
Porcine Epidemic Diarrhea in Europe: In-Detail Analyses of Disease Dynamics and Molecular Epidemiology

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Origin
2.2. Sequencing Protocol
2.2.1. Nucleic Acid Extraction
2.2.2. Synthesis of cDNA, Library Preparation, and Sequencing
2.2.3. Sequence Assembly and Mapping
2.3. Data Assessment, Generation of Phylogenetic Trees, Metagenomic Analyses
3. Results
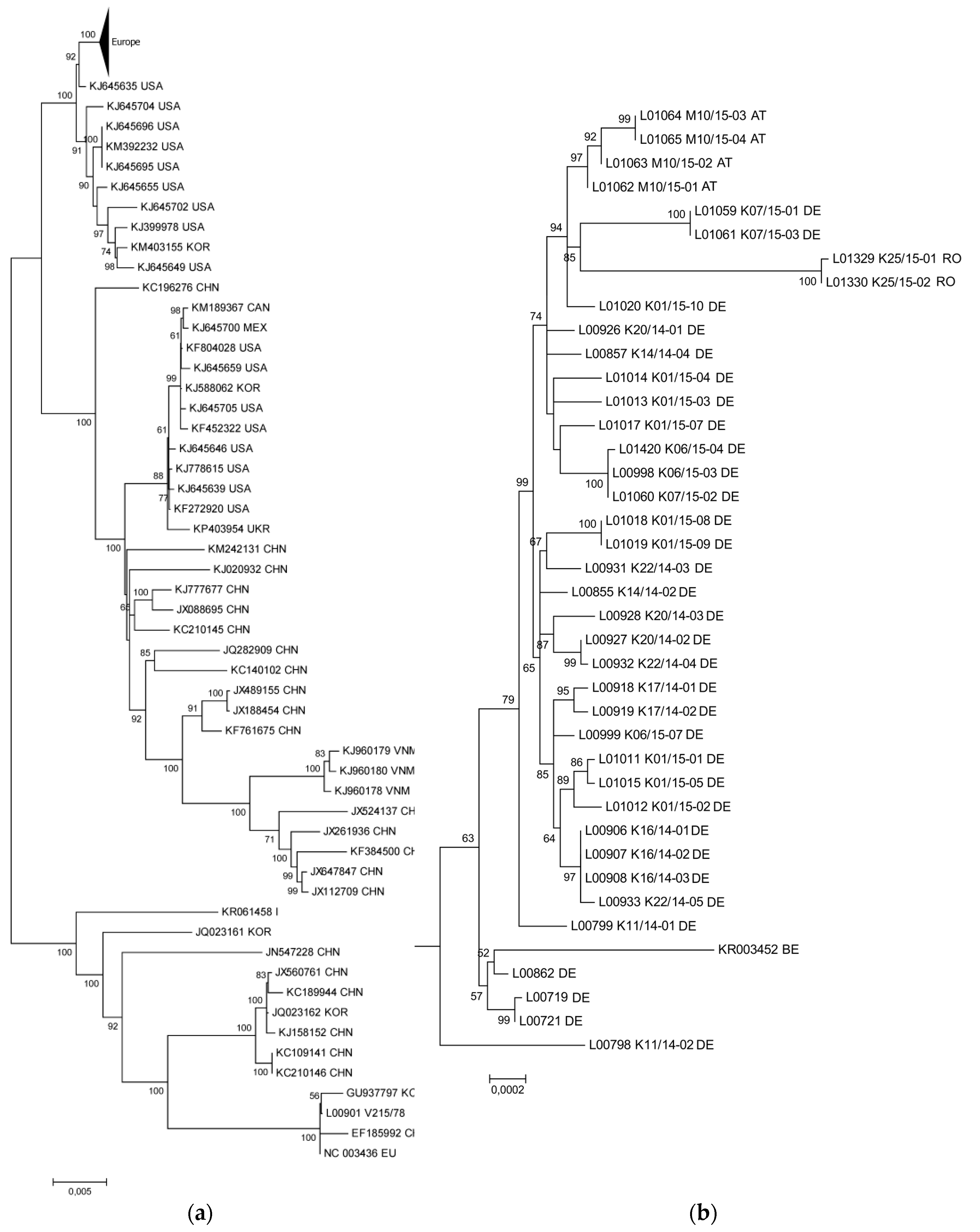
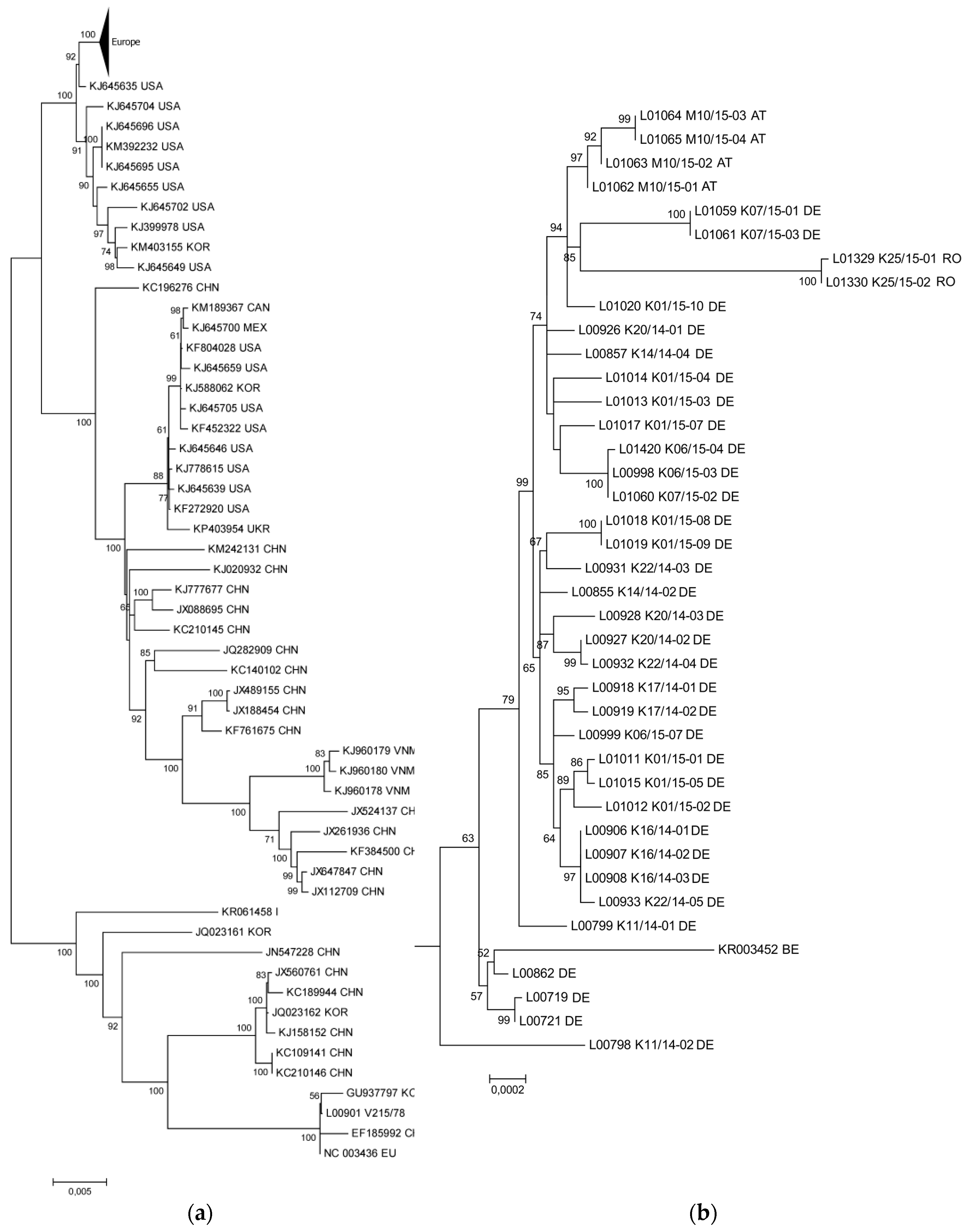
3.1. The New PEDV Strains from Germany, Austria, and Romania Cluster with S INDEL Strains from the USA and Asia
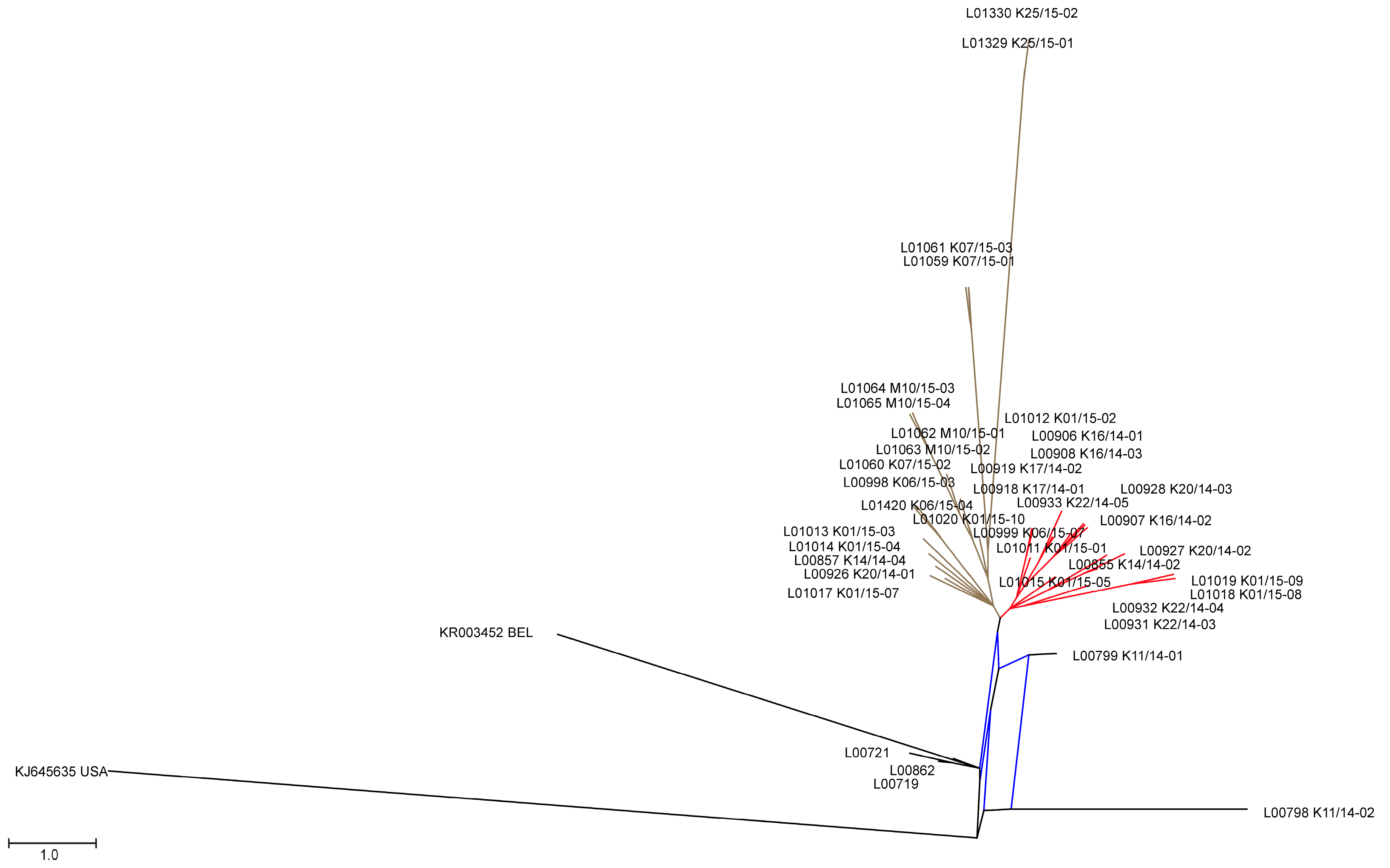
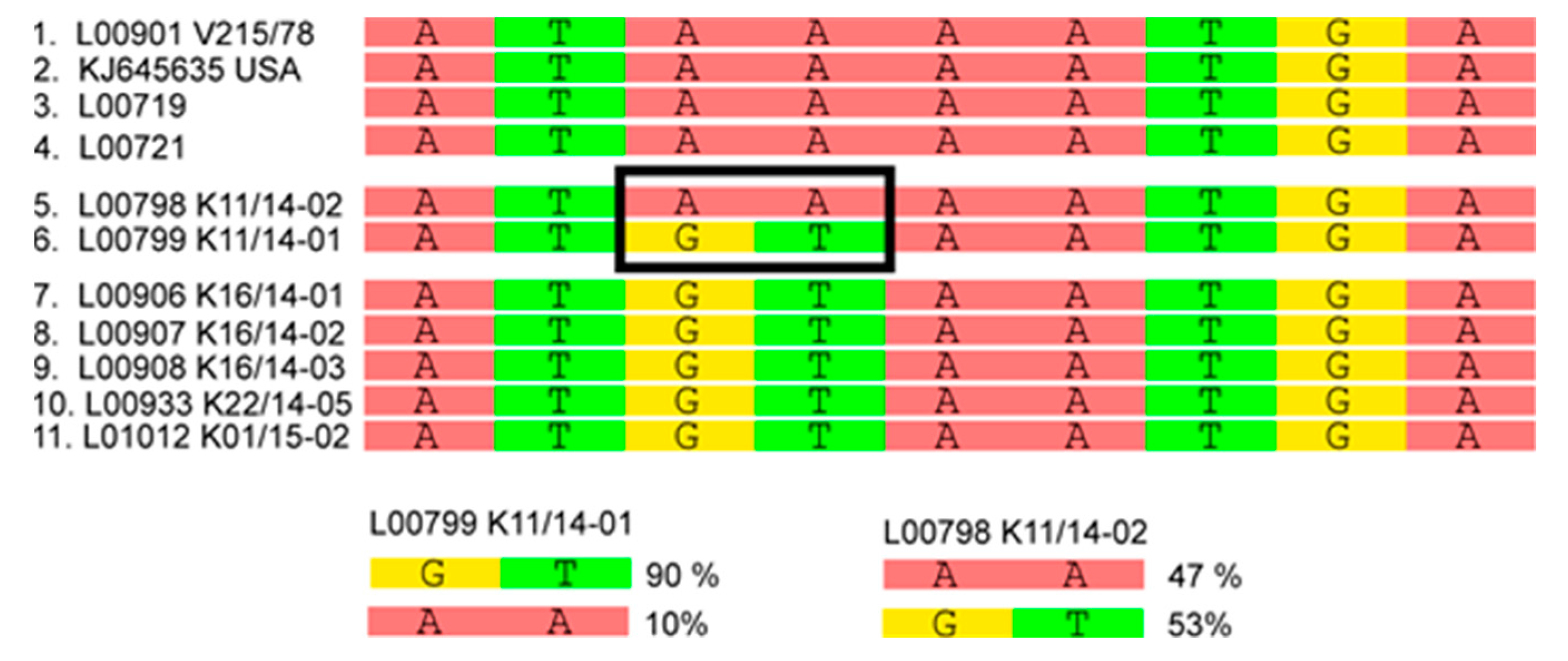
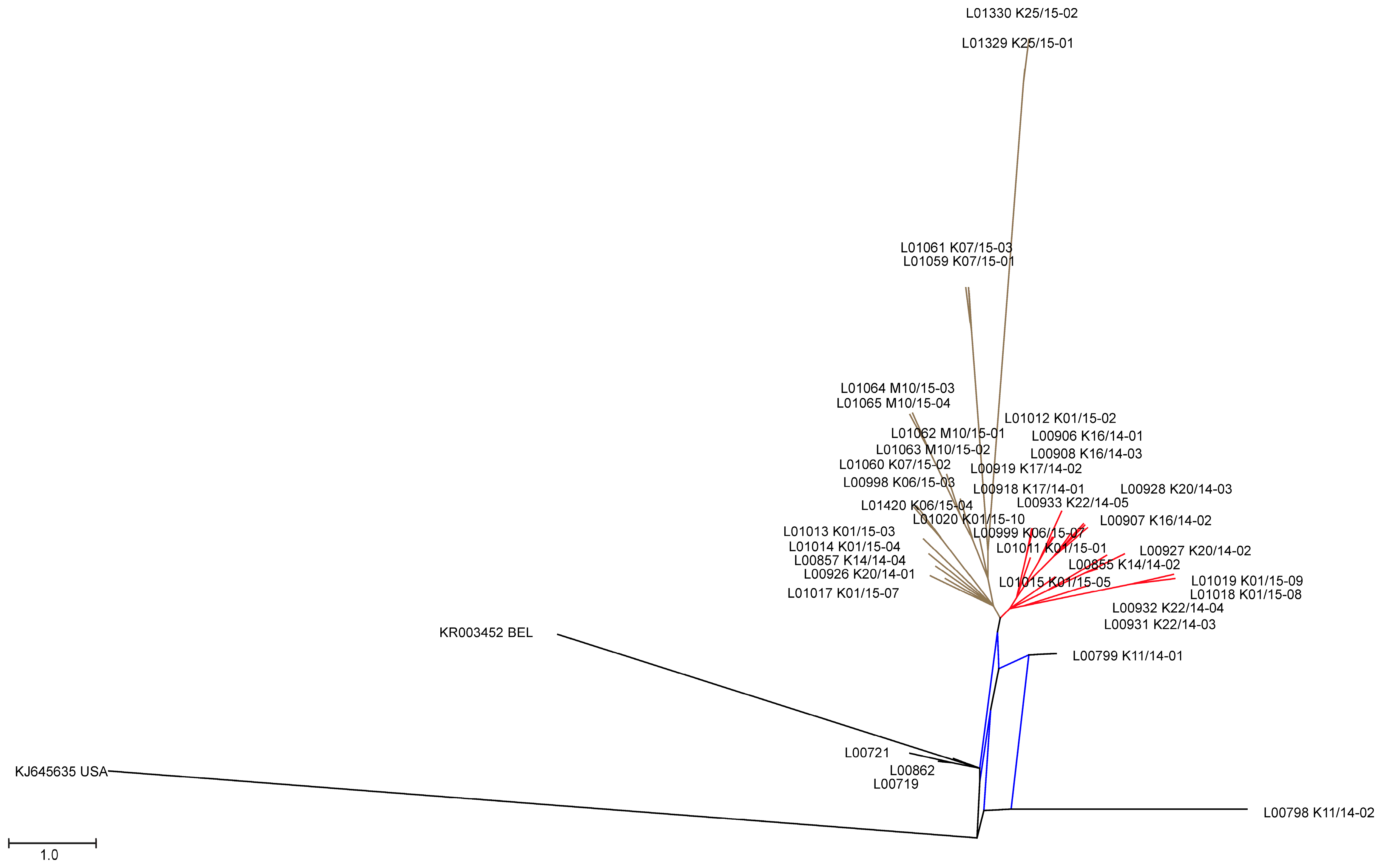
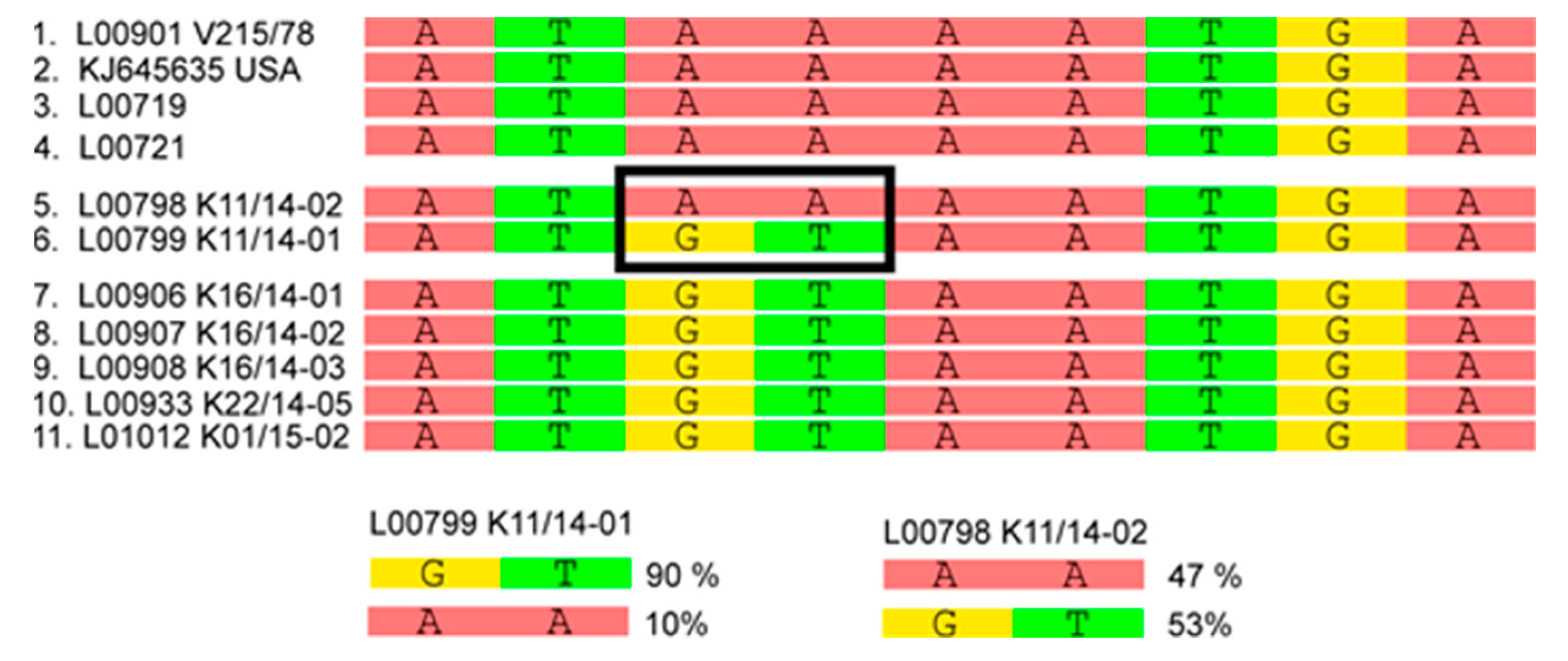
3.2. Phylogenetic Tree and Network Analyses Indicate Segregation into Two Clusters with Intermediate Stages in One Farm
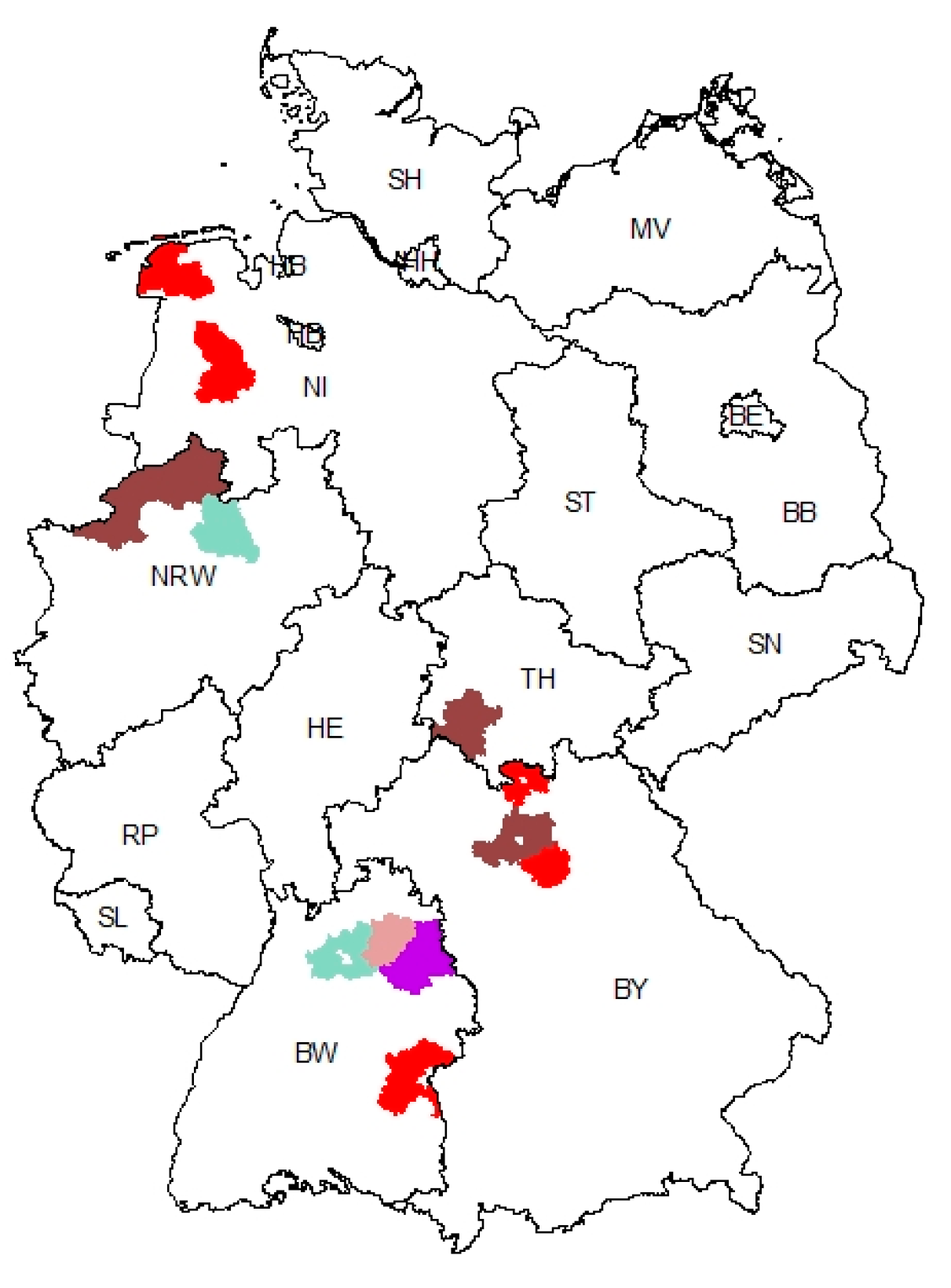
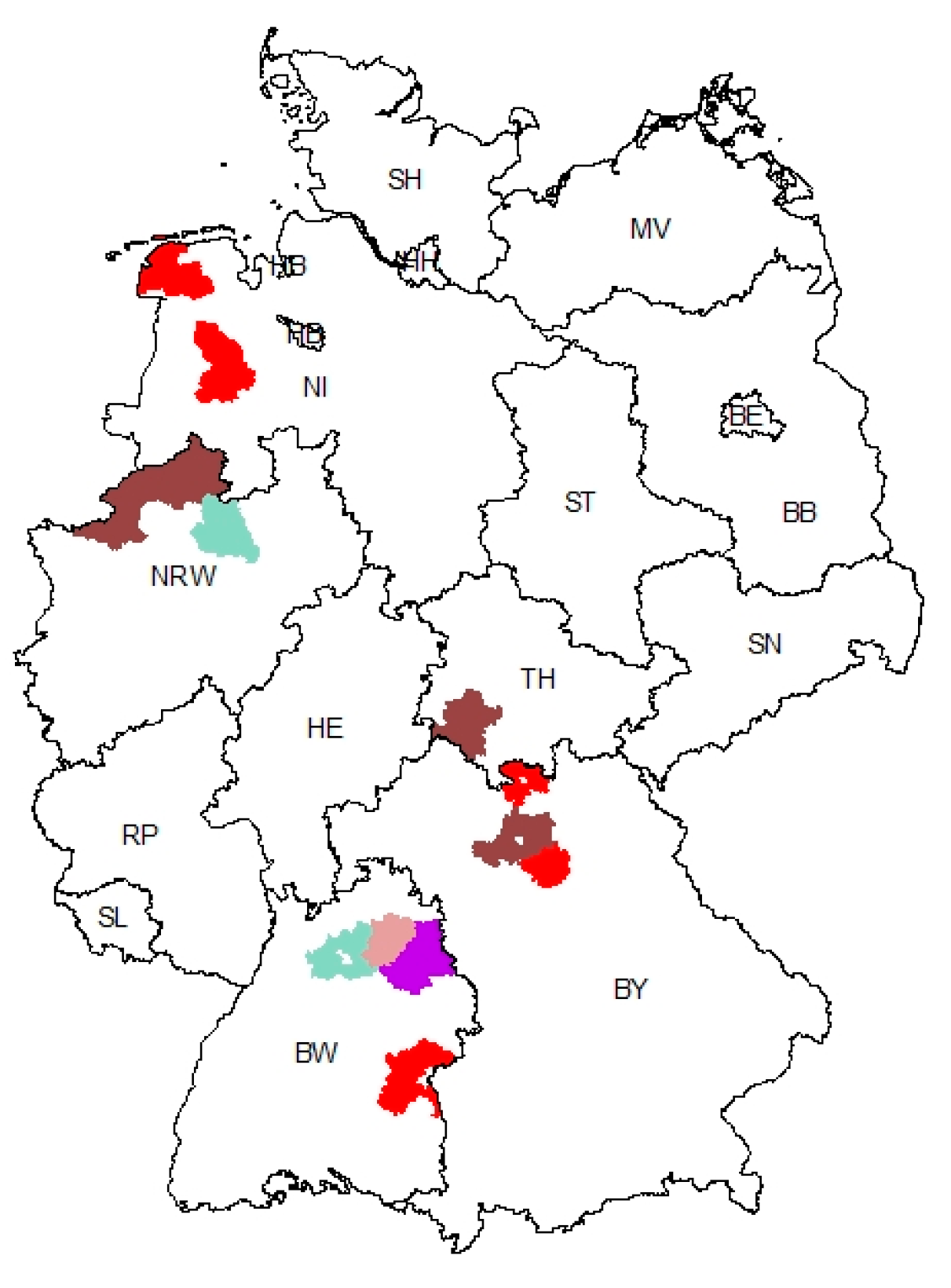
3.3. Geographic Clustering of PEDV Variants Reveals a Hotspot Region in Baden-Wuerttemberg
3.4. Metagenomic Analysis Indicates Several Co-Infections but No Correlation with Disease Severity
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Song, D.; Moon, H.; Kang, B. Porcine epidemic diarrhea: A review of current epidemiology and available vaccines. Clin. Exp. Vvaccine Res. 2015, 4, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. International Committee on Taxonomy of Viruses. 2012. Virus Taxonomy: 2012 Release. Available online: http://ictvonline.Org/virustaxonomy.Asp?Version (accessed on 5 May 2017).
- Debouck, P.; Pensaert, M. Experimental infection of pigs with a new porcine enteric coronavirus, CV 777. Am. J. Vet. Res. 1980, 41, 219–223. [Google Scholar] [PubMed]
