
Immune Tolerant Chronic Hepatitis B: The Unrecognized Risks

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Anti-Viral Therapy
3. When to Initiate Antiviral Therapy
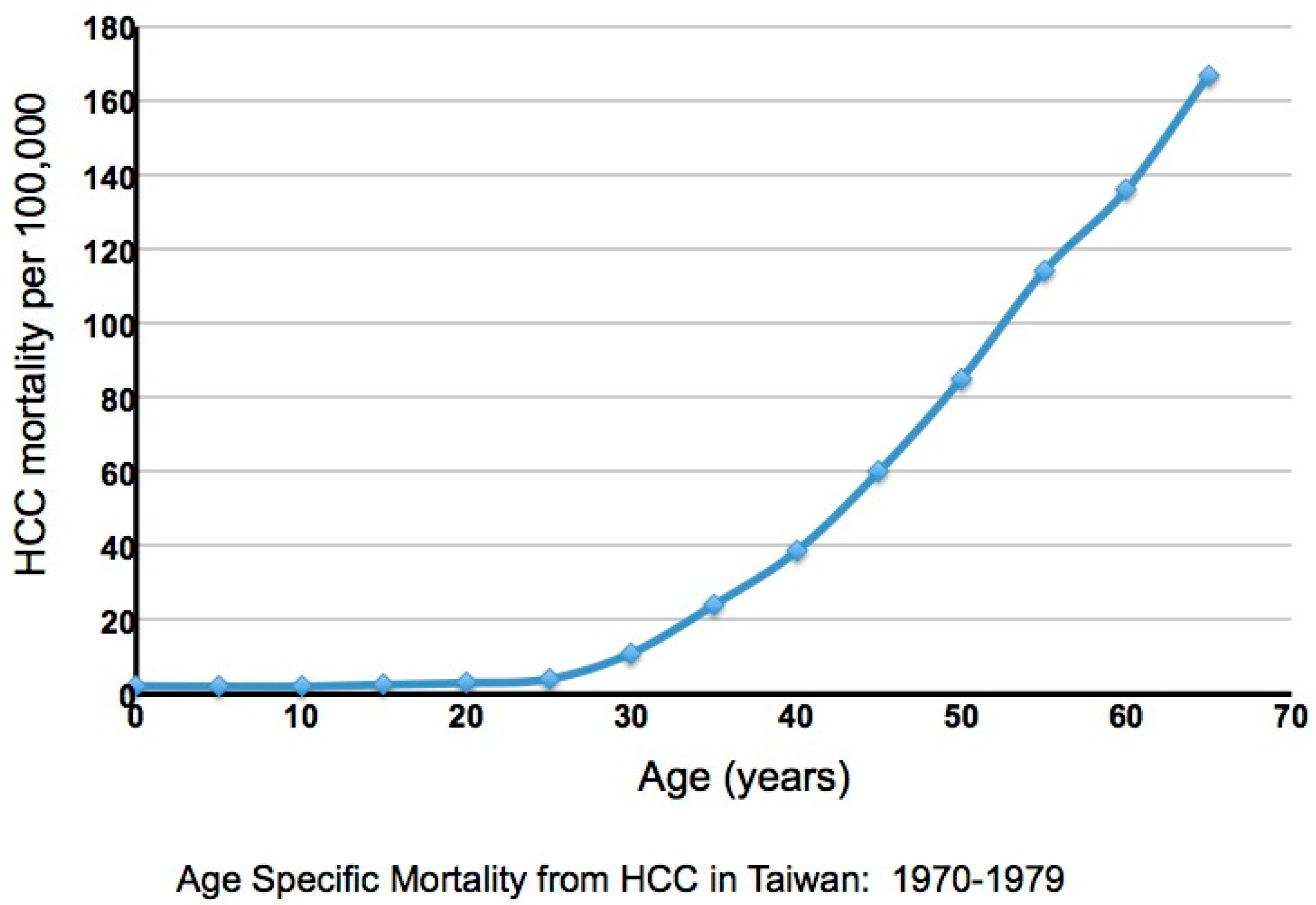
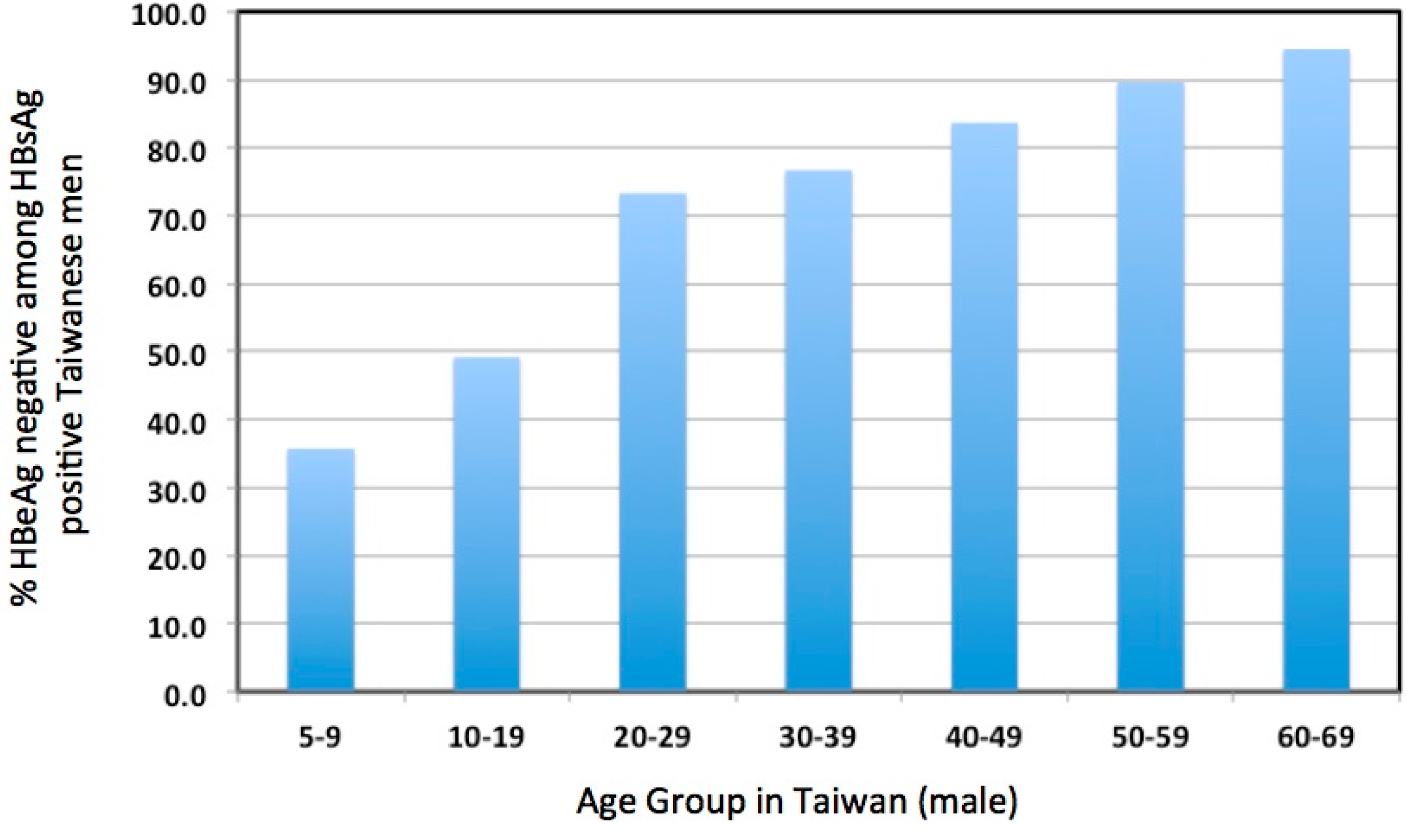
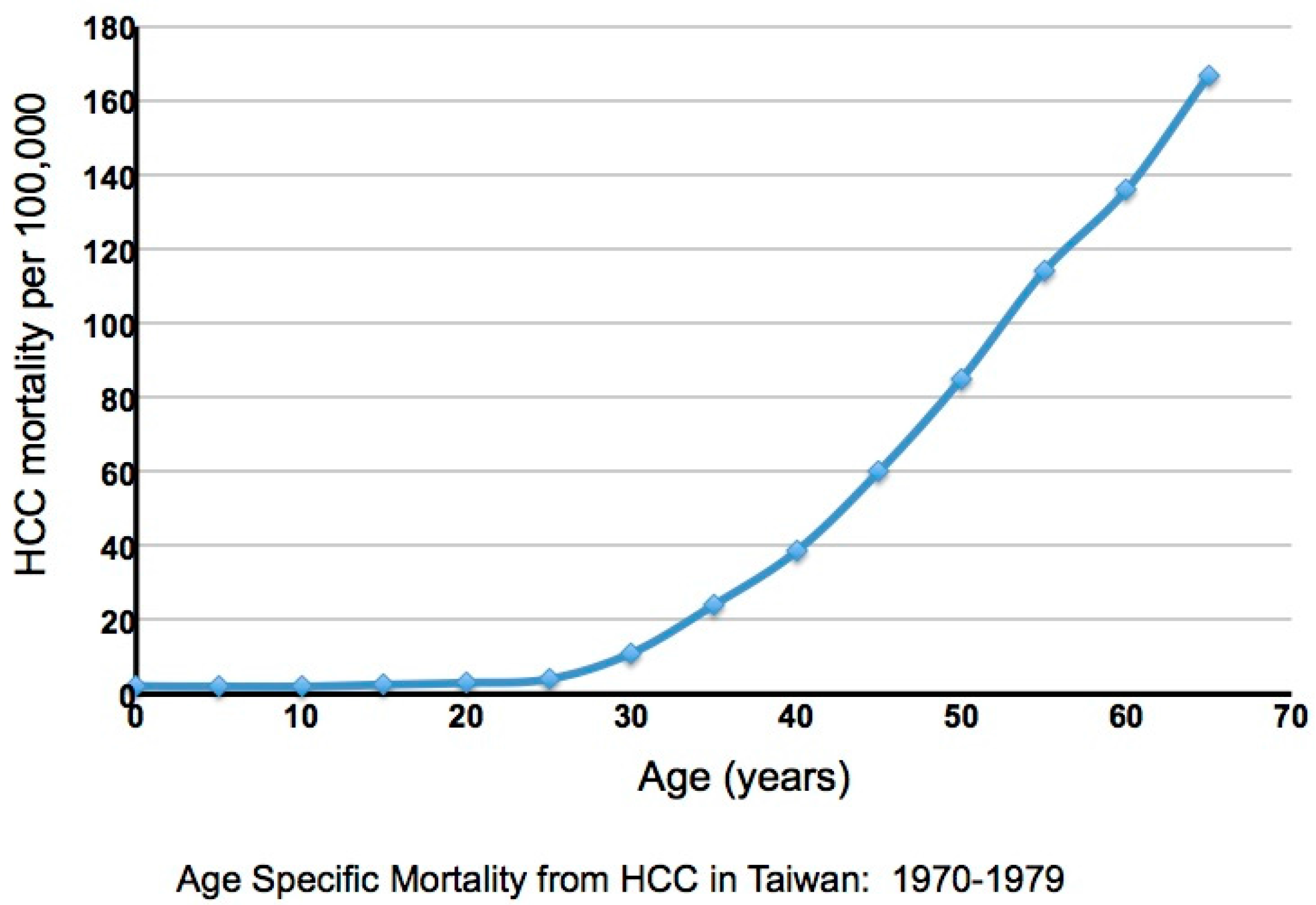
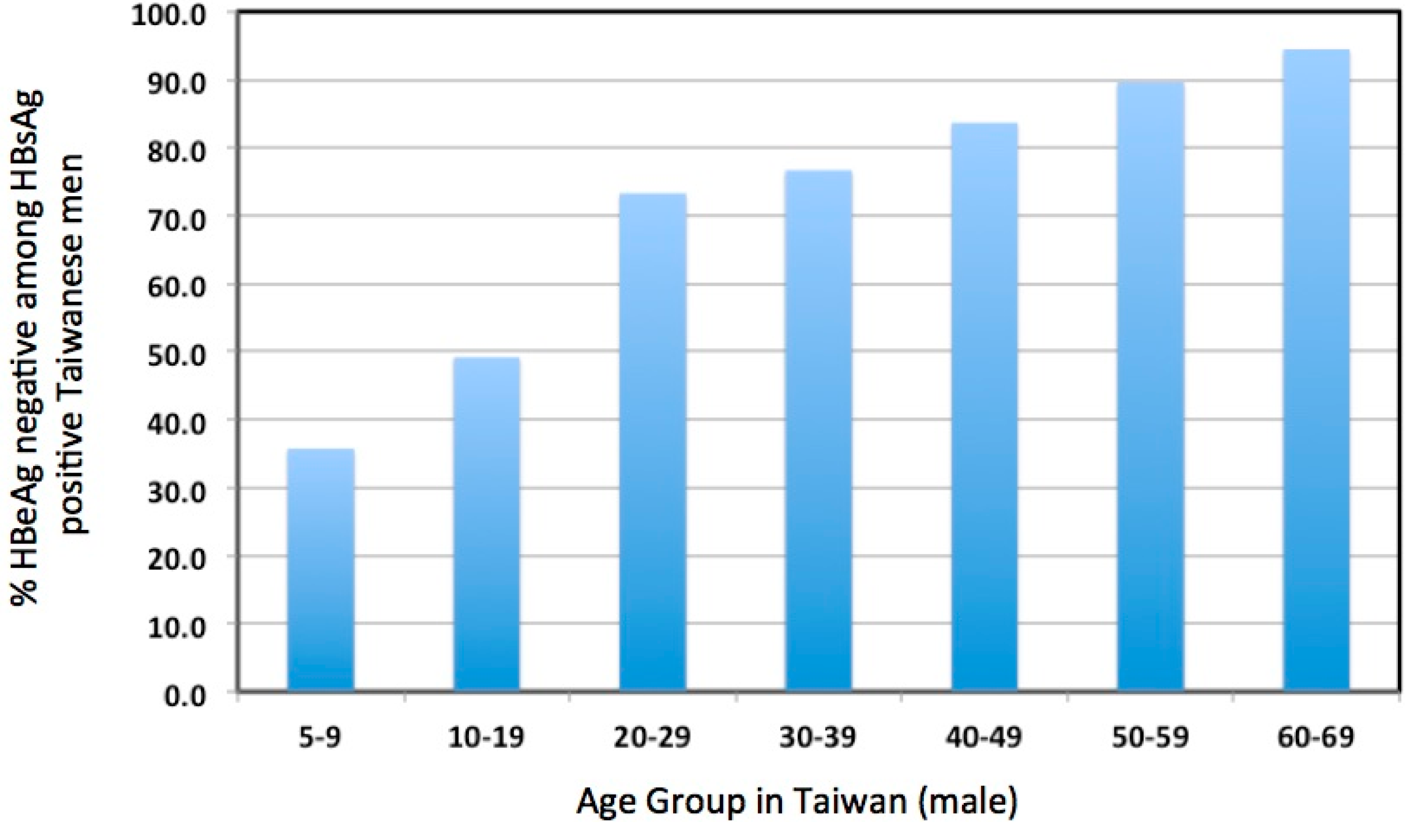
4. Evidence Consistent with Elevated Liver Damage in Immune Tolerant Patients
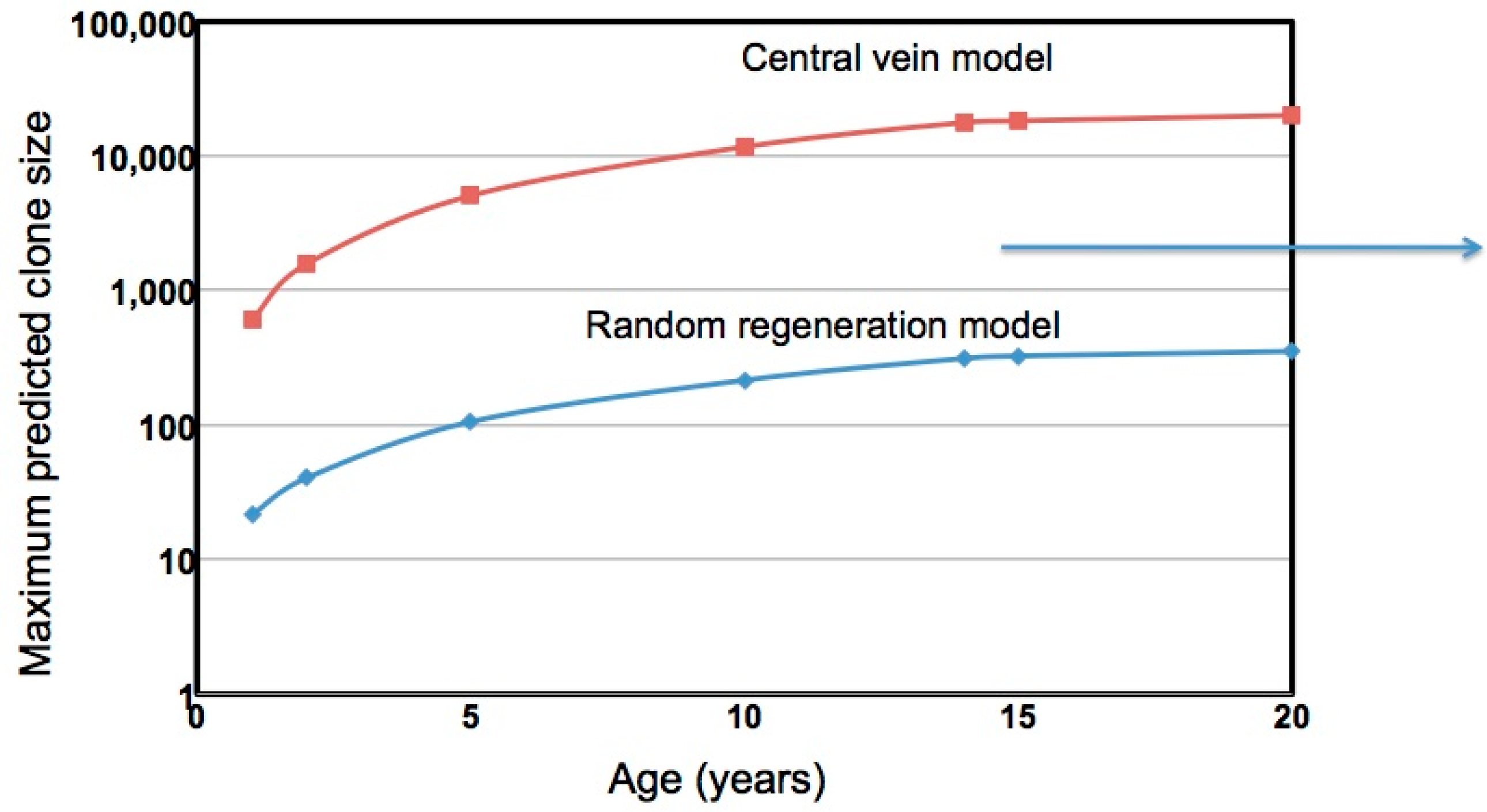
5. Clonal Hepatocyte Expansion and HCC
6. HBV Vaccine and Prospects for HBV Elimination
7. Summary and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lavanchy, D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 2004, 11, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.J.; Lok, A.S. Natural history of chronic hepatitis B virus infection: What we knew in 1981 and what we know in 2005. Hepatology 2006, 43, S173–S181. [Google Scholar] [CrossRef] [PubMed]
- Gish, R.G.; Given, B.D.; Lai, C.L.; Locarnini, S.A.; Lau, J.Y.; Lewis, D.L.; Schluep, T. Chronic hepatitis B: Virology, natural history, current management and a glimpse at future opportunities. Antivir. Res. 2015, 121, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Prati, D.; Taioli, E.; Zanella, A.; Della Torre, E.; Butelli, S.; Del Vecchio, E.; Vianello, L.; Zanuso, F.; Mozzi, F.; Milani, S.; et al. Updated definitions of healthy ranges for serum alanine aminotransferase levels. Ann. Intern. Med. 2002, 137, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sokal, E.M.; Paganelli, M.; Wirth, S.; Socha, P.; Vajro, P.; Lacaille, F.; Kelly, D.; Mieli-Vergani, G. Management of chronic hepatitis B in childhood: Espghan clinical practice guidelines: Consensus of an expert panel on behalf of the european society of pediatric gastroenterology, hepatology and nutrition. J. Hepatol. 2013, 59, 814–829. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.H.; Hwang, L.Y.; Hsu, H.C.; Lee, C.Y.; Beasley, R.P. Prospective study of asymptomatic HBsAg carrier children infected in the perinatal period: Clinical and liver histologic studies. Hepatology 1988, 8, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.M.; Karayiannis, P.; Fowler, M.J.; Monjardino, J.; Liaw, Y.F.; Thomas, H.C. Natural history of chronic hepatitis B virus infection in taiwan: Studies of hepatitis B virus DNA in serum. Hepatology 1985, 5, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Farber, E.; Sarma, D.S. Hepatocarcinogenesis: A dynamic cellular perspective. Lab. Investig. 1987, 56, 4–22. [Google Scholar] [PubMed]
