1. Introduction
Western equine encephalitis virus (WEEV) is a member of the alphavirus genus of the family
Togaviridae. Alphaviruses, Venezuelan equine encephalitis virus (VEEV), Eastern equine encephalitis virus (EEEV), and WEEV are grouped geographically as New World viruses capable of causing disease in both equids and humans, exhibiting overt encephalitic features in a significant number of cases. WEEV causes periodic epizootic outbreaks in Western and Central North America and is maintained in an enzootic cycle between mosquitos and birds or rodents [
1] Humans are usually infected as a result of close proximity to infected equines and by being bitten by an infected mosquito. In humans, WEEV infections are generally asymptomatic but may result in the onset of flu-like symptoms; fever, malaise, headaches, vomiting, and nausea. In a minority of cases, symptoms progress to weakness, confusion, seizures, and encephalitis, and may lead to coma and death. The case fatality rate is typically 3–4%, when infection is acquired from a natural enzootic cycle, particularly affecting infants and the elderly [
2]. Survivors of WEEV infection can be left with persisting neurological sequelae, requiring significant healthcare intervention [
3,
4,
5]. During epizootic outbreaks, the case fatality rate is reported to rise to 8–15% [
6], and after accidental laboratory exposures to WEEV by the aerosol route it has been documented to be as high as 40% (2/5) [
7]. Exposure by the aerosol route may facilitate neuroinvasion of WEEV, potentially the cause of a marked increase in mortality, and is in-part why WEEV is considered a potential biothreat agent [
8]. WEEV, as well as VEEV and EEEV, are listed as Category B agents by the National Institute of Allergy and Infectious Diseases, and the Centre for Disease Control [
9,
10]. Additionally, there are currently no licensed vaccines or antiviral therapies for the prevention or treatment of these viruses in humans. WEEV is therefore of significant concern in relation to both natural outbreaks and its potential use as a biothreat agent. Research directed at characterizing appropriate models of disease, understanding the pathogenesis of infection and ultimately identifying effective medical countermeasures to WEEV is required. The use of in vivo models is currently the most effective holistic approach to understanding the outcome or effect of medical countermeasures, offering well-defined experimental parameters. As with many in vivo models of human disease, a small animal model is very often the foundation for non-human primate studies, and progressively, human clinical trials.
A small number of studies have previously investigated rodents and non-human primates [
11] as models of WEEV infection and disease. In hamsters, the intranasal or intraperitoneal routes of infection were lethal with rapid neuroinvasion, and peak viral titres were observed in key tissues between 2–4 days post-challenge [
12]. A number of mouse strains have been investigated, utilizing both in-bred and out-bred strains of different ages and genders, and with parenteral and aerosol routes of exposure. Early investigations examined the use of Swiss Rockefeller and National Institute of Health Swiss mice and demonstrated age-dependent susceptibility [
13,
14]. More recently, detailed host factor assessments confirmed the importance of age on susceptibility to WEEV in mice and also demonstrated that gender and genetic background of the host were important factors for survival [
15]. As well as host factors, the route, virulence, and dose of virus also influences the outcome of infection. High- and low-virulence WEEV strains have been reported to infect Balb/c, C57BL/6, and CD1 mice, although spread to the central nervous system, severity of overt neurological signs, and mortality depended on the dose of virus and the route of exposure [
16,
17,
18,
19,
20].
The known hazard to humans from aerosol infection necessitates a model that uses this route. However, studies often use intra-nasal inoculation as a surrogate for aerosol exposure. In a recent study examining the clinical course of a closely-related Alphavirus, EEEV, adult Balb/c mice were infected by the intra-nasal and aerosol routes, yielding 100% lethality, with rapid neuroinvasion and acute onset of clinical signs [
21]. In this study, a number of parameters were comparable between the two exposure routes (weight, clinical scores, cytokine expression), although importantly, virus was detected in the brain of aerosol exposed mice at 6 h post-infection. In contrast, virus was not detected in the brain of intra-nasally exposed mice until 24 h post-infection. Such rapid neuroinvasion suggests that aerosol exposure may induce a precipitous onset of encephalitic disease. In this report, the course of WEEV infection in adult Balb/c mice exposed by the aerosol route is presented. This includes viral load and distribution, establishing a clinical scoring system and determination of the Median Lethal Dose (MLD), as well as histopathological and immuno-histochemistry findings.