- Martelli, P.; Lavazza, A.; Nigrelli, A.D.; Merialdi, G.; Alborali, L.G.; Pensaert, M.B. Epidemic of diarrhoea caused by porcine epidemic diarrhoea virus in Italy. Vet. Rec. 2008, 162, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, G.W.; Hoang, H.; Schwartz, K.J.; Burrough, E.R.; Sun, D.; Madson, D.; Cooper, V.L.; Pillatzki, A.; Gauger, P.; Schmitt, B.J.; et al. Emergence of porcine epidemic diarrhea virus in the United States: Clinical signs, lesions, and viral genomic sequences. J. Vet. Diagn. Investig. 2013, 25, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Cima, G. PED virus reinfecting U.S. herds. Virus estimated to have killed 7 million-plus pigs. J. Am. Vet. Med. Assoc. 2014, 245, 166–167. [Google Scholar] [PubMed]
- Vlasova, A.N.; Marthaler, D.; Wang, Q.; Culhane, M.R.; Rossow, K.D.; Rovira, A.; Collins, J.; Saif, L.J. Distinct characteristics and complex evolution of PEDV strains, North America, May 2013–February 2014. Emerg. Infect. Dis. 2014, 20, 1620–1628. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Y.; Byrum, B. Development and evaluation of a duplex real-time RT-PCR for detection and differentiation of virulent and variant strains of porcine epidemic diarrhea viruses from the United States. J. Virol. Methods 2014, 207, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Stadler, J.; Zoels, S.; Fux, R.; Hanke, D.; Pohlmann, A.; Blome, S.; Weissenbock, H.; Weissenbacher-Lang, C.; Ritzmann, M.; Ladinig, A. Emergence of porcine epidemic diarrhea virus in southern Germany. BMC Vet. Res. 2015, 11, 142. [Google Scholar] [CrossRef] [PubMed]