- Jilbert, A.R.; Wu, T.-T.; England, J.M.; de la, M.; Hall, P.M.; Carp, N.Z.; O‘Connell, A.P.; Mason, W.S. Rapid resolution of duck hepatitis B virus infections occurs after massive hepatocellular involvement. J. Virol. 1992, 66, 1377–1388. [Google Scholar] [PubMed]
- Kajino, K.; Jilbert, A.R.; Saputelli, J.; Aldrich, C.E.; Cullen, J.; Mason, W.S. Woodchuck hepatitis virus infections: Very rapid recovery after a prolonged viremia and infection of virtually every hepatocyte. J. Virol. 1994, 68, 5792–5803. [Google Scholar] [PubMed]
- Wieland, S.F.; Spangenberg, H.C.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Expansion and contraction of the hepatitis B virus transcriptional template in infected chimpanzees. Proc. Natl. Acad. Sci. USA 2004, 101, 2129–2134. [Google Scholar] [CrossRef] [PubMed]
- Asabe, S.; Wieland, S.F.; Chattopadhyay, P.K.; Roederer, M.; Engle, R.E.; Purcell, R.H.; Chisari, F.V. The size of the viral inoculum contributes to the outcome of hepatitis B virus infection. J. Virol. 2009, 83, 9652–9662. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.; Jilbert, A.R.; Yang, W.; Aldrich, C.E.; Saputelli, J.; Litwin, S.; Toll, E.; Mason, W.S. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc. Natl. Acad. Sci. USA 2003, 100, 11652–11659. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Stadler, D.; Lucifora, J.; Reisinger, F.; Webb, D.; Hosel, M.; Michler, T.; Wisskirchen, K.; Cheng, X.; Zhang, K.; et al. Interferon-gamma and tumor necrosis factor-alpha produced by T cells reduce the HBV persistence form, cccDNA, without cytolysis. Gastroenterology 2016, 150, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Sohn, J.A. Complete spectrum of crispr/cas9-induced mutations on HBV cccDNA. Mol. Ther. 2016, 24, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.M.; Wieland, S.F.; Purcell, R.H.; Chisari, F.V. Dynamics of hepatitis B virus clearance in chimpanzees. Proc. Natl. Acad. Sci. USA 2005, 102, 17780–17785. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Xu, C.; Low, H.C.; Saputelli, J.; Aldrich, C.E.; Scougall, C.; Grosse, A.; Colonno, R.; Litwin, S.; Jilbert, A.R. The amount of hepatocyte turnover that occurred during resolution of transient hepadnavirus infections was lower when virus replication was inhibited with entecavir. J. Virol. 2009, 83, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Le Mire, M.F.; Miller, D.S.; Foster, W.K.; Burrell, C.J.; Jilbert, A.R. Covalently closed circular DNA is the predominant form of duck hepatitis B virus DNA that persists following transient infection. J. Viroil. 2005, 79, 12242–12252. [Google Scholar] [CrossRef] [PubMed]
