
Hepatitis Delta Virus: Replication Strategy and Upcoming Therapeutic Options for a Neglected Human Pathogen


Abstract
:1. Introduction
2. Molecular Virology
2.1. Virus Structure
2.2. Viral Replication Cycle
3. HDV Infection Models
3.1. In Vitro Infection Models
3.2. In Vivo Infection Models
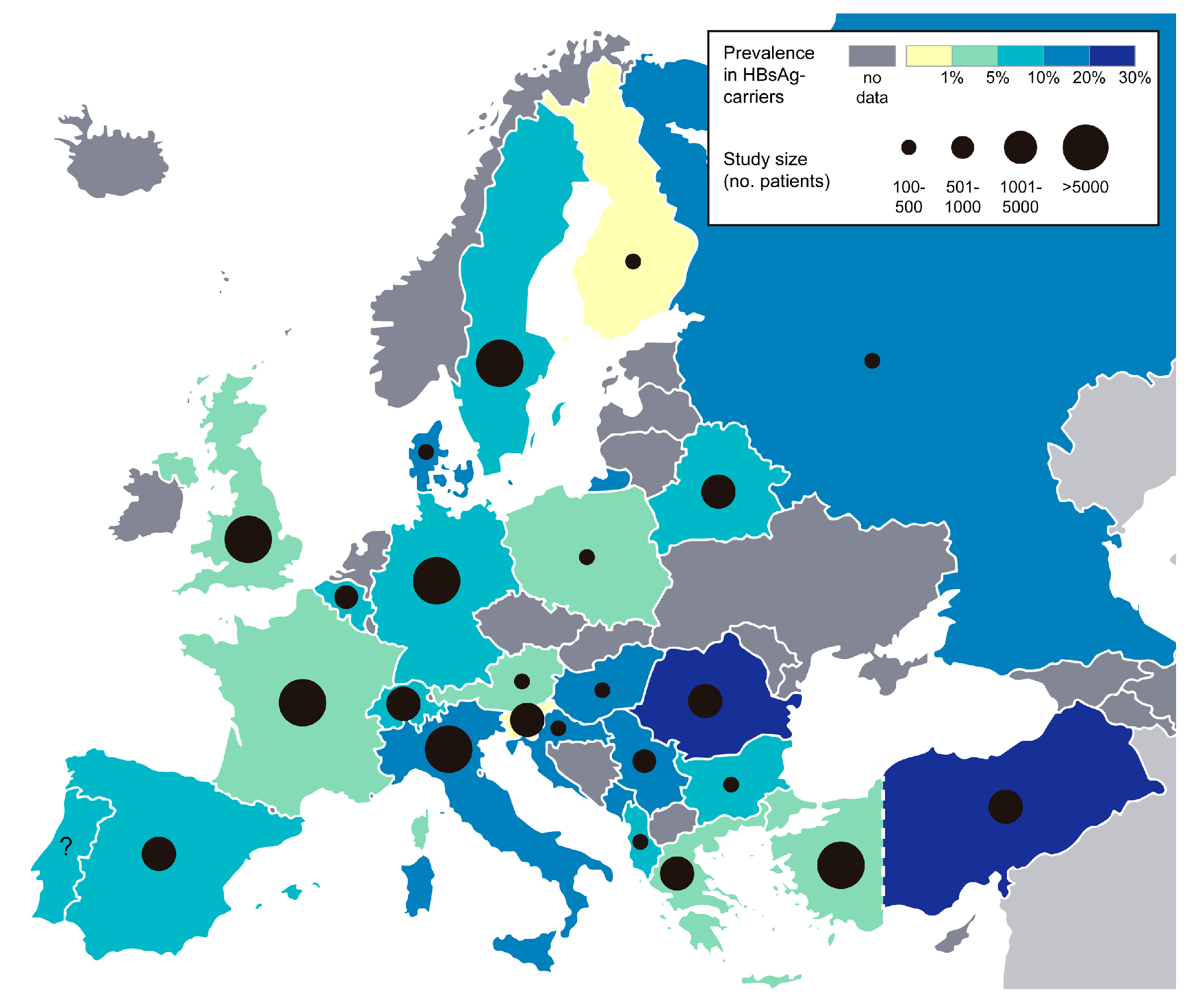
4. Epidemiology
5. Current Treatment Options and Drugs in Development
5.1. Current Treatment Options
5.2. Possible Targets to Interfere with HDV Replication
5.2.1. Viral factors
5.2.2. Host Factors
5.3. Novel Treatment Options for Chronic Hepatitis D Virus Infections
5.3.1. Lonafarnib
5.3.2. Nucleic Acid Polymers (REP2139Ca)
5.3.3. Myrcludex B
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rizzetto, M.; Canese, M.G.; Arico, S.; Crivelli, O.; Trepo, C.; Bonino, F.; Verme, G. Immunofluorescence detection of new antigen-antibody system (delta/anti-delta) associated to hepatitis B virus in liver and in serum of HBsAg carriers. Gut 1977, 18, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Montero, J.V.; Vispo, E.; Barreiro, P.; Sierra-Enguita, R.; de Mendoza, C.; Labarga, P.; Soriano, V. Hepatitis delta is a major determinant of liver decompensation events and death in HIV-infected patients. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2014, 58, 1549–1553. [Google Scholar] [CrossRef] [PubMed]
- Soriano, V.; Sherman, K.E.; Barreiro, P. Hepatitis delta and HIV infection. Aids 2017, 31, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Kalpana, G.; Goldberg, J.; Mason, W.; Werner, B.; Gerin, J.; Taylor, J. Structure and replication of the genome of the hepatitis delta virus. Proc. Natl. Acad. Sci. USA 1986, 83, 8774–8778. [Google Scholar] [CrossRef] [PubMed]
- Kos, A.; Dijkema, R.; Arnberg, A.C.; van der Meide, P.H.; Schellekens, H. The hepatitis delta (δ) virus possesses a circular RNA. Nature 1986, 323, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.S.; Choo, Q.L.; Weiner, A.J.; Ou, J.H.; Najarian, R.C.; Thayer, R.M.; Mullenbach, G.T.; Denniston, K.J.; Gerin, J.L.; Houghton, M. Structure, sequence and expression of the hepatitis delta (δ) viral genome. Nature 1986, 323, 508–514. [Google Scholar] [CrossRef] [PubMed]
- He, L.F.; Ford, E.; Purcell, R.H.; London, W.T.; Phillips, J.; Gerin, J.L. The size of the hepatitis delta agent. J. Med. Virol. 1989, 27, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Lamas Longarela, O.; Schmidt, T.T.; Schoneweis, K.; Romeo, R.; Wedemeyer, H.; Urban, S.; Schulze, A. Proteoglycans act as cellular hepatitis delta virus attachment receptors. PLoS ONE 2013, 8, e58340. [Google Scholar]
- Leistner, C.M.; Gruen-Bernhard, S.; Glebe, D. Role of glycosaminoglycans for binding and infection of hepatitis B virus. Cell. Microbiol. 2008, 10, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis b virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Sureau, C.; Salisse, J. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus A-determinant. Hepatology 2013, 57, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Verrier, E.R.; Colpitts, C.C.; Bach, C.; Heydmann, L.; Weiss, A.; Renaud, M.; Durand, S.C.; Habersetzer, F.; Durantel, D.; Abou-Jaoude, G.; et al. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis b and d viruses. Hepatology 2016, 63, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Wiedtke, E.; Qu, B.; Roques, P.; Chemin, I.; Vondran, F.W.; Le Grand, R.; Grimm, D.; Urban, S. Sodium taurocholate cotransporting polypeptide is the limiting host factor of hepatitis B virus infection in macaque and pig hepatocytes. Hepatology 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhuang, Q.; Wang, Y.; Zhang, T.; Zhao, J.; Zhang, Y.; Zhang, J.; Lin, Y.; Yuan, Q.; Xia, N.; et al. HBV life cycle is restricted in mouse hepatocytes expressing human NTCP. Cell. Mol. Immunol. 2014, 11, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Peng, B.; He, W.; Zhong, G.; Qi, Y.; Ren, B.; Gao, Z.; Jing, Z.; Song, M.; Xu, G.; et al. Molecular determinants of hepatitis B and D virus entry restriction in mouse sodium taurocholate cotransporting polypeptide. J. Virol. 2013, 87, 7977–7991. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.C.; Hsieh, T.Y.; Sheu, G.T.; Lai, M.M. Hepatitis delta antigen mediates the nuclear import of hepatitis delta virus RNA. J. Virol. 1998, 72, 3684–3690. [Google Scholar] [PubMed]
- Tavanez, J.P.; Cunha, C.; Silva, M.C.; David, E.; Monjardino, J.; Carmo-Fonseca, M. Hepatitis delta virus ribonucleoproteins shuttle between the nucleus and the cytoplasm. RNA 2002, 8, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Nie, X.; Chang, H.E.; Han, Z.; Taylor, J. Transcription of hepatitis delta virus RNA by RNA polymerase II. J. Virol. 2008, 82, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Greco-Stewart, V.S.; Miron, P.; Abrahem, A.; Pelchat, M. The human RNA polymerase II interacts with the terminal stem-loop regions of the hepatitis delta virus RNA genome. Virology 2007, 357, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.Y.; Sharmeen, L.; Dinter-Gottlieb, G.; Taylor, J. Characterization of self-cleaving RNA sequences on the genome and antigenome of human hepatitis delta virus. J. Virol. 1988, 62, 4439–4444. [Google Scholar] [PubMed]
- Wu, H.N.; Lin, Y.J.; Lin, F.P.; Makino, S.; Chang, M.F.; Lai, M.M. Human hepatitis delta virus RNA subfragments contain an autocleavage activity. Proc. Natl. Acad. Sci. USA 1989, 86, 1831–1835. [Google Scholar] [CrossRef] [PubMed]
- Gudima, S.; Wu, S.Y.; Chiang, C.M.; Moraleda, G.; Taylor, J. Origin of hepatitis delta virus mRNA. J. Virol. 2000, 74, 7204–7210. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.Y.; Chao, M.; Coates, L.; Taylor, J. Hepatitis delta virus genome replication: A polyadenylated mRNA for delta antigen. J. Virol. 1990, 64, 3192–3198. [Google Scholar] [PubMed]