- Grasland, B.; Bigault, L.; Bernard, C.; Quenault, H.; Toulouse, O.; Fablet, C.; Rose, N.; Touzain, F.; Blanchard, Y. Complete genome sequence of a porcine epidemic diarrhea S gene indel strain isolated in France in December 2014. Genome Announc. 2015, 3, e00535-15. [Google Scholar] [CrossRef] [PubMed]
- Theuns, S.; Conceicao-Neto, N.; Christiaens, I.; Zeller, M.; Desmarets, L.M.; Roukaerts, I.D.; Acar, D.D.; Heylen, E.; Matthijnssens, J.; Nauwynck, H.J. Complete genome sequence of a porcine epidemic diarrhea virus from a novel outbreak in Belgium, January 2015. Genome Announc. 2015, 3, e00506-15. [Google Scholar] [CrossRef] [PubMed]
- Boniotti, M.B.; Papetti, A.; Lavazza, A.; Alborali, G.; Sozzi, E.; Chiapponi, C.; Faccini, S.; Bonilauri, P.; Cordioli, P.; Marthaler, D. Porcine epidemic diarrhea virus and discovery of a recombinant swine enteric coronavirus, Italy. Emerg. Infect. Dis. 2016, 22, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Steinrigl, A.; Fernandez, S.R.; Stoiber, F.; Pikalo, J.; Sattler, T.; Schmoll, F. First detection, clinical presentation and phylogenetic characterization of porcine epidemic diarrhea virus in Austria. BMC Vet. Res. 2015, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- EFSA. Scientific report on the collection and review of updated scientific epidemiological data on porcine epidemic diarrhea. EFSA J. 2016, 1–52. [Google Scholar] [CrossRef]
- Dastjerdi, A.; Carr, J.; Ellis, R.J.; Steinbach, F.; Williamson, S. Porcine epidemic diarrhea virus among farmed pigs, Ukraine. Emerg. Infect. Dis. 2015, 21, 2235–2237. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Goede, D.P.; Morrison, R.B.; Davies, P.R.; Rovira, A.; Marthaler, D.G.; Torremorell, M. Evidence of infectivity of airborne porcine epidemic diarrhea virus and detection of airborne viral RNA at long distances from infected herds. Vet. Res. 2014, 45, 73. [Google Scholar] [CrossRef] [PubMed]
