of glutamate and beta-amyloid in rat hippocampal neurons. J. Neurochem. 2000, 74, 1968–1978. [Google Scholar] [CrossRef] [PubMed]
- Vis, J.C.; de Boer-van Huizen, R.T.; Verbeek, M.M.; de Waal, R.M.W.; ten Donkelaar, H.J.; Kremer, B. Creatine protects against 3-nitropropionic acid-induced cell death in murine corticostriatal slice cultures. Brain Res. 2004, 1024, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.T.; Yang, L.C.; Jenkins, B.G.; Ferrante, R.J.; Rosen, B.R.; Kaddurah-Daouk, R.; Beal, M.F. Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington’s disease. J. Neurosci. 1998, 18, 156–163. [Google Scholar] [PubMed]
- Matthews, R.T.; Ferrante, R.J.; Klivenyi, P.; Yang, L.C.; Klein, A.M.; Mueller, G.; Kaddurah-Daouk, R.; Kaddurah-Daouk, M.F. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp. Neurol. 1999, 157, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Juhn, M.S.; Tarnopolsky, M. Oral creatine supplementation and athletic performance: A critical review. Clin. J. Sport Med. 1998, 8, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Balsom, P.D.; Soderlund, K.; Sjodin, B.; Ekblom, B. Skeletal muscle metabolism during short duration high-intensity exercise: Influence of creatine supplementation. Acta Physiol. Scand. 1995, 154, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Greenhaff, P.L.; Bodin, K.; Soderlund, K.; Hultman, E. Effect of oral creatine supplementation on skeletal muscle phosphocreatine resynthesis. Am. J. Physiol. 1994, 266, E725–E730. [Google Scholar] [CrossRef] [PubMed]
- Gufford, B.T.; Sriraghavan, K.; Miller, N.J.; Miller, D.W.; Gu, X.C.; Vennerstrom, J.L.; Robinson, D.H. Physicochemical characterization of creatine N-methylguanidinium salts. J. Diet. Suppl. 2010, 7, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Deldicque, L.; Décombaz, J.; Foncea, H.Z.; Vuichoud, J.; Poortmans, J.R.; Francaux, M. Kinetics of creatine ingested as a food ingredient. Eur. J. Appl. Physiol. 2008, 102, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Dash, A.K.; Miller, D.W.; Han, H.Y.; Carnazzo, J.; Stout, J.R. Evaluation of creatine transport using Caco-2 monolayers as an in vitro model for intestinal absorption. J. Pharm. Sci. 2001, 90, 1593–1598. [Google Scholar] [CrossRef] [PubMed]
- Gufford, B.T.; Ezell, E.L.; Robinson, D.H.; Miller, D.W.; Miller, N.J.; Gu, X.C.; Vennerstrom, J.L. pH-dependent stability of creatine ethyl ester: Relevance to oral absorption. J. Diet. Suppl. 2013, 10, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Boenzi, S.; Rizzo, C.; Ciommo, V.M.D.; Martinelli, D.; Goffredo, B.M.; Marca, G.I.; Dionisi-Vici, C. Simultaneous determination of creatine and guanidinoacetate in plasma by liquid chromatography-tandem mass spectrometry (LC-MS/MS). J. Pharm. Biomed. Anal. 2011, 56, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Carling, R.S.; Hogg, S.L.; Wood, T.C.; Calvin, J. Simultaneous determination of guanidinoacetate, creatine and creatinine in urine and plasma by un-derivatized liquid chromatography-tandem mass spectrometry. Ann. Clin. Biochem. 2008, 45, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Chanutin, A. The Fate of Creatine When Administered in Man. J. Biol. Chem. 1925, 67, 29–41. [Google Scholar]
- Miller, D.W.; Augustine, S.; Robinson, D.H.; Vennerstrom, J.L.; Wagner, J.C. Oral Bioavailability of Creatine Supplements: Insights into Mechanism and Implications for Improved Absorption. In Nutrition and Enhanced Sports Performance: Muscle Biulding, Endurance, and Strength, 1st ed.; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- McCall, W.; Persky, A.M. Pharmacokinetics of creatine. Subcell. Biochem. 2007, 46, 261–273. [Google Scholar] [PubMed]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and creatinine metabolism. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; Takemoto, J.K.; Brocks, D.R.; Yáñez, J.A. Multiple peaking phenomena in pharmacokinetic disposition. Clin. Pharmacokinet. 2010, 49, 351–377. [Google Scholar] [CrossRef] [PubMed]