- Coffin, C.S.; Michalak, T.I. Persistence of infectious hepadnavirus in the offspring of woodchuck mothers recovered from viral hepatitis. J. Clin. Investig. 1999, 104, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B.; Ferrari, C.; Pasquinelli, C.; Chisari, F.V. The hepatitis B virus persists for decades after patients recovery from acute viral hepatitis despite active maintenance of a cytotoxic t-lymphocyte response. Nat. Med. 1996, 2, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, J.H. Reactivation of hepatitis B. Hepatology 2009, 49, S156–S165. [Google Scholar] [CrossRef] [PubMed]
- Protzer, U.; Maini, M.K.; Knolle, P.A. Living in the liver: Hepatic infections. Nat. Rev. Immunol. 2012, 12, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Thimme, R. Hepatic immune regulation and its involvement in viral hepatitis infection. Gastroenterology 2014, 146, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Milich, D.R.; Jones, J.; Hughes, J.; Maruyama, T. Role of T-cell tolerance in the persistence of hepatitis B virus infection. J. Immunother. Emphas. Tumor Immunol. 1993, 14, 226–233. [Google Scholar] [CrossRef]
- Tian, Y.; Kuo, C.F.; Akbari, O.; Ou, J.H. Maternal-derived hepatitis B virus e antigen alters macrophage function in offspring to drive viral persistence after vertical transmission. Immunity 2016, 44, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Yang, H.I.; Su, J.; Jen, C.L.; You, S.L.; Lu, S.N.; Huang, G.T.; Iloeje, U.H.; REVEAL-HBV Study Group. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006, 295, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.S.; Chien, R.N.; Yeh, C.T.; Sheen, I.S.; Chiou, H.Y.; Chu, C.M.; Liaw, Y.F. Long-term outcome after spontaneous HBeAg seroconversion in patients with chronic hepatitis B. Hepatology 2002, 35, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Seto, W.K.; Lai, C.L.; Ip, P.P.; Fung, J.; Wong, D.K.; Yuen, J.C.; Hung, I.F.; Yuen, M.F. A large population histology study showing the lack of association between ALT elevation and significant fibrosis in chronic hepatitis B. PLoS ONE 2012, 7, e32622. [Google Scholar] [CrossRef] [PubMed]
- Beasley, R.P.; Lin, C.C.; Hwang, L.Y.; Chien, C.S. Hepatocellular carcinoma and hepatitis B virus. Lancet 1981, 2, 1129–1133. [Google Scholar] [CrossRef]
- Beasley, R.P. Hepatitis B virus as the etiologic agent in hepatocellular carcinoma: Epidemiologic considerations. Hepatology 1982, 2, 21S–26S. [Google Scholar]