- Lo, K.; Hwang, S.B.; Duncan, R.; Trousdale, M.; Lai, M.M. Characterization of mRNA for hepatitis delta antigen: Exclusion of the full-length antigenomic RNA as an mRNA. Virology 1998, 250, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.X.; Chao, M.; Hsieh, S.Y.; Sureau, C.; Nishikura, K.; Taylor, J. A specific base transition occurs on replicating hepatitis delta virus RNA. J. Virol. 1990, 64, 1021–1027. [Google Scholar] [PubMed]
- Polson, A.G.; Bass, B.L.; Casey, J.L. RNA editing of hepatitis delta virus antigenome by dsRNA-adenosine deaminase. Nature 1996, 380, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Lazinski, D.W. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc. Natl. Acad. Sci. USA 2002, 99, 15118–15123. [Google Scholar] [CrossRef] [PubMed]
- Glenn, J.S.; Watson, J.A.; Havel, C.M.; White, J.M. Identification of a prenylation site in delta virus large antigen. Science 1992, 256, 1331–1333. [Google Scholar] [CrossRef] [PubMed]
- Otto, J.C.; Casey, P.J. The hepatitis delta virus large antigen is farnesylated both in vitro and in animal cells. J. Biol. Chem. 1996, 271, 4569–4572. [Google Scholar] [PubMed]
- Hwang, S.B.; Lai, M.M. Isoprenylation mediates direct protein-protein interactions between hepatitis large delta antigen and hepatitis B virus surface antigen. J. Virol. 1993, 67, 7659–7662. [Google Scholar] [PubMed]
- Hwang, S.B.; Lai, M.M. Isoprenylation masks a conformational epitope and enhances trans-dominant inhibitory function of the large hepatitis delta antigen. J. Virol. 1994, 68, 2958–2964. [Google Scholar] [PubMed]
- Huang, H.C.; Lee, C.P.; Liu, H.K.; Chang, M.F.; Lai, Y.H.; Lee, Y.C.; Huang, C. Cellular nuclear export factors TAP and ALY are required for HDAG-l-mediated assembly of hepatitis delta virus. J. Biol. Chem. 2016, 291, 26226–26238. [Google Scholar] [CrossRef] [PubMed]
- Freitas, N.; Cunha, C.; Menne, S.; Gudima, S.O. Envelope proteins derived from naturally integrated hepatitis B virus DNA support assembly and release of infectious hepatitis delta virus particles. J. Virol. 2014, 88, 5742–5754. [Google Scholar] [CrossRef] [PubMed]
- Sureau, C.; Guerra, B.; Lanford, R.E. Role of the large hepatitis B virus envelope protein in infectivity of the hepatitis delta virion. J. Virol. 1993, 67, 366–372. [Google Scholar] [PubMed]
- Sureau, C.; Guerra, B.; Lee, H. The middle hepatitis B virus envelope protein is not necessary for infectivity of hepatitis delta virus. J. Virol. 1994, 68, 4063–4066. [Google Scholar] [PubMed]
- Lempp, F.A.; Ni, Y.; Urban, S. Hepatitis delta virus: Insights into a peculiar pathogen and novel treatment options. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Freitas, N.; Abe, K.; Cunha, C.; Menne, S.; Gudima, S.O. Support of the infectivity of hepatitis delta virus particles by the envelope proteins of different genotypes of hepatitis B virus. J. Virol. 2014, 88, 6255–6267. [Google Scholar] [CrossRef] [PubMed]
- Gudima, S.; He, Y.; Meier, A.; Chang, J.; Chen, R.; Jarnik, M.; Nicolas, E.; Bruss, V.; Taylor, J. Assembly of hepatitis delta virus: Particle characterization, including the ability to infect primary human hepatocytes. J. Virol. 2007, 81, 3608–3617. [Google Scholar] [CrossRef] [PubMed]
- Jaoude, G.A.; Sureau, C. Role of the antigenic loop of the hepatitis B virus envelope proteins in infectivity of hepatitis delta virus. J. Virol. 2005, 79, 10460–10466. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.Y.; Chao, M.; Taylor, J. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: Role of delta antigen. J. Virol. 1989, 63, 1945–1950. [Google Scholar] [PubMed]
- Glenn, J.S.; Taylor, J.M.; White, J.M. In vitro-synthesized hepatitis delta virus RNA initiates genome replication in cultured cells. J. Virol. 1990, 64, 3104–3107. [Google Scholar] [PubMed]
- Sureau, C.; Moriarty, A.M.; Thornton, G.B.; Lanford, R.E. Production of infectious hepatitis delta virus in vitro and neutralization with antibodies directed against hepatitis B virus pre-S antigens. J. Virol. 1992, 66, 1241–1245. [Google Scholar] [PubMed]
- Wu, J.C.; Chen, P.J.; Kuo, M.Y.; Lee, S.D.; Chen, D.S.; Ting, L.P. Production of hepatitis delta virus and suppression of helper hepatitis B virus in a human hepatoma cell line. J. Virol. 1991, 65, 1099–1104. [Google Scholar] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Kakinuma, S.; Asahina, Y.; Kamiya, A.; Miyoshi, M.; Tsunoda, T.; Nitta, S.; Asano, Y.; Nagata, H.; Otani, S.; et al. Human induced pluripotent stem cell-derived hepatic cell lines as a new model for host interaction with hepatitis B virus. Sci. Rep. 2016, 6, 29358. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Urban, S. Stem cell-derived hepatocytes: A promising novel tool to study hepatitis B virus infection. J. Hepatol. 2017, 66, 473–475. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, F.; Mitani, S.; Yamamoto, T.; Takayama, K.; Tachibana, M.; Watashi, K.; Wakita, T.; Iijima, S.; Tanaka, Y.; Mizuguchi, H. Human induced-pluripotent stem cell-derived hepatocyte-like cells as an in vitro model of human hepatitis B virus infection. Sci. Rep. 2017, 7, 45698. [Google Scholar] [CrossRef] [PubMed]
- Shlomai, A.; Schwartz, R.E.; Ramanan, V.; Bhatta, A.; de Jong, Y.P.; Bhatia, S.N.; Rice, C.M. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc. Natl. Acad. Sci. USA 2014, 111, 12193–12198. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Carpentier, A.; Cheng, X.; Block, P.D.; Zhao, Y.; Zhang, Z.; Protzer, U.; Jake Liang, T. Human stem cell-derived hepatocytes as a model for hepatitis B virus infection, spreading and virus-host interactions. J. Hepatol. 2016, 66, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Alfaiate, D.; Lucifora, J.; Abeywickrama-Samarakoon, N.; Michelet, M.; Testoni, B.; Cortay, J.C.; Sureau, C.; Zoulim, F.; Deny, P.; Durantel, D. Hdv RNA replication is associated with HBV repression and interferon-stimulated genes induction in super-infected hepatocytes. Antivir. Res. 2016, 136, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Nußbaum, L.; Rieble, L.; Ni, Y.; Urban, S. Screening an FDA-approved drug library on a cell line that supports the full lifecycle of hepatitis deltavirus. J. Hepatol. 2016, 64, S388. [Google Scholar] [CrossRef]