3. Discussion
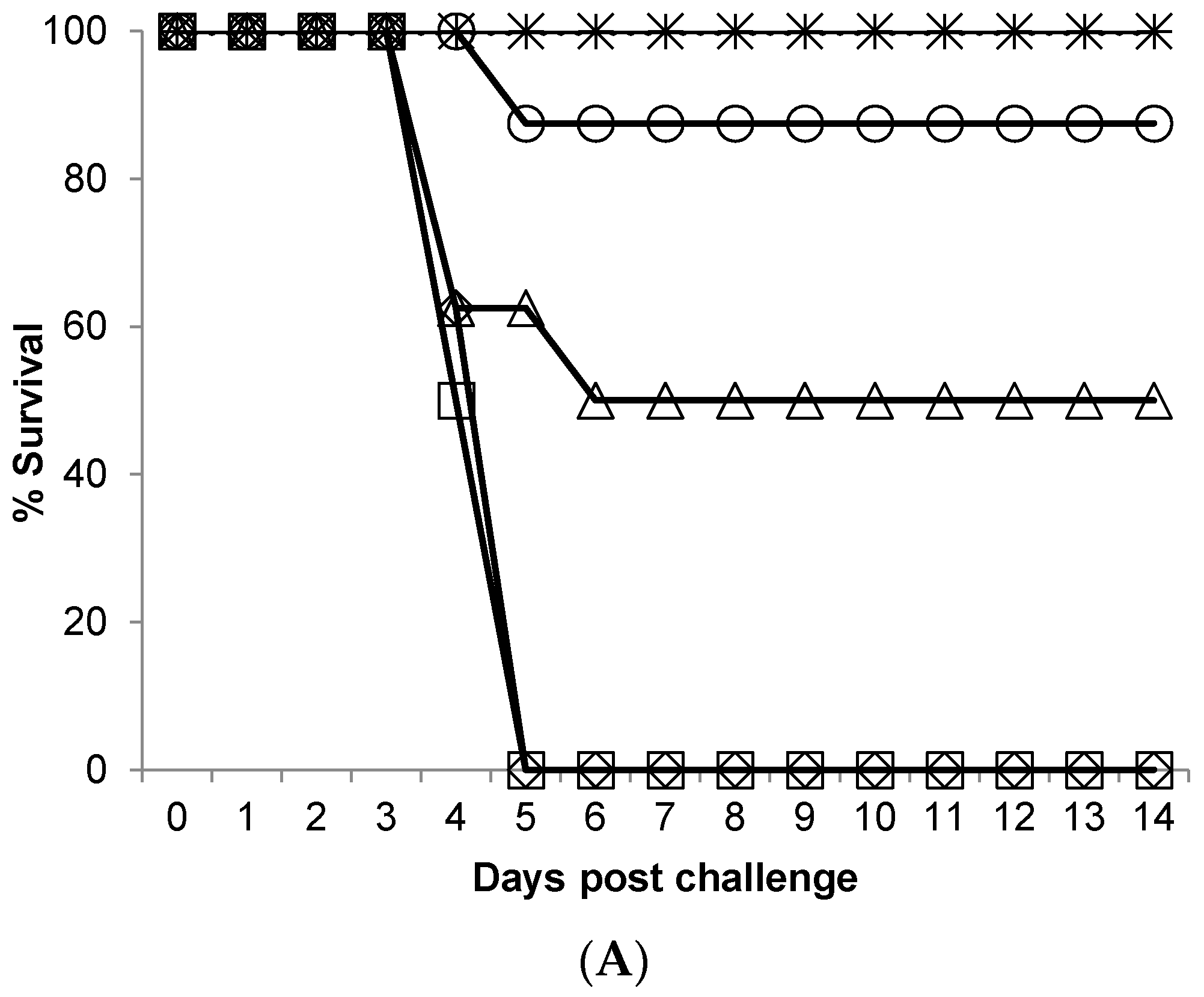
Aerosol infection is the likely route of exposure to WEEV in a biowarfare scenario, and one way in which laboratory personnel may be accidentally exposed. It is therefore critical to understand the susceptibility, lethality and pathogenesis of infection when exposed by this route. In this study, aerosol exposure to WEEV Fleming was lethal in adult female Balb/c mice. The disease course was acute (with mice succumbing abruptly 4–5 days post-challenge), with severe clinical signs, rapid neuroinvasion and 100% mortality observed when challenged with ≥1.7 × 10
2 pfu presented dose. This compares with another study [
18] using intranasal infection with a high and low virulence strains of WEEV, including Fleming. The mean time to death for high virulence strains was 5–6 days post-challenge, as opposed to 4–5 days by the aerosol route seen here. This is also in-line with a related encephalitic alphavirus, EEEV, where virus was first found in the brain at 6 h after aerosol challenge, and 24 h after intranasal challenge [
21].