- You, S.L.; Yang, H.I.; Chen, C.J. Seropositivity of hepatitis B e antigen and hepatocellular carcinoma. Ann. Med. 2004, 36, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.I.; Lu, S.N.; Liaw, Y.F.; You, S.L.; Sun, C.A.; Wang, L.Y.; Hsiao, C.K.; Chen, P.J.; Chen, D.S.; Chen, C.J. Hepatitis B e antigen and the risk of hepatocellular carcinoma. NEJM 2002, 347, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.; van Zonneveld, M.; Senturk, H.; Zeuzem, S.; Akarca, U.S.; Cakaloglu, Y.; Simon, C.; So, T.M.; Gerken, G.; de Man, R.A.; et al. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: A randomised trial. Lancet 2005, 365, 123–129. [Google Scholar] [CrossRef]
- Lau, G.K.; Piratvisuth, T.; Luo, K.X.; Marcellin, P.; Thongsawat, S.; Cooksley, G.; Gane, E.; Fried, M.W.; Chow, W.C.; Paik, S.W.; et al. Peginterferon alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. NEJM 2005, 352, 2682–2695. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Bonino, F.; Lau, G.K.; Farci, P.; Yurdaydin, C.; Piratvisuth, T.; Jin, R.; Gurel, S.; Lu, Z.M.; Wu, J.; et al. Sustained response of hepatitis B e antigen-negative patients 3 years after treatment with peginterferon alpha-2a. Gastroenterology 2009, 136, 2169–2179. [Google Scholar] [CrossRef] [PubMed]
- Liaw, Y.F. Does chemotherapy prevent HBV-related hepatocellular carcinoma? Pros. Dig. Liver Dis. 2010, 42 (Suppl. 3), S293–S297. [Google Scholar] [CrossRef]
- Liaw, Y.F.; Sung, J.J.; Chow, W.C.; Farrell, G.; Lee, C.Z.; Yuen, H.; Tanwandee, T.; Tao, Q.M.; Shue, K.; Keene, O.N.; et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. NEJM 2004, 351, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Ahn, S.H.; Chen, D.S.; Chen, P.J.; Dusheiko, G.M.; Hou, J.L.; Maddrey, W.C.; Mizokami, M.; Seto, W.K.; Zoulim, F.; et al. Chronic hepatitis B virus infection: Disease revisit and management recommendations. J. Clin. Gastroenterol. 2016, 50, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Tenney, D.J.; Rose, R.E.; Baldick, C.J.; Pokornowski, K.A.; Eggers, B.J.; Fang, J.; Wichroski, M.J.; Xu, D.; Yang, J.; Wilber, R.B.; et al. Long-term monitoring shows hepatitis B virus resistance to entecavir in nucleoside-naive patients is rare through 5 years of therapy. Hepatology 2009, 49, 1503–1514. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, T.; Suzuki, F.; Kumada, H. Long-term entecavir treatment reduces hepatocellular carcinoma incidence in patients with hepatitis B virus infection. Hepatology 2013, 58, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Tanaka, Y.; Fong, D.Y.; Fung, J.; Wong, D.K.; Yuen, J.C.; But, D.Y.; Chan, A.O.; Wong, B.C.; Mizokami, M.; et al. Independent risk factors and predictive score for the development of hepatocellular carcinoma in chronic hepatitis B. J. Hepatol. 