- Rizzetto, M.; Canese, M.G.; Gerin, J.L.; London, W.T.; Sly, D.L.; Purcell, R.H. Transmission of the hepatitis B virus-associated delta antigen to chimpanzees. J. Infect. Dis. 1980, 141, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Negro, F.; Bergmann, K.F.; Baroudy, B.M.; Satterfield, W.C.; Popper, H.; Purcell, R.H.; Gerin, J.L. Chronic hepatitis D virus (HDV) infection in hepatitis b virus carrier chimpanzees experimentally superinfected with HDV. J. Infect. Dis. 1988, 158, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Sureau, C.; Taylor, J.; Chao, M.; Eichberg, J.W.; Lanford, R.E. Cloned hepatitis delta virus cDNA is infectious in the chimpanzee. J. Virol. 1989, 63, 4292–4297. [Google Scholar] [PubMed]
- Ponzetto, A.; Cote, P.J.; Popper, H.; Hoyer, B.H.; London, W.T.; Ford, E.C.; Bonino, F.; Purcell, R.H.; Gerin, J.L. Transmission of the hepatitis B virus-associated delta agent to the eastern Woodchuck. Proc. Natl. Acad. Sci. USA 1984, 81, 2208–2212. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Hu, H.; Liu, Y.; Jing, Z.; Li, W. Woodchuck sodium taurocholate cotransporting polypeptide supports low-level hepatitis B and D virus entry. Virology 2017, 505, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, M.; Kosinska, A.; Schumann, A.; Brovko, O.; Walker, A.; Lu, M.; Johrden, L.; Mayer, A.; Wildner, O.; Roggendorf, M. Prime/boost immunization with DNA and adenoviral vectors protects from hepatitis D virus (HDV) infection after simultaneous infection with HDV and Woodchuck hepatitis virus. J. Virol. 2013, 87, 7708–7716. [Google Scholar] [CrossRef] [PubMed]
- Guilhot, S.; Huang, S.N.; Xia, Y.P.; La Monica, N.; Lai, M.M.; Chisari, F.V. Expression of the hepatitis delta virus large and small antigens in transgenic mice. J. Virol. 1994, 68, 1052–1058. [Google Scholar] [PubMed]
- Polo, J.M.; Jeng, K.S.; Lim, B.; Govindarajan, S.; Hofman, F.; Sangiorgi, F.; Lai, M.M. Transgenic mice support replication of hepatitis delta virus RNA in multiple tissues, particularly in skeletal muscle. J. Virol. 1995, 69, 4880–4887. [Google Scholar] [PubMed]
- Chang, J.; Sigal, L.J.; Lerro, A.; Taylor, J. Replication of the human hepatitis delta virus genome is initiated in mouse hepatocytes following intravenous injection of naked DNA or RNA sequences. J. Virol. 2001, 75, 3469–3473. [Google Scholar] [CrossRef] [PubMed]
- Bordier, B.B.; Ohkanda, J.; Liu, P.; Lee, S.Y.; Salazar, F.H.; Marion, P.L.; Ohashi, K.; Meuse, L.; Kay, M.A.; Casey, J.L.; et al. In vivo antiviral efficacy of prenylation inhibitors against hepatitis delta virus. J. Clin. Investig. 2003, 112, 407–414. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Cao, Z.; Mao, F.; Ren, B.; Li, Y.; Li, D.; Li, H.; Peng, B.; Yan, H.; Qi, Y.; et al. Modification of three amino acids in sodium taurocholate cotransporting polypeptide renders mice susceptible to infection with hepatitis D virus in vivo. J. Virol. 2016, 90, 8866–8874. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Ren, B.; Mao, F.; Jing, Z.; Li, Y.; Liu, Y.; Peng, B.; Yan, H.; Qi, Y.; Sun, Y.; et al. Hepatitis D virus infection of mice expressing human sodium taurocholate co-transporting polypeptide. PLoS Pathog. 2015, 11, e1004840. [Google Scholar] [CrossRef] [PubMed]
- Dandri, M.; Lutgehetmann, M. Mouse models of hepatitis B and delta virus infection. J. Immunol. Methods 2014, 410, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Giersch, K.; Helbig, M.; Volz, T.; Allweiss, L.; Mancke, L.V.; Lohse, A.W.; Polywka, S.; Pollok, J.M.; Petersen, J.; Taylor, J.; et al. Persistent hepatitis D virus mono-infection in humanized mice is efficiently converted by hepatitis B virus to a productive co-infection. J. Hepatol. 2014, 60, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Giersch, K.; Allweiss, L.; Volz, T.; Helbig, M.; Bierwolf, J.; Lohse, A.W.; Pollok, J.M.; Petersen, J.; Dandri, M.; Lutgehetmann, M. Hepatitis delta co-infection in humanized mice leads to pronounced induction of innate immune responses in comparison to HBV mono-infection. J. Hepatol. 2015, 63, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.L.; Gerin, J.L. The woodchuck model of HDV infection. Curr. Top. Microbiol. Immunol. 2006, 307, 211–225. [Google Scholar] [PubMed]
- Suarez-Amaran, L.; Usai, C.; Di Scala, M.; Godoy, C.; Ni, Y.; Hommel, M.; Palomo, L.; Segura, V.; Olague, C.; Vales, A.; et al. A new HDV mouse model identifies mitochondrial antiviral signaling protein (MAVS) as a key player in IFN-β induction. J. Hepatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Protzer, U. Viral hepatitis: The bumpy road to animal models for HBV infection. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 327–328. [Google Scholar] [CrossRef] [PubMed]
- Francois-Souquiere, S.; Makuwa, M.; Bisvigou, U.; Kazanji, M. Epidemiological and molecular features of hepatitis B and hepatitis delta virus transmission in a remote rural community in central Africa. Infect. Genet. Evol. 2016, 39, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Ramia, S.; Bahakim, H. Perinatal transmission of hepatitis B virus-associated hepatitis D virus. Ann. Inst. Pasteur Virol. 1988, 139, 285–290. [Google Scholar] [CrossRef]
- Rizzetto, M.; Ciancio, A. Epidemiology of hepatitis D. Semin. Liver Dis. 2012, 32, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Oidovsambuu, O.; Liu, P.; Grosely, R.; Elazar, M.; Winn, V.D.; Fram, B.; Boa, Z.; Dai, H.; Dashtseren, B.; et al. A novel quantitative microarray antibody capture (q-MAC) assay identifies an extremely high HDV prevalence amongst HBV infected mongolians. Hepatology 2016. [Google Scholar] [CrossRef] [PubMed]
- Smedile, A.; Lavarini, C.; Farci, P.; Arico, S.; Marinucci, G.; Dentico, P.; Giuliani, G.; Cargnel, A.; Del Vecchio Blanco, C.; Rizzetto, M. Epidemiologic patterns of infection with the hepatitis B virus-associated delta agent in Italy. Am. J. Epidemiol. 1983, 117, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, G.; Giuberti, T.; Verucchi, G.; Levantesi, M.; Sacchini, D.; Fattovich, G.; Madonia, S.; Fasano, M.; Gavrila, C.; Nardi, A.; et al. Epidemiological evolution of chronic hepatitis delta in italy. An analysis of the master-B cohort. Dig. Liver Dis. 46, e12–e13. [CrossRef]