The MLD in this model of WEEV Fleming was calculated to be 11 pfu/mouse presented dose, by the aerosol route. The MLD for WEEV Fleming has not previously been determined for rodents infected by the aerosol route, although intraperitoneal, subcutaneous, and intra-nasal challenges of C57/BL6 mice with the CBA 87 strain, suggest that these routes require at least a 10-fold greater challenge dose to cause lethality [
15]. Luciferase-expressing WEEV has previously been used to track infection in CD-1 outbred mice exposed by the intra-nasal route, revealing that neuroinvasion occurs primarily via cranial nerves, chiefly in the olfactory tract [
16]. This may explain the rapid neuroinvasion of WEEV into the brains of mice exposed to ~10 × MLD in this study, where viable virus was detected at near-peak levels as early as three days after aerosol challenge (
Figure 3). The 3-jet collision nebulizer generates particles in the range of 1–3 microns, which is in the respirable range for deep lung inhalation [
22,
23,
24]. Our data suggest that despite the particle size being suited to deposition in the deep lung, the virus preferentially utilizes the olfactory processes as a route to the brain. If so, the disparity between the different sides of the brain with respect to number of lesions may be the result of uneven uptake of virus by the olfactory epithelium, and therefore uneven distribution of virus between the left and right olfactory bulbs. However, we do not have quantitative data to support this theory.
Phillips et al. summarized bioluminescent WEEV pathogenesis as occurring in three phases; (1) extraneural viral lesions, within 24 h post-challenge (2) neuroinvasion, within 24–48 h post-challenge, and (3) central nervous system (CNS) dissemination, 48–72 h post-challenge [
25]. IHC analysis of both WEEV antigen and luciferase in their study demonstrated that infection was almost entirely limited to neurons and that dissemination was probably via neuronal connectivity. Our viral load and dissemination data, as well as histopathological findings, were in general agreement with this published data from intranasal challenge, although the timings of the phases were delayed in comparison. The altered kinetics may be due to differences in challenge dose, challenge route, strain of virus, and/or host factors.
In Balb/c mice challenged with WEEV Fleming by the aerosol route, extraneural lesions were detected three days post-challenge, although as samples were not taken two days post-challenge, it is possible that lesions appeared earlier than this. There was clear evidence of neuroinvasion at three days post-challenge, as demonstrated by the viral load and virus detection by IHC, although lesions were very limited. This appears to be representative of the early clinical phase of the disease, and could possibly offer a window of opportunity for the administration of any medical countermeasure. By day four post-challenge, focused lesions were observed in the encephalon of mice culled on welfare grounds, and extensive bilateral lesions were found throughout the encephalon in mice that had succumbed to lethal disease. Of interest was the detection of WEEV antigen in the retina of a single mouse culled four days post-challenge; a finding that has not been reported previously, and may in-part be why these animals were observed to have eye issues. By day five post-challenge, extensive bilateral lesions in the encephalon were also present in animals that had succumbed to lethal disease. By design, a number of mice survived the aerosol challenge and they were all free of clinical signs and free of noteworthy pathological findings at the point at which they were sacrificed (≥7 days post-challenge). This suggests that adult Balb/c mice were either not exposed to sufficient virus to cause infection, or were able to mount an effective immune response to challenge with ~10 × MLD of WEEV by the aerosol route. The former is unlikely, as cage-mates exposed concurrently to the same aerosol challenge exhibited pronounced clinical signs of disease, and succumbed to infection. The latter, therefore, would be the most probable explanation, although the immune response to infection was not characterized in these studies as it was beyond the scope of the program of work.
In a Sindbis virus (SINV) model of alphavirus-induced encephalitis, Metcalf et al. [
26] IFNγdemonstrated that viral clearance from intra-cerebral inoculation in mice occurred in three phases: clearance of infectious virus (from three days post-challenge), clearance of viral RNA (from eight days post-challenge), and retention of low levels of viral RNA (>60 days post-challenge). The adaptive immune response was initiated in the draining cervical lymph nodes, with CD8
+ T cells being the first to enter the brain, rapidly followed by other immune cells. The viral clearance phase correlated with SINV-specific immunoglobulin (Ig)M secretion, and viral RNA clearance correlated with an abundance of inflammatory cells, SINV-specific IgG, and memory B cell production. Non-cytolytic markers of viral clearance included interferon (IFN)γ (likely to be produced by local CD8
+ T cells) and IgG, both of which are also important in other CNS virus infections [
27,
28,
29,
30], and may account for the absence of histopathology findings in surviving mice in this study. It has been shown [
20] that passive transfer of serum from glycoprotein E1-immunized mice to naïve mice, conferred protection against WEEV challenge. This and other research identified virus-specific antibody as being an important component in recovery from a lethal challenge of WEEV [
31], although such rapid production in naïve mice post-challenge was unlikely. [
32] examined the immunological profile of Balb/c mice protected from aerosol VEEV challenge following vaccination with a live attenuated vaccine, which demonstrated protection despite the absence of detectable levels of virus-specific IgG or IgA antibody.