- University of Minnesota. New Rapid Semi-Quantitative RT-PCR Assay Developed to Detect Porcine Epidemic Diarrhea Virus. 2014. Available online: https://www.cahfs.umn.edu/sites/cahfs.umn.edu/files/rt-pcr-assay-veterinay-diagnostic-lab.pdf (accessed on 22 May 2017).
- Hanke, D.; Jenckel, M.; Petrov, A.; Ritzmann, M.; Stadler, J.; Akimkin, V.; Blome, S.; Pohlmann, A.; Schirrmeier, H.; Beer, M.; et al. Comparison of porcine epidemic diarrhea viruses from Germany and the United States, 2014. Emerg. Infect. Dis. J. 2015, 21, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Henniger, T.; Schwarz, B.A. Porcine epidemic diarrhoea (PED)—Neuausbrüche in deutschen Mastschweinebeständen. Tierärztliche Umschau 2014, 69, 15. [Google Scholar]
- Pensaert, M.B.; de Bouck, P. A new coronavirus-like particle associated with diarrhea in swine. Arch. Virol. 1978, 58, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using clustalw and clustalx. Curr. Protoc. Bioinform. 2002. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Boil. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Boil. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Scheuch, M.; Höper, D.; Beer, M. RIEMS: A software pipeline for sensitive and comprehensive taxonomic classification of reads from metagenomics datasets. BMC Bioinform. 2015, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, V.; Ar Gouilh, M.; Cheval, J.; Muth, E.; Pariente, K.; Burguiere, A.; Caro, V.; Manuguerra, J.C.; Eloit, M. A member of a new picornaviridae genus is shed in pig feces. J. Virol. 2012, 86, 10036–10046. [Google Scholar] [CrossRef] [PubMed]