- Heidrich, B.; Deterding, K.; Tillmann, H.L.; Raupach, R.; Manns, M.P.; Wedemeyer, H. Virological and clinical characteristics of delta hepatitis in central Europe. J. Viral Hepat. 2009, 16, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Alfaiate, D.; Deny, P.; Durantel, D. Hepatitis delta virus: From biological and medical aspects to current and investigational therapeutic options. Antivir.Res. 2015, 122, 112–129. [Google Scholar] [CrossRef] [PubMed]
- Gish, R.G.; Yi, D.H.; Kane, S.; Clark, M.; Mangahas, M.; Baqai, S.; Winters, M.A.; Proudfoot, J.; Glenn, J.S. Coinfection with hepatitis B and D: Epidemiology, prevalence and disease in patients in northern California. J. Gastroenterol. Hepatol. 2013, 28, 1521–1525. [Google Scholar] [CrossRef] [PubMed]
- Kushner, T.; Serper, M.; Kaplan, D.E. Delta hepatitis within the veterans affairs medical system in the United States: Prevalence, risk factors, and outcomes. J. Hepatol. 2015, 63, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.; Zhang, F.; Lin, S.; He, H.; Liu, Y.; Zhang, J.; Xu, Y.; Yi, J.; Chen, Y.; Liu, H.; et al. Epidemiological, clinical and histological characteristics of HBV/HDV co-infection: A retrospective cross-sectional study in Guangdong, China. PLoS ONE 2014, 9, e115888. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Gu, Y.; Sun, L.; Yang, Y.; Wang, F.; Li, Y.; Bi, S. Development of a hepatitis delta virus antibody assay for study of the prevalence of HDV among individuals infected with hepatitis B virus in China. J. Med. Virol. 2012, 84, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.J.; Li, L.; Zhang, Y.Y.; Song, P.H. Hepatitis D virus infection in liver tissues of patients with hepatitis B in china. Chin. Med. J. (Engl.) 1992, 105, 204–208. [Google Scholar] [PubMed]
- Zhang, J.Y.; Jin, Z.H.; Wang, C.J. A seroepidemiological study on hepatitis D virus (HDV) infection in Henan province, China. Zhonghua Liu Xing Bing Xue Za Zhi 1995, 16, 365–368. [Google Scholar] [PubMed]
- Te, H.S.; Jensen, D.M. Epidemiology of hepatitis B and C viruses: A global overview. Clin. Liver Dis. 2010, 14, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Hayakawa, E.; Sminov, A.V.; Rossina, A.L.; Ding, X.; Huy, T.T.; Sata, T.; Uchaikin, V.F. Molecular epidemiology of hepatitis B, C, D and E viruses among children in Moscow, Russia. J. Clin. Virol. 2004, 30, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Kozhanova, T.V.; Il’chenko, L.; Mikhailov, M.I. Viral hepatitis delta. Is there the delta infection problem in the Russian Federation? Eksp. Naia Klin. Gastroenterol. Exp. Clin. Gastroenterol. 2014, 4–12. [Google Scholar]
- Semenov, A.V. Prevalence of seronegative hepatitis D among patients with chronic viral hepatitis B. Zhurnal Mikrobiol. Epidemiol. I Immunobiol. 2012, 106–109. [Google Scholar]
- Manesis, E.K.; Vourli, G.; Dalekos, G.; Vasiliadis, T.; Manolaki, N.; Hounta, A.; Koutsounas, S.; Vafiadis, I.; Nikolopoulou, G.; Giannoulis, G.; et al. Prevalence and clinical course of hepatitis delta infection in Greece: A 13-year prospective study. J. Hepatol. 2013, 59, 949–956. [Google Scholar] [CrossRef] [PubMed]
- El Bouzidi, K.; Elamin, W.; Kranzer, K.; Irish, D.N.; Ferns, B.; Kennedy, P.; Rosenberg, W.; Dusheiko, G.; Sabin, C.A.; Smith, B.C.; et al. Hepatitis delta virus testing, epidemiology and management: A multicentre cross-sectional study of patients in London. J. Clin. Virol. 2015, 66, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, M.; Rosina, F.; Saracco, G.; Bellando, P.C.; Actis, G.C.; Bonino, F.; Smedile, A.; Trinchero, P.; Sansalvadore, F.; Pintus, C.; et al. Treatment of chronic delta hepatitis with α-2 recombinant interferon. J. Hepatol. 1986, 3, S229–S233. [Google Scholar] [CrossRef]
- Wedemeyer, H.; Yurdaydin, C.; Dalekos, G.N.; Erhardt, A.; Cakaloglu, Y.; Degertekin, H.; Gurel, S.; Zeuzem, S.; Zachou, K.; Bozkaya, H.; et al. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N. Engl. J. Med. 2011, 364, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Lampertico, P.; Agarwal, K.; Berg, T.; Buti, M.; Janssen, H.L.A.; Papatheodoridis, G.; Zoulim, F.; Tacke, F. EASL 2017 clinical practice guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Soriano, V.; Vispo, E.; Sierra-Enguita, R.; Mendoza, C.; Fernandez-Montero, J.V.; Labarga, P.; Barreiro, P. Efficacy of prolonged tenofovir therapy on hepatitis delta in HIV-infected patients. AIDS 2014, 28, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.A.; Wedemeyer, H.; Harrison, P.M. Hepatitis delta virus. Lancet 2011, 378, 73–85. [Google Scholar] [CrossRef]
- Wedemeyer, H.; Manns, M.P. Epidemiology, pathogenesis and management of hepatitis D: Update and challenges ahead. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Wranke, A.; Heidrich, B.; Hardtke, S.; Wedemeyer, H. Current management of HBV/HDV coinfection and future perspectives. Curr. Hepatol. Rep. 2015, 14, 284–292. [Google Scholar] [CrossRef]
- Zoulim, F.; Carosi, G.; Greenbloom, S.; Mazur, W.; Nguyen, T.; Jeffers, L.; Brunetto, M.; Yu, S.; Llamoso, C. Quantification of HBsAg in nucleos(t)ide-naive patients treated for chronic hepatitis B with entecavir with or without tenofovir in the be-low study. J. Hepatol. 2015, 62, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Heidrich, B.; Yurdaydin, C.; Kabacam, G.; Ratsch, B.A.; Zachou, K.; Bremer, B.; Dalekos, G.N.; Erhardt, A.; Tabak, F.; Yalcin, K.; et al. Late HDV RNA relapse after peginterferon α-based therapy of chronic hepatitis delta. Hepatology 2014, 60, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, H.; Yurdaydin, C.; Ernst, S.; Caruntu, F.A.; Curescu, M.G.; Yalcin, K.; Akarca, U.S.; Gürel, S.; Zeuzem, S.; Erhardt, A.; et al. O4 prolonged therapy of hepatitis delta for 96 weeks with pegylated-interferon-α-2A plus tenofovir or placebo does not prevent HDV RNA relapse after treatment: The HIDIT-2 study. J. Hepatol. 2014, 60, S2–S3. [Google Scholar] [CrossRef]