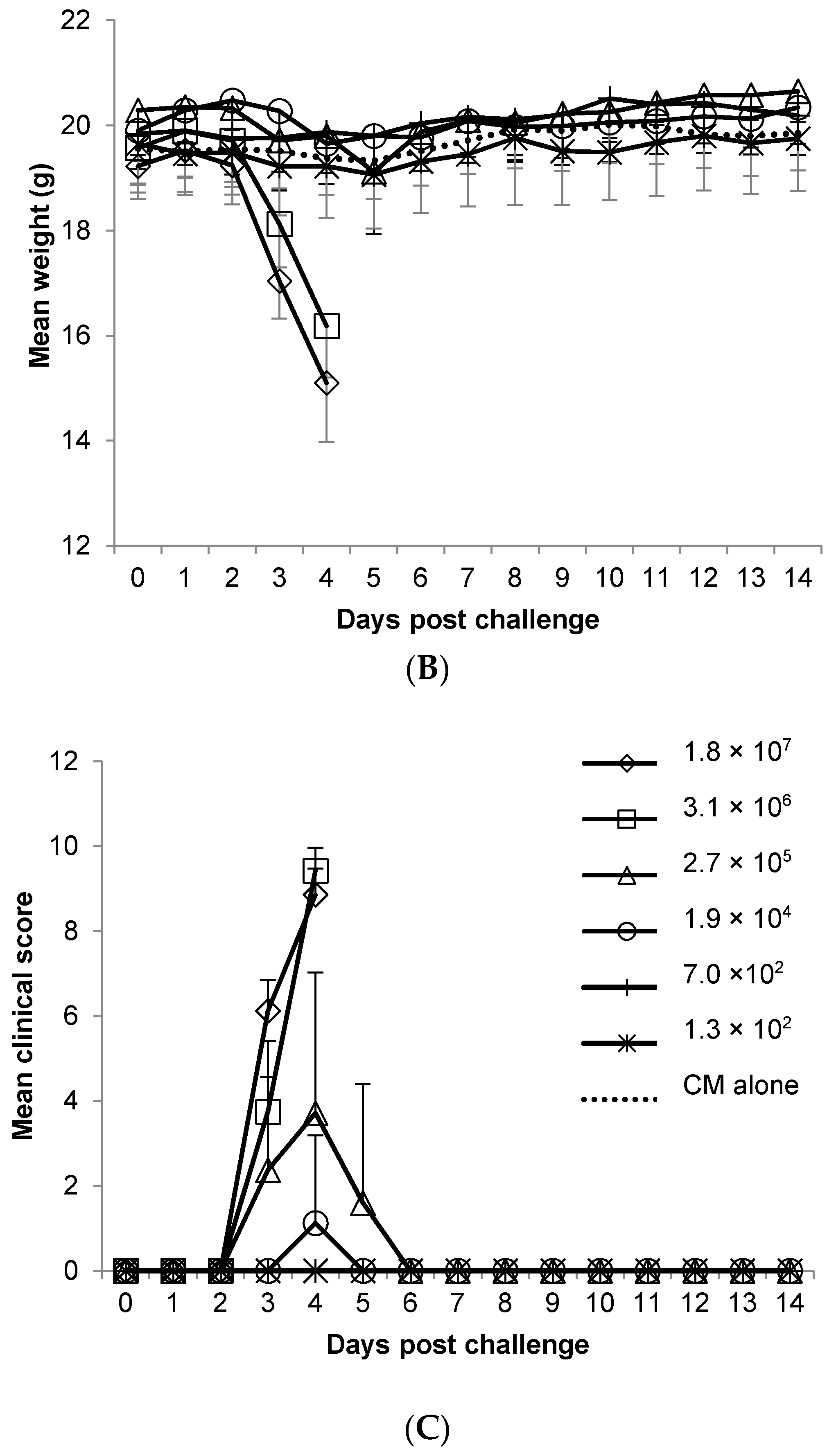
In this study, we have expanded a clinical scoring system previously described for the low-virulence WEEV 71V-1658 intra-nasal challenge model [
33], for use with high-virulence WEEV Fleming strain, in aerosol challenges in mice. The weight loss observed in infected mice was a good indicator, but not a good predictor of outcome as the disease course was acute. Any mouse observed to have pronounced mobility issues (unable to reach food and water) was culled on welfare grounds in order to comply with UK Home Office Licensing requirements. Further to this, mice found to have observable mobility issues and altered behaviour, or one of these signs in combination with observable respiratory issues, rapidly succumbed to infection (within 12–24 h). This combination of clinical signs may be a useful way to determine a humane end-point. Pronounced altered behaviour (spinning, obsessive grooming, lack of coordinated motor control) coincided with high viral titres of WEEV in the brain at three days post-challenge. It seems likely that viral colonisation of the brain was the cause of this behaviour, although this could be tissue-dependent and/or viral load-dependent within the brain, as one mouse positive for WEEV in the brain three days post-challenge was free of all clinical signs, including altered behaviours. This particular animal had a titre of 7.24 Log
10 pfu/g, compared to 9.01 Log
10 pfu/g in an animal sacrificed at the same time but that displayed signs of altered behaviour (and other clinical signs). In addition, in mice that succumbed to disease, as opposed to being sacrificed for time points, the viral load in the brain appeared to be independent of the challenge dose (
Table 1). This suggests that severe clinical signs are associated with a high level of virus replication in the brain, and that a major component of recovery may be mechanisms that limit viral replication in brain tissue. If this is the case, the critical difference between low and high dose challenges is a greater likelihood of achieving a high viral load in the brain in high dose challenges, and the important issue for development of medical countermeasures may be to limit virus replication in the brain, rather than prevent neuroinvasion completely.