- Anbalagan, S.; Peterson, J.; Wassman, B.; Elston, J.; Schwartz, K. Genome sequence of torovirus identified from a pig with porcine epidemic diarrhea virus from the United States. Genome Announc. 2014, 2, e01291-14. [Google Scholar] [CrossRef] [PubMed]
- Shan, T.; Li, L.; Simmonds, P.; Wang, C.; Moeser, A.; Delwart, E. The fecal virome of pigs on a high-density farm. J. Virol. 2011, 85, 11697–11708. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.W.; Kim, M.S.; Lee, J.S.; Kim, H.; Park, S.J. Changes in the swine gut microbiota in response to porcine epidemic diarrhea infection. Microbes Environ. 2015, 30, 284–287. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Country | Federal State | Comments |
|---|---|---|---|
| L00719 BH76/14_1 | Germany | Baden-Wuerttemberg | Fecal material from fattening pigs, sample referred to as farm 4 in metagenomic analyses |
| L00721 BH76/14_2 | Germany | Baden-Wuerttemberg | Fecal material from fattening pigs, referred to as farm 4 in metagenomic analyses |
| L00798 K11/14-02 | Germany | Baden-Wuerttemberg | Fecal material, sample referred to as farm 5 in metagenomic analyses |
| L00799 K11/14-01 | Germany | Baden-Wuerttemberg | Fecal material, sample referred to as farm 5 in metagenomic analyses |
| L00855 K14/14-02 | Germany | Baden-Wuerttemberg | Fecal material from piglets and weaners, referred to as farm 2 in metagenomic analyses |
| L00857 K14/14-04 | Germany | Baden-Wuerttemberg | Fecal material from fattening pigs |
| L00862 | Germany | Border Lower-Saxony North Rhine-Westphalia | Nucleic acids derived from fecal material of fattening pigs |