- Deminice, R.; Jordao, A.A. Creatine supplementation reduces oxidative stress biomarkers after acute exercise in rats. Amino Acids 2012, 43, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C.; Nevill, M.; Harris, D.B.; Fallowfield, J.L.; Bogdanis, G.C.; Wise, J.A. Absorption of creatine supplied as a drink, in meat or in solid form. J. Sports Sci. 2002, 20, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Schedel, J.M.; Tanaka, H.; Kiyonaga, A.; Shindo, M.; Schutz, Y. Acute creatine ingestion in human: Consequences on serum creatine and creatinine concentrations. Life Sci. 1999, 65, 2463–2470. [Google Scholar] [CrossRef]
- Sale, C.; Harris, R.C.; Florance, J.; Kumps, A.; Sanvura, R.; Poortmans, J.R. Urinary creatine and methylamine excretion following 4 × 5 g day−1 or 20 × 1 g day−1 of creatine monohydrate for 5 days. J. Sports Sci. 2009, 27, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Atassi, N.; Ratai, E.M.; Greenblatt, D.J.; Pulley, D.; Zhao, Y.L.; Bombardier, J.; Wallace, S.; Eckenrode, J.; Cudkowicz, M.; Dibernardo, A. A phase I, pharmacokinetic, dosage escalation study of creatine monohydrate in subjects with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2010, 11, 508–513. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Snow, R.J.; Murphy, R.M. Creatine and the creatine transporter: A review. Mol. Cell. Biochem. 2001, 224, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Braissant, O. Creatine and guanidinoacetate transport at blood-brain and blood-cerebrospinal fluid barriers. J. Inherit. Metab. Dis. 2012, 35, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Nash, S.R.; Giros, B.; Kingsmore, S.F.; Rochelle, J.M.; Suter, S.T.; Gregor, P.; Seldin, M.F.; Caron, M.G. Cloning, pharmacological characterization, and genomic localization of the human creatine transporter. Recept. Channels 1994, 2, 165–174. [Google Scholar] [PubMed]
- Sora, I.; Richman, J.; Santoro, G.; Wei, H.B.; Wang, Y.; Vanderah, T.; Horvath, R.; Nguyen, M.; Waite, S.; Roeske, W.R.; et al. The cloning and expression of a human creatine transporter. Biochem. Biophys. Res. Commun. 1994, 204, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Salomons, G.S.; van Dooren, S.J.M.; Verhoeven, N.M.; Cecil, K.M.; Ball, W.S.; Degrauw, T.J.; Jakobs, C. X-linked creatine-transporter gene (SLC6A8) defect: A new creatine-deficiency syndrome. Am. J. Hum. Genet. 2001, 68, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Tachikawa, M.; Tachikawa, M.; Fukaya, M.; Terasaki, T.; Ohtsuki, S.; Watanabe, M. Distinct cellular expressions of creatine synthetic enzyme GAMT and creatine kinases uCK-Mi and CK-B suggest a novel neuron-glial relationship for brain energy homeostasis. Eur. J. Neurosci. 2004, 20, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Tachikawa, M.; Takanaga, H.; Shimizu, H.; Watanabe, M.; Hosoya, K.; Terasaki, T. The blood-brain barrier creatine transporter is a major pathway for supplying creatine to the brain. J. Cereb. Blood Flow Metab. 2002, 22, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Salomons, G.S.; Wyss, M. Creatine and Creatine Kinase in Health and Disease: From Cell Deconstruction to System Reconstruction; Subcellular Biochemistry; Springer: Berlin, Germany, 2007; Volume 46, pp. 99–113. [Google Scholar]
- Jager, R.; Harris, R.C.; Purpura, M.; Francaux, M. Comparison of new forms of creatine in raising plasma creatine levels. J. Int. Soc. Sports Nutr. 2007, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Allan, G.; Davis, J.; Dickins, M.; Garder, I.; Jenkins, T.; Jones, H.; Webster, R.; Westgate, H. Pre-clinical pharmacokinetics of UK-453,061, a novel non-nucleoside reverse transcriptase inhibitor (NNRTI), and use of in silico physiologically based prediction tools to predict the oral pharmacokinetics of UK-453,061 in man. Xenobiotica 2008, 38, 620–640. [Google Scholar] [CrossRef] [PubMed]