The above analysis is supported by the lack of viable WEEV in blood, the majority of spleens, and all but a single liver sample at the time-points chosen. This suggests that inhaled virus can cause lethal disease with little apparent replication in peripheral tissues. Ref. [
15] made the same observation in intra-nasally infected WEEV CBA 87 mice, where WEEV was not detected in the spleen, liver, lung, kidney, or heart at the time-points studied. In contrast, mice infected by the sub-cutaneous or intra-peritoneal route with WEEV CBA 87 were positive in all extraneural tissues tested. In humans, WEEV is rarely isolated from blood or cerebro-spinal fluid when patients present with overt clinical signs, although virus has been isolated from brain tissue in biopsy or at autopsy [
2].
The Balb/c mouse displays overt clinical signs of WEEV disease that reproduce the most severe clinical symptoms observed in humans. In humans, clinical features may include abrupt onset of fever, headache, nausea, vomiting, anorexia, viremia, and general malaise. A minority of those infected may also go on to have an altered mental state (confusion, lack of co-ordination), seizures, and encephalitis, leading to coma and possibly death. In the present mouse model of aerosol induced infection, the non-specific clinical features present abruptly as a combination of clinical signs; ruffled fur, hunched posture, a change in respiratory rate, weight loss, and one or both eyes closed. This very rapidly leads to altered behaviour, mobility issues and in some cases, tremors/seizures, all of which were lethal in this model of disease. An aerosol challenge of WEEV Fleming by the aerosol route provided a set of clinical signs that were consistent with those reported in human disease. The mortality rate however was higher in mice than in naturally occurring epizootic episodes, and this was likely, among other factors, to be attributed to the route of infection, and the relative susceptibility of the Balb/c mouse to WEEV. Human cases of aerosol infection are rare with limited detail surrounding accidental exposures of laboratory personnel, although a single report by [
7] registers mortality as being 40% (2/5).
This report has detailed the susceptibility of adult Balb/c mice to WEEV Fleming by the aerosol route and the associated presented MLD. The pathogenesis of disease has also been described, particularly detailing viral load and distribution, histopathology within target neuronal tissues, and a provisional clinical scoring system for use in assessing animal condition and to determine a humane end-point. Reproduction of the most severe clinical features of human WEEV disease in mice should allow researchers to robustly assess antiviral therapies or vaccines for this Hazard Group 3 (HG 3) bio-threat agent. Understanding the pathogenesis of disease in an animal model is critical to being able to target key tissues and key intervention times in disease progression, and to further understand the full effects of any clinical interventions. This in vivo mouse model may be a useful tool in assessing the utility of putative medical countermeasure against WEEV, both prophylactically and therapeutically, and provide key data pertaining to the efficacy of any medical countermeasures against a rapidly neuroinvasive aerosol challenge. Clinical features of disease and viral load analysis will be key parameters in this acute model, and a program of work is required to elucidate the immunological profile in Balb/c mice infected with WEEV Fleming.