2009, 50, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Ahn, S.H. Prediction models of hepatocellular carcinoma development in chronic hepatitis B patients. World J. Gastroenterol. 2016, 22, 8314–8321. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.I.; Sherman, M.; Su, J.; Chen, P.J.; Liaw, Y.F.; Iloeje, U.H.; Chen, C.J. Nomograms for risk of hepatocellular carcinoma in patients with chronic hepatitis B virus infection. J. Clin. Oncol. 2010, 28, 2437–2444. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Chan, S.L.; Mo, F.; Chan, T.C.; Loong, H.H.; Wong, G.L.; Lui, Y.Y.; Chan, A.T.; Sung, J.J.; Yeo, W.; et al. Clinical scoring system to predict hepatocellular carcinoma in chronic hepatitis B carriers. J. Clin. Oncol. 2010, 28, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, S.A.; Do, S.Y.; Kim, B.J. Precore/core region mutations of hepatitis B virus related to clinical severity. World J. Gastroenterol. 2016, 22, 4287–4296. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.; Freitas, N.; Kallakury, B.V.; Menne, S.; Gudima, S.O. Super-infection with woodchuck hepatitis virus (WHV) strain WHVNY of the livers chronically infected with the strain WHV7. J. Virol. 2014, 89, 384–405. [Google Scholar] [CrossRef] [PubMed]
- Frelin, L.; Wahlstrom, T.; Tucker, A.E.; Jones, J.; Hughes, J.; Lee, B.O.; Billaud, J.N.; Peters, C.; Whitacre, D.; Peterson, D.; et al. A mechanism to explain the selection of the hepatitis e antigen-negative mutant during chronic hepatitis B virus infection. J. Virol. 2009, 83, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Arama, V.; Leblebicioglu, H.; Simon, K.; Zarski, J.P.; Niederau, C.; Habersetzer, F.; Vermehren, J.; Bludzin, W.; Jinga, M.; Ulusoy, S.; et al. Chronic hepatitis B monitoring and treatment patterns in five european countries with different access and reimbursement policies. Antivir. Ther. 2014, 19, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.; Burak, K.; Maroun, J.; Metrakos, P.; Knox, J.J.; Myers, R.P.; Guindi, M.; Porter, G.; Kachura, J.R.; Rasuli, P.; et al. Multidisciplinary Canadian consensus recommendations for the management and treatment of hepatocellular carcinoma. Curr. Oncol. 2011, 18, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Tedder, R.S.; Rodger, A.J.; Fries, L.; Ijaz, S.; Thursz, M.; Rosenberg, W.; Naoumov, N.; Banatvala, J.; Williams, R.; Dusheiko, G.; et al. The diversity and management of chronic hepatitis B virus infections in the United Kingdom: A wake-up call. Clin. Infect. Dis. 2013, 56, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Allain, J.P.; Opare-Sem, O. Screening and diagnosis of HBV in low-income and middle-income countries. Nat. Rev. Gastroenterol Hepatol. 