- Lai, M.M. RNA replication without RNA-dependent RNA polymerase: Surprises from hepatitis delta virus. J. Virol. 2005, 79, 7951–7958. [Google Scholar] [CrossRef] [PubMed]
- Yen, L.; Magnier, M.; Weissleder, R.; Stockwell, B.R.; Mulligan, R.C. Identification of inhibitors of ribozyme self-cleavage in mammalian cells via high-throughput screening of chemical libraries. RNA 2006, 12, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Wooddell, C.I.; Rozema, D.B.; Hossbach, M.; John, M.; Hamilton, H.L.; Chu, Q.; Hegge, J.O.; Klein, J.J.; Wakefield, D.H.; Oropeza, C.E.; et al. Hepatocyte-targeted RNAi therapeutics for the treatment of chronic hepatitis B virus infection. Mol. Ther. 2013, 21, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Chan, H.L.; Liu, S.; Given, B.; Schluep, T.; Hamilton, J.; Lai, C.L.; Locarnini, S.; Lau, J.Y.; Ferrari, C.; et al. Arc-520 produces deep and durable knockdown of viral antigens and DNA in a phase II study in patients with chronic hepatitis B. Hepatology 2015, 62, 1385A. [Google Scholar]
- Sureau, C.; Negro, F. The hepatitis delta virus: Replication and pathogenesis. J. Hepatol 2016, 64, S102–S116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ni, Y.; Filzmayer, C.; Urban, S. MDA5 mediated activation of innate immune responses by Hepatitis D virus infection. In Proceedings of the International Meeting on the Molecular Biology of Hepatitis B Viruses, Seoul, South Korea, 21–24 September 2016. submitted. [Google Scholar]
- Elazar, M.; Glenn, J.S. Emerging concepts for the treatment of hepatitis delta. Curr. Opin. Virol. 2017, 24, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.; Canini, L.; Dahari, H.; Zhao, X.; Uprichard, S.L.; Haynes-Williams, V.; Winters, M.A.; Subramanya, G.; Cooper, S.L.; Pinto, P.; et al. Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: A proof-of-concept randomised, double-blind, placebo-controlled phase 2a trial. Lancet. Infect. Dis. 2015, 15, 1167–1174. [Google Scholar] [CrossRef]
- Yurdaydin, C.; Idilman, R.; Choong, I.; Kalkan, C.; Keskin, O.; Karakaya, M.F.; Tuzun, A.E.; Karatayli, E.; Bozdayi, M.; Cory, D.; et al. O118: Optimizing the prenylation inhibitor lonafarnib using ritonavir boosting in patients with chronic delta hepatitis. J. Hepatol. 2015, 62, S252. [Google Scholar] [CrossRef]
- Koh, C.; Yurdaydin, C.; Cooper, S.L.; Cory, D.; Dahari, H.; Haynes-Williams, V.; Winters, M.; Bys, M.; Choong, I.; Idilman, R.; et al. Prenylation inhibition with lonafarnib decreases hepatitis D levels in humans. Hepatology 2014, 60, 1092A. [Google Scholar]
- Bazinet, M.; Pantea, V.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Vaillant, A. Hdv2 o-09: Rep 2139 monotherapy and combination therapy with pegylated interferon: Safety and potent reduction of HBsAg and HDV RNA in caucasian patients with chronic HBV/HDV co-infection. J. Viral Hepat. 2015, 22, 5–6. [Google Scholar]
- Poutay, D.; Sabra, M.; Abou-Jaoude, G.; Chemin, I.; Trepo, C.; Vaillant, A.; Sureau, C. P177: Nucleic acid polymers are efficient in blocking hepatitis delta virus entry in vitro. J. Viral Hepat. 2015, 22, 107. [Google Scholar]
- Bazinet, M.; Pantea, V.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Albrecht, J.; Schmid, P.; Karimzadeh, H.; Roggendorf, M.; Vaillant, A. Update on the safety and efficacy of rep 2139 mono-therapy and subsequent combination therapy with pegylated interferon α-2a in chronic HBV/HDV co-infection in caucasian patients. Hepatology 2015, 62, 222A. [Google Scholar]
- Vaillant, A. Nucleic acid polymers: Broad spectrum antiviral activity, antiviral mechanisms and optimization for the treatment of hepatitis B and hepatitis D infection. Antivir. Res. 2016, 133, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Yust-Katz, S.; Liu, D.; Yuan, Y.; Liu, V.; Kang, S.; Groves, M.; Puduvalli, V.; Levin, V.; Conrad, C.; Colman, H.; et al. Phase 1/1b study of lonafarnib and temozolomide in patients with recurrent or temozolomide refractory glioblastoma. Cancer 2013, 119, 2747–2753. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, M.; Ciancio, A. The prenylation inhibitor, lonafarnib: A new therapeutic strategy against hepatitis delta. Lancet Infect. Dis. 2015, 15, 1119–1120. [Google Scholar] [CrossRef]
- Bordier, B.B.; Marion, P.L.; Ohashi, K.; Kay, M.A.; Greenberg, H.B.; Casey, J.L.; Glenn, J.S. A prenylation inhibitor prevents production of infectious hepatitis delta virus particles. J. Virol. 2002, 76, 10465–10472. [Google Scholar] [CrossRef] [PubMed]
- Mijimolle, N.; Velasco, J.; Dubus, P.; Guerra, C.; Weinbaum, C.A.; Casey, P.J.; Campuzano, V.; Barbacid, M. Protein farnesyltransferase in embryogenesis, adult homeostasis, and tumor development. Cancer Cell 2005, 7, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef] [PubMed]
- Palsuledesai, C.C.; Distefano, M.D. Protein prenylation: Enzymes, therapeutics, and biotechnology applications. ACS Chem. Biol. 2015, 10, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Noordeen, F.; Scougall, C.A.; Grosse, A.; Qiao, Q.; Ajilian, B.B.; Reaiche-Miller, G.; Finnie, J.; Werner, M.; Broering, R.; Schlaak, J.F.; et al. Therapeutic antiviral effect of the nucleic acid polymer rep 2055 against persistent duck hepatitis B virus infection. PLoS ONE 2015, 10, e0140909. [Google Scholar] [CrossRef] [PubMed]
- Noordeen, F.; Vaillant, A.; Jilbert, A.R. Nucleic acid polymers prevent the establishment of duck hepatitis B virus infection in vivo. Antimicrob. Agents Chemother. 2013, 57, 5299–5306. [Google Scholar] [CrossRef] [PubMed]
- Al-Mahtab, M.; Bazinet, M.; Vaillant, A. Safety and efficacy of nucleic acid polymers in monotherapy and combined with immunotherapy in treatment-naive Bangladeshi patients with HBeAg+ chronic hepatitis B infection. PLoS ONE 2016, 11, e0156667. [Google Scholar] [CrossRef] [PubMed]
- Replicor. Available online: http://replicor.com/science/conference-presentations/ (accessed on 28 June 2017).