4. Materials and Methods
4.1. Cells and Virus
WEEV strain Fleming was kindly supplied by Les P. Nagata (Defense Research and Development Canada). The preparation of virulent stocks in suckling mice has been described previously [
34], and titre determined by standard plaque assay methods utilising a carboxymethyl cellulose overlay. Briefly, suckling mouse pups were inoculated intra-cranially with WEEV Fleming and were moribund within 24 h, at which point they were culled with an overdose of sodium pentobarbital, and stored at −70 °C. Virus was harvested by aspirating the brain through the dorsal cranium with a large-bore syringe needle and expelled into Leibovitz L-15 media supplemented with 2% (
v/v) foetal calf serum (Gibco, ThermoFisher Scientific, Loughborough, UK). This preparation was homogenised by passing through a 70 µm nylon cell strainer, and clarified at 10,000 rpm for 10 min in a SW28 rotor (Beckman Coulter, High Mycombe, UK). The supernatant was titrated and stored at −80 °C until required. All work with WEEV was carried out under UK Advisory Committee on Dangerous Pathogens Level 3 (BSL3) containment. Fleming is a pathogenic, neurotropic strain of WEEV from a human isolate obtained during a Californian outbreak, with 98.6% nucleotide identity to the closely related, and commonly studied California strain of WEEV (horse isolate) [
18]. When compared to one another in an intranasal model of infection in mice, both Fleming and California were highly comparable in disease outcome and grouped along with the McMillan strain, as pathotype A [
18].
Vero cells were obtained from the European Collection of Animal Cell Cultures (ECACC, Salisbury, UK) and propagated in Dulbecco’s minimal essential medium with 10% (v/v) foetal calf serum, 2 mM l-glutamine, 50 IU/mL penicillin and 50 μg/mL streptomycin, at 37 °C, in a 5% CO2 humidified atmosphere. For virus culture, Vero cells were maintained in Leibovitz L-15 media supplemented with 2% (v/v) foetal calf serum, 2 mM l-glutamine, 50 IU/mL penicillin and 50 μg/mL streptomycin (Maintenance Media; MM), at 37 °C. Aerosol challenge preparations were made in L-15 media supplemented with 2mM l-glutamine only (Challenge Media; CM). Media and supplements came from two sources; Sigma Aldrich, Poole, UK and Gibco, ThermoFisher Scientific, Loughborough, UK.
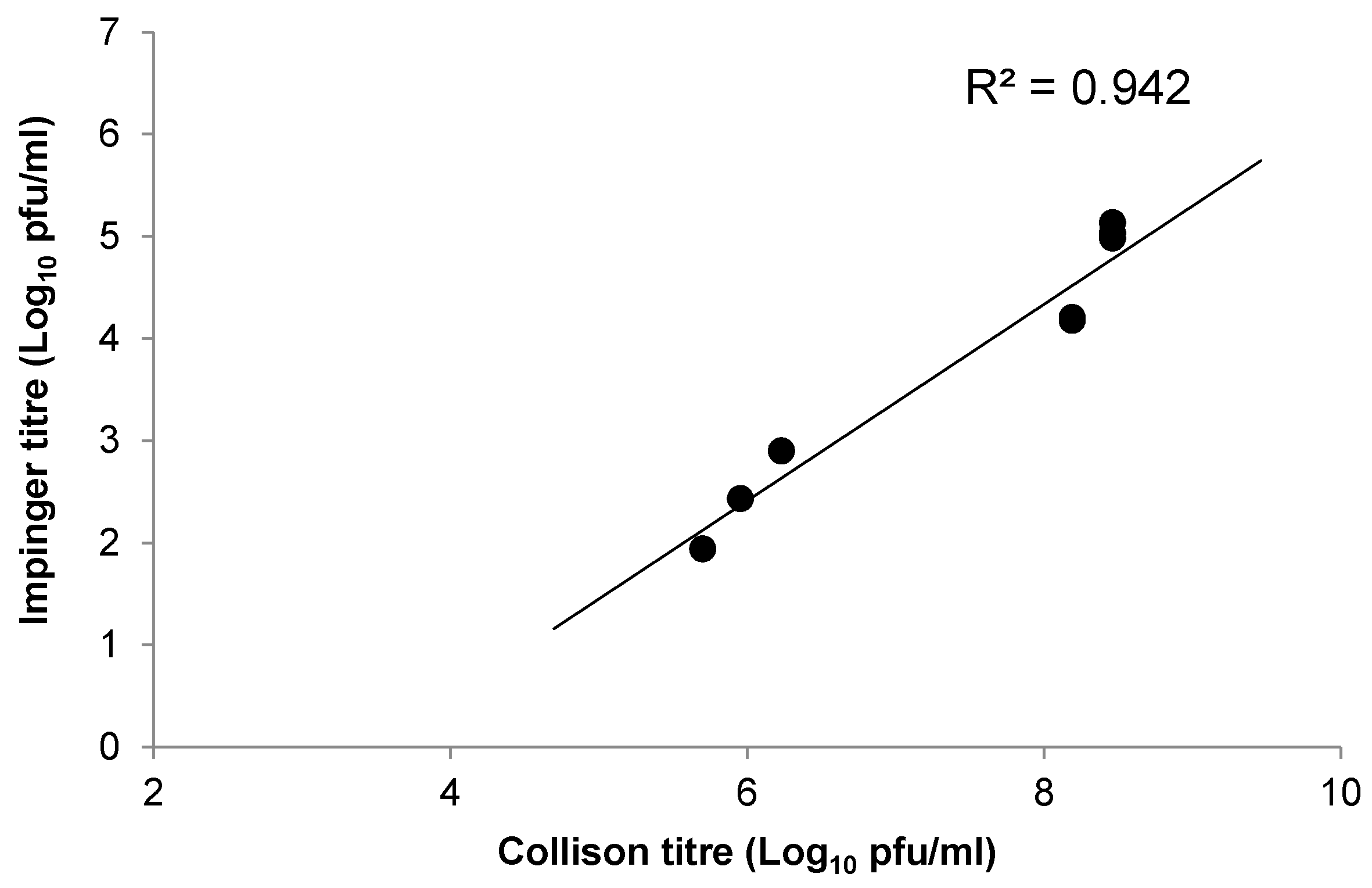
Plaque assays were routinely performed in a 24-well plate format with duplicate wells for each dilution step, and virus inoculum applied to wells in 100 µL/well. The limit of detection in this assay is 10 pfu/mL of original sample. The exceptions were aerosol samples collected in Impingers (output virus), which were performed in a 6-well plate format with duplicate wells for each dilution step, and virus inoculum applied to wells in 500 µL/well. The limit of detection for Impinger samples is 2 pfu/mL.
4.2. Aerosol Generation and Challenge