2016, 13, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Spradling, P.R.; Rupp, L.; Moorman, A.C.; Lu, M.; Teshale, E.H.; Gordon, S.C.; Nakasato, C.; Boscarino, J.A.; Henkle, E.M.; Nerenz, D.R.; et al. Hepatitis B and C virus infection among 1.2 million persons with access to care: Factors associated with testing and infection prevalence. Clin. Infect. Dis. 2012, 55, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Gill, U.S.; Zissimopoulos, A.; Al-Shamma, S.; Burke, K.; McPhail, M.J.; Barr, D.A.; Kallis, Y.N.; Marley, R.T.; Kooner, P.; Foster, G.R.; et al. Assessment of bone mineral density in tenofovir-treated patients with chronic hepatitis B: Can the fracture risk assessment tool identify those at greatest risk? J. Infect. Dis. 2015, 211, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.; McMahon, B.J. Chronic hepatitis B: Update 2009. Hepatology 2009, 50, 661–662. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.F.; McMahon, B.J. Chronic hepatitis B. Hepatology 2007, 45, 507–539. [Google Scholar] [CrossRef] [PubMed]
- Yapali, S.; Talaat, N.; Lok, A.S. Management of hepatitis B: Our practice and how it relates to the guidelines. Clin. Gastroenterol. Hepatol. 2014, 12, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Schall, R.A.; et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013, 381, 468–475. [Google Scholar] [CrossRef]
- Tana, M.M.; Hoofnagle, J.H. Scar undone: Long-term therapy of hepatitis B. Lancet 2013, 381, 433–434. [Google Scholar] [CrossRef]
- Mason, W.S.; Low, H.C.; Xu, C.; Aldrich, C.E.; Scougall, C.A.; Grosse, A.; Clouston, A.; Chavez, D.; Litwin, S.; Peri, S.; et al. Detection of clonally expanded hepatocytes in chimpanzees with chronic hepatitis B virus infection. J. Virol. 2009, 83, 8396–8408. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Mason, W.S.; Clouston, A.D.; Shackel, N.A.; McCaughan, G.W.; Yeh, M.M.; Schiff, E.R.; Ruszkiewicz, A.R.; Chen, J.W.; Harley, H.A.J.; et al. Clonal expansion of hepatocytes with a selective advantage occurs during all stages of chronic hepatitis B virus infection. J. Viral Hepat. 2015, 22, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Liu, C.; Aldrich, C.E.; Litwin, S.; Yeh, M.M. Clonal expansion of normal-appearing human hepatocytes during chronic hepatitis B virus infection. J. Virol. 2010, 84, 8308–8315. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.L.