| L00901 V215/78 | Germany | not provided | Cell culture virus, closely related to CV777 [20] from 1978 |
| L00906 K16/14-01 | Germany | Baden-Wuerttemberg | Fecal material from a sow, high mortality in piglets, referred to as farm 6 |
| L00907 K16/14-02 | Germany | Baden-Wuerttemberg | Fecal material from a sow and their suckling piglets, high mortality in piglets, referred to as farm 6 |
| L00908 K16/14-03 | Germany | Baden-Wuerttemberg | Fecal material from a sow and their suckling piglets, high mortality in piglets, referred to as farm 6 |
| L00918 K17/14-01 | Germany | Baden-Wuerttemberg | Fecal material from a sow, referred to as farm 3 |
| L00,919 K17/14-02 | Germany | Baden-Wuerttemberg | Fecal material from suckling pigs, referred to as farm 3 |
| L00926 K20/14-01 | Germany | Bavaria | No details provided |
| L00927 K20/14-02 | Germany | Baden-Wurttemberg | No details provided |
| L00928 K20/14-03 | Germany | Baden-Wuerttemberg | No details provided |
| L00929 K22/14-01 | Germany | Baden-Wuerttemberg | Re-introduction into farm 2, no complete PEDV genome |
| L00931 K22/14-03 | Germany | Baden-Wuerttemberg | No details provided |
| L00932 K22/14-04 | Germany | Baden-Wuerttemberg | No details provided |
| L00933 K22/14-05 | Germany | Baden-Wuerttemberg | No details provided |
| L00998 K06/15-03 | Germany | North Rhine-Westphalia | Fecal material from sows, high mortality in suckling pigs, referred to as farm 1 |
| L00999 K06/15-07 | Germany | Schleswig-Holstein | Fecal material from fattening pigs |
| L01011 K01/15-01 | Germany | Lower Saxony | Fecal material from a farm with affected fattening and breeding animals |
| L01012 K01/15-02 | Germany | Lower Saxony | Fecal material from fattening pigs |