- Bungay, P.J.; Tweedy, S.; Howe, D.C.; Gibson, K.R.; Jones, H.M.; Mount, N.M. Preclinical and clinical pharmacokinetics of PF-02413873, a nonsteroidal progesterone receptor antagonist. Drug Metab. Dispos. 2011, 39, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- De Buck, S.S.; Sinha, V.K.; Fenu, L.A.; Nijsen, M.J.; Mackie, C.E.; Gilissen, R.A.H.J. Prediction of human pharmacokinetics using physiologically based modeling: A retrospective analysis of 26 clinically tested drugs. Drug Metab. Dispos. 2007, 35, 1766–1780. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.M.; Gardner, I.B.; Collard, W.T.; Stanley, P.J.; Oxley, P.; Hosea, N.A.; Plowchalk, D.; Gernhardt, S.; Lin, J.; Dickins, M.; et al. Simulation of human intravenous and oral pharmacokinetics of 21 diverse compounds using physiologically based pharmacokinetic modelling. Clin. Pharmacokinet. 2011, 50, 331–347. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.M.; Dickins, M.; Youdim, K.; Gosset, J.R.; Attkins, N.; Hay, T.L.; Gurrell, I.K.; Logan, Y.R.; Bungay, P.J.; Jones, B.C.; et al. Application of PBPK modelling in drug discovery and development at Pfizer. Xenobiotica 2012, 42, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.K.; Snoeys, J.; Osselaer, N.V.; Peer, A.V.; Mackie, C.; Heald, D. From preclinical to human—Prediction of oral absorption and drug-drug interaction potential using physiologically based pharmacokinetic (PBPK) modeling approach in an industrial setting: A workflow by using case example. Biopharm. Drug Dispos. 2012, 33, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Heimbach, T.; He, H.; Lin, T.H. Nilotinib preclinical pharmacokinetics and practical application toward clinical projections of oral absorption and systemic availability. Biopharm. Drug Dispos. 2012, 33, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, S.; Skaptason, J.; Romero, D.; Vekich, S.; Jones, H.M.; Tan, W.W.; Wilner, K.D.; Koudriakova, T. Prediction of oral pharmacokinetics of cMet kinase inhibitors in humans: Physiologically based pharmacokinetic model versus traditional one-compartment model. Drug Metab. Dispos. 2011, 39, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Tuckfield, C. First use of creatine hydrochloride in premanifest Huntington disease. Med. J. Aust. 2015, 202, 378–380. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | IV Injection of CM (10 mg/kg) | Oral Suspension of CM (70 mg/kg) | ||
|---|---|---|---|---|
| Simulated | Observed | Simulated | Observed | |
| Cmax (μg/mL) | 122 | 76.18 ± 15.23 | 14.07 | 13.72 ± 3.57 |
| Tmax (min) | 3 | 3 | 87 | 60 |
| Vd (L/kg) | 0.208 | 0.304 ± 0.087 | - | - |
| T1/2 (min) | 64.08 | 69.3 ± 3.7 | - | - |
| CL (L/hr) | 0.039 | 0.051 ± 0.01 | - | - |
| AUC0–∞ (μg·h/mL) | 2279.36 | 2450.01 ± 110 | 2286.1 | 2501.33 ± 378 |
| F (%) | - | - | 16.8 | 15.69 ± 4.3 |
| R2 value (model vs. observed) | 0.99 | - | 0.84 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alraddadi, E.A.; Lillico, R.; Vennerstrom, J.L.; Lakowski, T.M.; Miller, D.W. Absolute Oral Bioavailability of Creatine Monohydrate in Rats: Debunking a Myth. Pharmaceutics 2018, 10, 31. https://doi.org/10.3390/pharmaceutics10010031
Alraddadi EA, Lillico R, Vennerstrom JL, Lakowski TM, Miller DW. Absolute Oral Bioavailability of Creatine Monohydrate in Rats: Debunking a Myth. Pharmaceutics. 2018; 10(1):31. https://doi.org/10.3390/pharmaceutics10010031
Chicago/Turabian StyleAlraddadi, Eman A., Ryan Lillico, Jonathan L. Vennerstrom, Ted M. Lakowski, and Donald W. Miller. 2018. "Absolute Oral Bioavailability of Creatine Monohydrate in Rats: Debunking a Myth" Pharmaceutics 10, no. 1: 31. https://doi.org/10.3390/pharmaceutics10010031
APA StyleAlraddadi, E. A., Lillico, R., Vennerstrom, J. L., Lakowski, T. M., & Miller, D. W. (2018). Absolute Oral Bioavailability of Creatine Monohydrate in Rats: Debunking a Myth. Pharmaceutics, 10(1), 31. https://doi.org/10.3390/pharmaceutics10010031