Three experiments were conducted to assess the effect of aerosolisation on the viability and calculated presented dose of WEEV Fleming. The aerosol was generated using a 3-jet Collison nebulizer, containing 10 mL of virus (input virus), controlled and conditioned to 50% (±5%) relative humidity by an AeroMP platform system (Biaera Technologies, Hagerstown, MD, USA). Aerosols were generated for a total of 9 min, with aerosol sampling achieved using an all-glass impinger (AGI-30; Ace Glass, Vineland, NJ, USA) containing 10 mL phosphate buffered saline (output virus). A total of three samples were taken during aerosolisation for 1 min each, at a flow rate of 12 L/min. All virus samples were kept on ice until used or titrated.
After characterising virus output from aerosols, the aerosol challenge for in vivo experiments was generated as described above; mice were physically restrained in holding tubes and nose-only exposed to the aerosol for 10 min. A single sample of each aerosol exposure was taken using an AGI-30 as described. A maximum of 20 mice were challenged in any single aerosol exposure, with fresh virus preparations used for each exposure. The mean calculated, presented challenge dose was determined using the viral titres obtained from the Impingers (output virus), and Guyton’s formula for the respiratory volumes of laboratory animals [
35].
4.3. Mouse Studies
Female, Balb/c mice (aged 7–9 weeks), with micro-chip; (Charles River Laboratories, Margate, UK) weighing between 18–20 g were appropriately housed with access to food and water ad libitum in a rigid-walled BSL3 containment isolator. All studies were performed in accordance with the UK Scientific Procedures (Animals) Act 1986 and the UK Codes of Practice for the Housing and Care of Animals Used in Scientific Procedures 1989 (as well the Animal Care and Use Review Office, Fort Detrick, MD, USA ). Prior to commencing the study, mice were acclimatised in the BSL3 isolator for a minimum of five days and were weighed prior to aerosol challenge. Post-challenge, a clinical scoring system was employed based on our previous experience of exposing Balb/c mice to the closely related Venezuelan equine encephalitis virus, by the aerosol route [
36]. Scores were assigned on a truncated scale of 0 (absent), 1 (observable), and 2 (pronounced), and were based on observations of coat condition, hunched posture, respiratory state, eye condition, tremors, changes in behaviour, ability to move, and paralysis (
Table 2). After challenge, all animals were weighed daily and observed a minimum of twice daily for clinical signs of infection by an independent observer who was unaware of group allocation. As clinical scoring is a subjective measure, it is important to ensure the use of independent personnel to conduct the scoring and monitoring where possible. Personnel must also have sufficient experience in understanding the normal behaviour and condition of a Balb/c mouse, to then be able to make consistent judgements about diseased animals. Any mouse observed to be paralysed or to have pronounced mobility issues (unable to reach food and water) was culled on welfare grounds. At the onset of severe clinical signs the frequency of observations was increased, in accordance with UK Home Office requirements. All culls were performed using a UK Schedule 1 method (cervical dislocation followed by confirmation of cessation of heart beat).