| L01013 K01/15-03 | Germany | North Rhine-Westphalia | Fecal material from fattening pigs |
| L01014 K01/15-04 | Germany | North Rhine-Westphalia | Fecal material from fattening pigs |
| L01015 K01/15-05 | Germany | Lower Saxony | Fecal material from a farm with affected fattening and breeding animals |
| L01017 K01/15-07 | Germany | Bavaria | Fecal material from gilts |
| L01018 K01/15-08 | Germany | Bavaria | Fecal material from fattening pigs |
| L01019 K01/15-09 | Germany | Bavaria | Fecal material from fattening pigs |
| L01020 K01/15-10 | Germany | Thuringia | Fecal material from a farm with affected fattening and breeding animals |
| L01059 K07/15-01 | Germany | Thuringia | Fecal material from young fattening pigs that had been affected by PED as suckling pigs |
| L01060 K07/15-02 | Germany | North Rhine-Westphalia | Fecal material from a sow and their piglets, high mortality in suckling pigs, corresponds to farm 1 |
| L01061 K07/15-03 | Germany | Thuringia | Fecal material from fattening pigs, possible re-infection |
| L01062 M10/15-01 | Austria | Upper Austria | Fecal material from fattening pigs |
| L01063 M10/15-02 | Austria | Upper Austria | Fecal material from fattening pigs |
| L01064 M10/15-03 | Austria | Upper Austria | Fecal material from fattening pigs |
| L01065 M10/15-04 | Austria | Upper Austria | Fecal material from fattening pigs |
| L01329 K25/15-01 | Romania | not applicable | No details provided |
| L01330 K25/15-02 | Romania | not applicable | No details provided |
| L01420 K06/15-04 | Germany | North Rhine-Westphalia | Fecal material from fattening pigs, corresponds to farm 1 (high piglet mortality) |
| Sample ID | Virus |
|---|---|
| L00721 Pa | Pasivirus A |