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA integration and clonal hepatocyte expansion in chronic hepatitis B patients considered immune tolerant. Gastroenterology 2016, 151, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Milich, D.R. The concept of immune tolerance in chronic hepatitis B virus infection is alive and well. Gastroenterology 2016, 151, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Kennedy, P.T. The immune tolerant phase of chronic HBV infection: New perspectives on an old concept. Cell. Mol. Immunol. 2015, 12, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Maini, M.K.; Boni, C.; Lee, C.K.; Larrubia, J.R.; Reignat, S.; Ogg, G.S.; King, A.S.; Herberg, J.; Gilson, R.; Alisa, A.; et al. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. NEJM 2000, 191, 1269–1280. [Google Scholar] [CrossRef]
- Kennedy, P.T.; Sandalova, E.; Jo, J.; Gill, U.; Ushiro-Lumb, I.; Tan, A.T.; Naik, S.; Foster, G.R.; Bertoletti, A. Preserved T-cell function in children and young adults with immune-tolerant chronic hepatitis B. Gastroenterology 2012, 143, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Bill, C.; Summers, J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc. Natl. Acad. Sci. USA 2004, 101, 11135–11140. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.; Marucci, L.; Benedetti, A.; Jezequel, A.M.; Orlandi, F. Immunohistochemical analysis of S-phase cells in normal human and rat liver by pc10 monoclonal antibody. Liver 1994, 14, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhao, L.L.; Fish, M.; Logan, C.Y.; Nusse, R. Self-renewing diploid axin2+ cells fuel homeostatic renewal of the liver. Nature 2015, 524, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Bralet, M.P.; Branchereau, S.; Brechot, C.; Ferry, N. Cell lineage study in the liver using retroviral mediated gene transfer. Evidence against the streaming of hepatocytes in normal liver. Am. J. Pathol. 1994, 144, 896–905. [Google Scholar] [PubMed]
- Kennedy, S.; Rettinger, S.; Flye, M.W.; Ponder, K.P. Experiments in transgenic mice show that hepatocytes are the source for postnatal liver growth and do not stream. Hepatology 1995, 22, 160–168. [Google Scholar] [PubMed]
- Gong, S.S.; Jensen, A.D.; Chang, C.J.; Rogler, C.E. Double-stranded linear duck hepatitis B virus (DHBV) stably integrates at a higher frequency than wild-type DHBV in lmh chicken hepatoma cells. J. Virol. 1999, 73, 1492–1502. [Google Scholar] [PubMed]
- Yang, W.; Summers, J. Integration of hepadnavirus DNA in infected liver: Evidence from a linear precursor. J. Virol. 1999, 73, 9710–9717. [Google Scholar] [PubMed]