4.4. Median Lethal Dose of Western Equine Encephalitis Virus by the Aerosol Route in Balb/c Mice
Six groups of eight mice were challenged by the aerosol route with a dilution range of WEEV Fleming. Virus was diluted in CM to yield 5 × 10
7 pfu/mL, and 10-fold dilutions were prepared to 5 × 10
2 pfu/mL for use as input virus in the Collison nebulizer. A control group of seven mice were challenged with CM only (no virus) by the aerosol route, and placed in the first challenge run. All mice were weighed daily and scored for clinical signs of disease at least twice daily. Tissues of mice which succumbed to infection were excised to determine viral load. The MLD was calculated using the 50% end point calculation of Reed & Muench [
37] and was based on the number of surviving animals from each exposure group 14 days post-challenge.
4.5. Pathogenesis of Western Equine Encephalitis Virus by the Aerosol Route in Balb/c Mice
Mice were challenged with ~10 × MLD WEEV Fleming by the aerosol route as described above, and groups culled on successive days (1, 3, 4, 5, 7, 10, and 14 days post-challenge). At the scheduled time-points, groups were anaesthetized with gaseous halothane and exsanguinated by cardiac puncture prior to cull by cervical dislocation. Brain, lung, and spleen were separately prepared for virus enumeration by homogenisation through a 40 µm cell sieve (Corning Falcon cell strainer, Fisher Scientific, Loughborough, UK) into 1 mL MM. Serial dilutions were prepared from homogenates in MM and added to duplicate cell monolayers for plaque titration in 24-well tissue culture plates under a carboxymethyl cellulose (CMC) overlay. Neat samples of blood obscure the cell monolayer in plaque titration experiments and were not assayed. Blood samples therefore have a higher limit of detection (100 pfu/mL). All plates were incubated for 3–4 days at 37 °C in a humidified atmosphere before fixation of monolayers with 10% (v/v) formal saline solution and staining with 0.1% (w/v) crystal violet to visualise plaques.
At least 3 whole mice per scheduled cull were terminally anaesthetized with gaseous halothane (confirmation of cull by cessation of heart beat), and prepared for immersion in 10% neutral buffered formalin (NBF; SigmaPoole, UK) for histological analysis. Carcass preparation for immersion in NBF required full abdominal, thoracic, and cranial cavity access to ensure complete virus inactivation prior to removal of carcasses from high containment. A control group of 10 mice, challenged with CM only, was included and culled one day post-challenge.
Following preservation in 10% NBF, the following tissues were dissected from each individual carcass and routinely processed for histopathology: brain (six coronal sections as described by Hicks et al. [
38], to allow examination of the olfactory bulbs, cortex, hippocampus, thalamus, hypothalamus, pons, cerebellum, and medulla), lung, heart, trachea, mediastinal structures including thymus and mediastinal lymph nodes, mandibular lymph nodes, and salivary glands. The left femur and the facial and cranial structures were subjected to a minimum of 48 h decalcification in Gooding and Stewart’s fluid, a 1:1:18 solution of 96–100% formic acid (VWR Chemicals, Leighton Buzzard, UK), 36% formaldehyde (VWR Chemicals) and purified water, prior to preparation of cross sections to allow the examination of the bone structures of femur, face, oral cavity and teeth, nasal cavity, nasal associated lymphoid tissues, and eyes. All samples were routinely processed and embedded in paraffin wax. 4 µm thick sections were cut and either stained with haematoxylin and eosin, or used for IHC detection of viral antigen. WEEV was detected by IHC using automated protocols optimised for use on the Ventana Discovery XT staining module (Ventana Medical Systems, Tucson, AZ, USA).
Tissue sections were de-waxed prior to antigen retrieval, using a standard CC2 (Ventana Medical Systems, Tucson, AZ, USA) cell conditioning regime for rabbit anti-WEEV E2 glycoprotein (Integrated Biotherapeutics, Rockville, MD, USA) polyclonal antibody (pAb) protocol. Primary anti-WEEV pAb (4 µg/mL) diluted in Ventana Ab Diluent (Ventana Medical Systems) was applied for 60 min at room temperature. Antibody–antigen interaction was detected and amplified using anti-rabbit HRP multimer (Ventana Medical Systems) for 16 min and visualised using a ChromoMap DAB kit (Ventana Medical Systems). Formalin-fixed, paraffin-embedded tissue sections were counterstained in haematoxylin (Ventana Reagents, Tucson, AZ, USA) for 8 min, before being permanently mounted for interpretation. Concentration-matched (4 µg/mL) rabbit antibody isotype (Vector Laboratories, Peterborough, UK) controls diluted in Ventana Ab diluent were included as test controls.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}