- Guillot, C.; Martel, N.; Berby, F.; Bordes, I.; Hantz, O.; Blanchet, M.; Sureau, C.; Vaillant, A.; Chemin, I. Inhibition of hepatitis B viral entry by nucleic acid polymers in HepaRG cells and primary human hepatocytes. PLoS ONE 2017, 12, e0179697. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Cannie, I.; Urban, S. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J. Virol. 2005, 79, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Dandri, M.; Mier, W.; Lutgehetmann, M.; Volz, T.; von Weizsacker, F.; Haberkorn, U.; Fischer, L.; Pollok, J.M.; Erbes, B.; et al. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat. Biotechnol. 2008, 26, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Schieck, A.; Ni, Y.; Mier, W.; Urban, S. Fine mapping of pre-s sequence requirements for hepatitis B virus large envelope protein-mediated receptor interaction. J. Virol. 2010, 84, 1989–2000. [Google Scholar] [CrossRef] [PubMed]
- Meier, A.; Mehrle, S.; Weiss, T.S.; Mier, W.; Urban, S. Myristoylated PRES1-domain of the hepatitis B virus l-protein mediates specific binding to differentiated hepatocytes. Hepatology 2013, 58, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Schieck, A.; Schulze, A.; Gahler, C.; Muller, T.; Haberkorn, U.; Alexandrov, A.; Urban, S.; Mier, W. Hepatitis B virus hepatotropism is mediated by specific receptor recognition in the liver and not restricted to susceptible hosts. Hepatology 2013, 58, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Nkongolo, S.; Ni, Y.; Lempp, F.A.; Kaufman, C.; Lindner, T.; Esser-Nobis, K.; Lohmann, V.; Mier, W.; Mehrle, S.; Urban, S. Cyclosporin a inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J. Hepatol. 2014, 60, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Haag, M.; Hofmann, U.; Murdter, T.E.; Heinkele, G.; Leuthold, P.; Blank, A.; Haefeli, W.E.; Alexandrov, A.; Urban, S.; Schwab, M. Quantitative bile acid profiling by liquid chromatography quadrupole time-of-flight mass spectrometry: Monitoring hepatitis B therapy by a novel Na(+)-taurocholate cotransporting polypeptide inhibitor. Anal. Bioanal. Chem. 2015, 407, 6815–6825. [Google Scholar] [CrossRef] [PubMed]
- Slijepcevic, D.; Kaufman, C.; Wichers, C.G.; Gilglioni, E.H.; Lempp, F.A.; Duijst, S.; de Waart, D.R.; Elferink, R.P.; Mier, W.; Stieger, B.; et al. Impaired uptake of conjugated bile acids and hepatitis B virus PRES1-binding in Na(+) -taurocholate cotransporting polypeptide knockout mice. Hepatology 2015, 62, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Vaz, F.M.; Paulusma, C.C.; Huidekoper, H.; de Ru, M.; Lim, C.; Koster, J.; Ho-Mok, K.; Bootsma, A.H.; Groen, A.K.; Schaap, F.G.; et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: Conjugated hypercholanemia without a clear clinical phenotype. Hepatology 2015, 61, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.; Mao, M.; Guo, L.; Chen, F.P.; Wen, W.R.; Song, Y.Z. Clinical and molecular study of a pediatric patient with sodium taurocholate cotransporting polypeptide deficiency. Exp. Ther. Med. 2016, 12, 3294–3300. [Google Scholar] [CrossRef] [PubMed]
- Blank, A.; Markert, C.; Hohmann, N.; Carls, A.; Mikus, G.; Lehr, T.; Alexandrov, A.; Haag, M.; Schwab, M.; Urban, S.; et al. First-in-human application of the first-in-class hepatitis B and hepatitis D virus entry inhibitor myrcludex B. J. Hepatol. 2016, 65, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Bogomolov, P.; Voronkova, N.; Allweiss, L.; Dandri, M.; Schwab, M.; Lempp, F.A.; Haag, M.; Wedemeyer, H.; Alexandrov, A.; Urban, S. A proof-of-concept phase 2a clinical trial with HBV/HDV entry inhibitor myrcludex b. Hepatology 2014, 60, 1279A–1280A. [Google Scholar]
- Bogomolov, P.; Alexandrov, A.; Voronkova, N.; Macievich, M.; Kokina, K.; Petrachenkova, M.; Lehr, T.; Lempp, F.A.; Wedemeyer, H.; Haag, M.; et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex b—First results of a phase Ib/IIa study. J. Hepatol. 2016, 65, 490–498. [Google Scholar] [CrossRef] [PubMed]
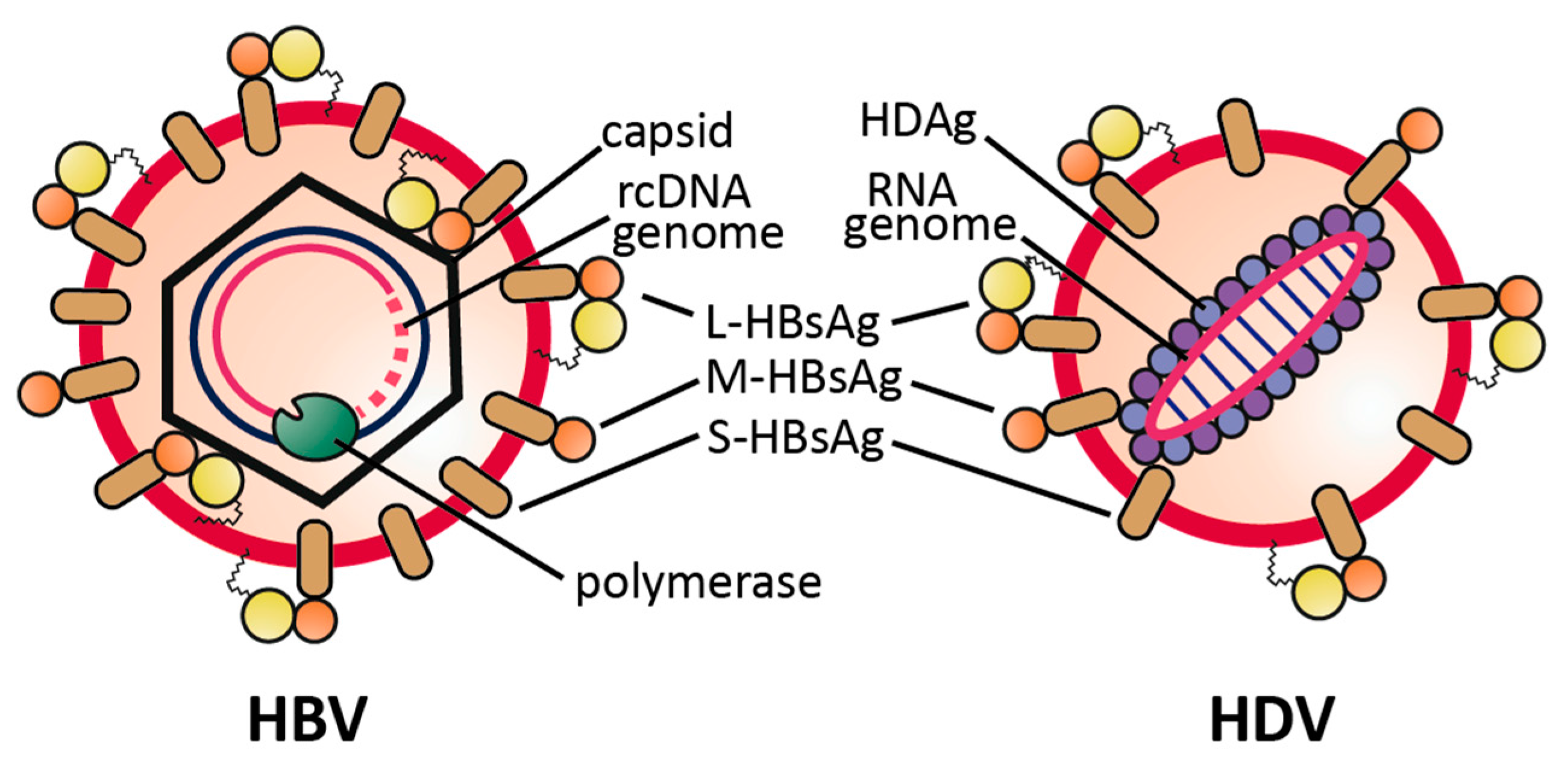

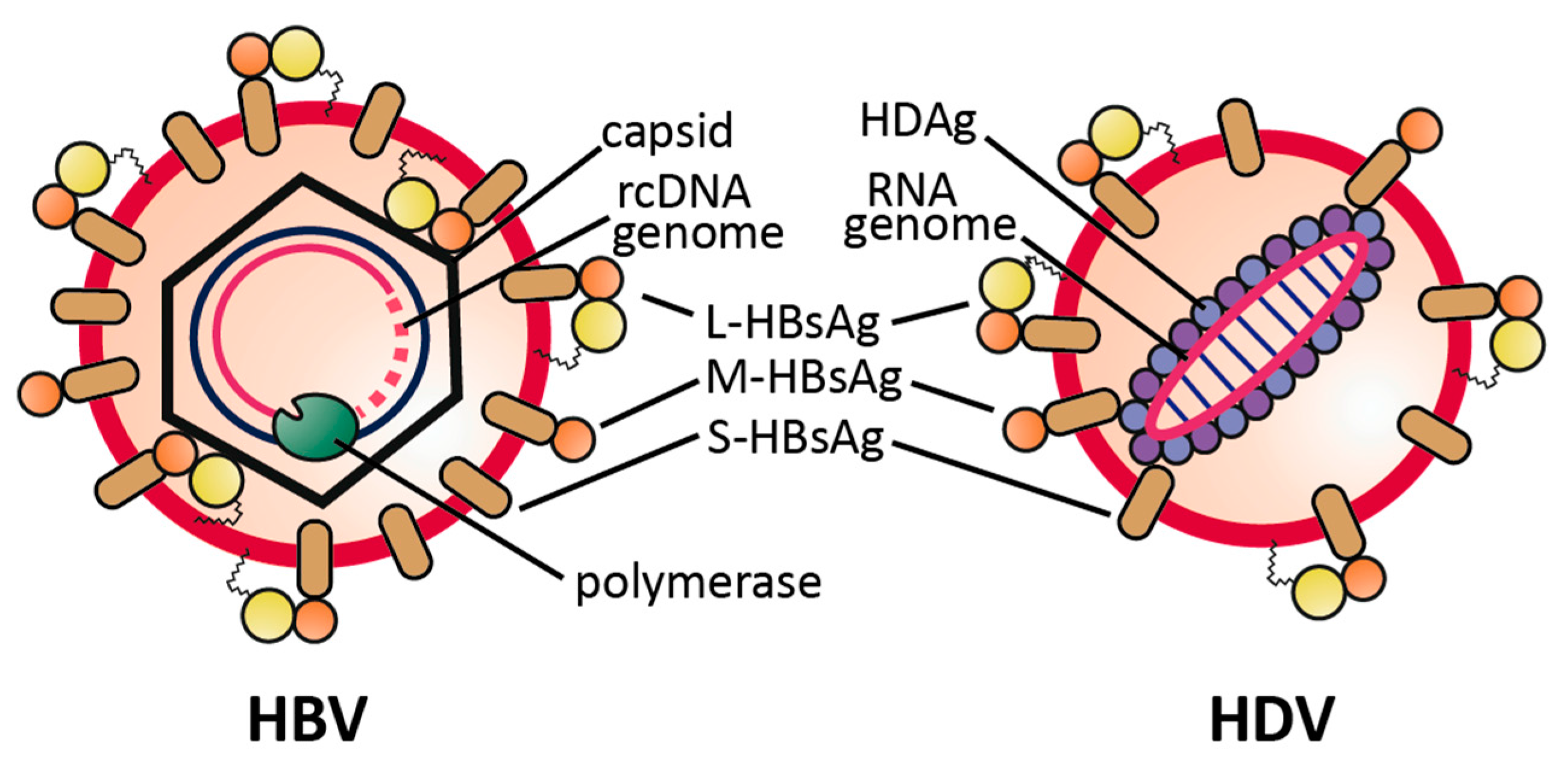{kind=link}
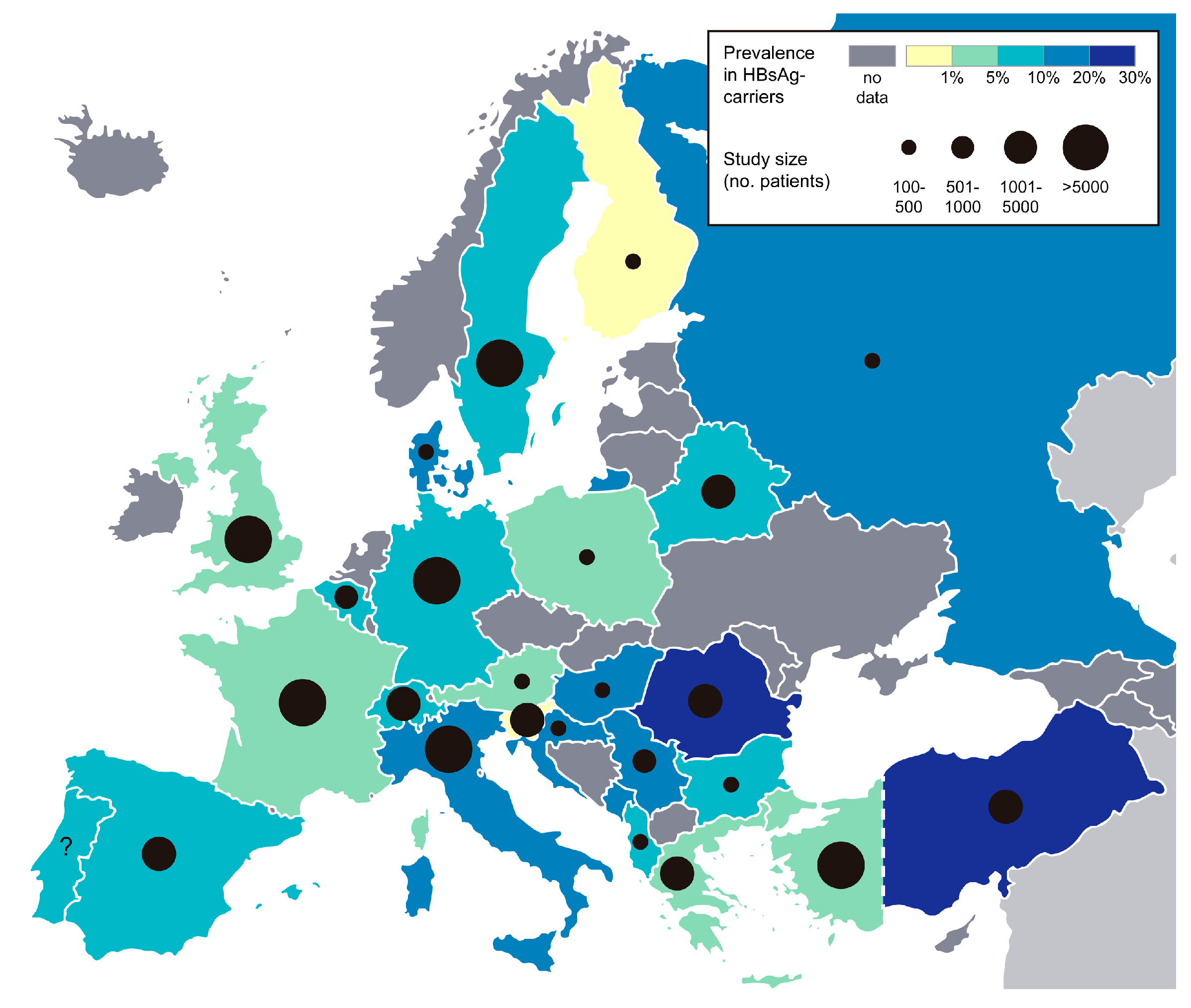{kind=link}
| Hepatitis B Virus (HBV) | Hepatitis Delta Virus (HDV) | |
|---|---|---|
| Family Genus | Hepadnaviridae Orthohepadnaviruses | Unassigned Deltavirus |
| Genome | relaxed circular partially double-stranded DNA ca. 3.2 kbp | single-stranded (−) RNA 1.7 kb |
| Virus-Encoded Proteins | HBcAg, HBeAg, pol, HBx, L-/M-/S-HBsAg | L-/S-HDAg |
| Cellular Receptors | HSPG, hNTCP | HSPG, hNTCP |
| Chronically Infected Individuals Worldwide | 240 million | 15–20 million (co-infected with HBV) |
| Vaccine Available | Yes | HBV vaccine |
| Curative Therapy Available | No | No |
| Model | Entry | Repli-Cation | Progeny Release | Pros | Cons | Ref. |
|---|---|---|---|---|---|---|
| Transfected HuH7 | − | + | + 1 | Easy access/handling | Does not reflect authentic infection | [42,43,44,45] |
| PHH | + | + | + 2 | Natural host; most physiological | Limited availability; High donor-to-donor variability | [39,40,41] |
| HepaRG | + | + | + 2 | Exhibit some hepatic function; fully support innate immunity | Requires elaborate differentiation protocol; only hepatocyte-like cells are susceptible | [46] |
| HuH7/HepG2-hNTCP | + | + | + 2 | Easy access/handling; efficient infection | Only partially resemble hepatocytes | [13] |