| L00798 K11/14-02 S | Porcine sapelovirus A |
| L00799 K11/14-01 Pa | Pasivirus A |
| L00855 K14/14-02 A | Porcine astrovirus |
| L00919 K17/14-02 A | Porcine astrovirus |
| L00919 K17/14-02 K | Porcine kobuvirus |
| L00926 K20/14-01 T | Porcine torovirus |
| L00928 K20/14-03 E | Enterobacteria Phage MS2 |
| L01017 K01/15-07 Po | Posavirus 1 |
| L01061 K07/15-03 Pa | Pasivirus A |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanke, D.; Pohlmann, A.; Sauter-Louis, C.; Höper, D.; Stadler, J.; Ritzmann, M.; Steinrigl, A.; Schwarz, B.-A.; Akimkin, V.; Fux, R.; et al. Porcine Epidemic Diarrhea in Europe: In-Detail Analyses of Disease Dynamics and Molecular Epidemiology. Viruses 2017, 9, 177. https://doi.org/10.3390/v9070177
Hanke D, Pohlmann A, Sauter-Louis C, Höper D, Stadler J, Ritzmann M, Steinrigl A, Schwarz B-A, Akimkin V, Fux R, et al. Porcine Epidemic Diarrhea in Europe: In-Detail Analyses of Disease Dynamics and Molecular Epidemiology. Viruses. 2017; 9(7):177. https://doi.org/10.3390/v9070177
Chicago/Turabian StyleHanke, Dennis, Anne Pohlmann, Carola Sauter-Louis, Dirk Höper, Julia Stadler, Mathias Ritzmann, Adi Steinrigl, Bernd-Andreas Schwarz, Valerij Akimkin, Robert Fux, and et al. 2017. "Porcine Epidemic Diarrhea in Europe: In-Detail Analyses of Disease Dynamics and Molecular Epidemiology" Viruses 9, no. 7: 177. https://doi.org/10.3390/v9070177
APA StyleHanke, D., Pohlmann, A., Sauter-Louis, C., Höper, D., Stadler, J., Ritzmann, M., Steinrigl, A., Schwarz, B.-A., Akimkin, V., Fux, R., Blome, S., & Beer, M. (2017). Porcine Epidemic Diarrhea in Europe: In-Detail Analyses of Disease Dynamics and Molecular Epidemiology. Viruses, 9(7), 177. https://doi.org/10.3390/v9070177