| Stem-cell derived hepatocytes | + | + | ? | Same donor can be used for different experiments; physiologically close to PHH | Requires elaborate differentiation protocol; ethical concerns in some countries | [47,48,49,50,51] |
| Model | Entry | HBV Co-Infection | Immuno-Competent | Pros | Cons | Ref. |
|---|---|---|---|---|---|---|
| Chimpanzee | + | + | + | Immunocompetent infection model | Ethical considerations | [54,55,56] |
| Woodchuck | +/- | + 1 | + | Immunocompetent infection model | Relies on WHV rather than HBV envelope | [57,59,69] |
| HDV/HDAg-transgenic mice | - | - | + | Stable mouse lines; tissue-specific expression can be analyzed | No virus infection/spread | [60,61] |
| Hydrodynamic injection | - | - | + | Fast and easy way to deliver nucleic acids to the liver | No virus infection/spread; harmful to the animal | [62,63] |
| AAV-HDV transduction | - | - | + | Allows studies of host virus interactions in vivo | No authentic infection system | [70] |
| Liver-chimeric mice | + | + | - | Authentic HBV/HDV infection; allows for long-term infections | No adaptive immunity; very sophisticated model | [66,67] |
| hNTCP mice | + | - | + | Immunocompetent transgenic infection system | Low infection rates, transient infection | [64,65] |
| Macaque/pig hNTCP-transduced | + | + | + | Immunocompetent models allowing authentic infection | Only in vitro data available so far; sophisticated animal models | [15,71] |
| Drug | Target | Mechanism | Clinical Trial Identifier(s) | Company |
|---|---|---|---|---|
| Lonafarnib | Farnesyl transferase | Assembly inhibition | NCT02430181 NCT02430194 | Eiger Bio (Palo Alto, CA, USA) |
| Nucleic acid polymers | HBsAg? | HDV release inhibition? | NCT02233075 NCT02876419 | Replicor (Montreal, QC, Canada) |
| Myrcludex B | hNTCP | Entry inhibition | NCT02637999 | Myr GmbH (Burgwedel, NI, Germany) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lempp, F.A.; Urban, S. Hepatitis Delta Virus: Replication Strategy and Upcoming Therapeutic Options for a Neglected Human Pathogen. Viruses 2017, 9, 172. https://doi.org/10.3390/v9070172
Lempp FA, Urban S. Hepatitis Delta Virus: Replication Strategy and Upcoming Therapeutic Options for a Neglected Human Pathogen. Viruses. 2017; 9(7):172. https://doi.org/10.3390/v9070172
Chicago/Turabian StyleLempp, Florian A., and Stephan Urban. 2017. "Hepatitis Delta Virus: Replication Strategy and Upcoming Therapeutic Options for a Neglected Human Pathogen" Viruses 9, no. 7: 172. https://doi.org/10.3390/v9070172
APA StyleLempp, F. A., & Urban, S. (2017). Hepatitis Delta Virus: Replication Strategy and Upcoming Therapeutic Options for a Neglected Human Pathogen. Viruses, 9(7), 172. https://doi.org/10.3390/v9070172