- Mason, W.S.; Jilbert, A.R.; Summers, J. Clonal expansion of hepatocytes during chronic woodchuck hepatitis virus infection. Proc. Natl. Acad. Sci. USA 2005, 102, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, S.; Conrad, A.; Lim, B.; Valinluck, B.; Kim, A.M.; Schmid, P. Study of preneoplastic changes in liver cells by immunohistochemical and molecular hybridization techniques. Arch. Pathol. Lab. Med. 1990, 114, 1042–1045. [Google Scholar] [PubMed]
- Bannasch, P.; Haertel, T.; Su, Q. Significance of hepatic preneoplasia in risk identification and early detection of neoplasia. Toxicol. Pathol. 2003, 31, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Alt, E.; Rogler, C.E. Coordinate expression of N-myc 2 and insulin-like growth factor II in pre-cancerous altered hepatic foci in woodchuck hepatitis virus carriers. Cancer Res. 1993, 53, 2020–2027. [Google Scholar] [PubMed]
- Xu, C.; Yamamoto, T.; Zhou, T.; Aldrich, C.E.; Frank, K.; Cullen, J.M.; Jilbert, A.R.; Mason, W.S. The liver of woodchucks chronically infected with the woodchuck hepatitis virus contains foci of virus core antigen-negative hepatocytes with both altered and normal morphology. Virology 2007, 359, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Grompe, M. Liver stem cells, where art thou? Cell Stem Cell. 2014, 15, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.J.; Chen, F.; Li, J.X.; Liu, C.C.; Zhang, H.B.; Xia, Y.; Yu, B.; You, P.; Xiang, D.; Lu, L.; et al. Reversal of hepatocyte senescence after continuous in vivo cell proliferation. Hepatology 2014, 60, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Schaub, J.R.; Malato, Y.; Gormond, C.; Willenbring, H. Evidence against a stem cell origin of new hepatocytes in a common mouse model of chronic liver injury. Cell Rep. 2014, 8, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Yanger, K.; Knigin, D.; Zong, Y.; Maggs, L.; Gu, G.; Akiyama, H.; Pikarsky, E.; Stanger, B.Z. Adult hepatocytes are generated by self-duplication rather than stem cell differentiation. Cell Stem Cell. 2014, 15, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Marongiu, F.; Serra, M.P.; Sini, M.; Marongiu, M.; Contini, A.; Laconi, E. Cell turnover in the repopulated rat liver: Distinct lineages for hepatocytes and the biliary epithelium. Cell Tissue Res. 2014, 356, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Tarlow, B.D.; Pelz, C.; Naugler, W.E.; Wakefield, L.; Wilson, E.M.; Finegold, M.J.; Grompe, M. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell. 2014, 15, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.S.; Litwin, S.; Jilbert, A.R. Immune selection during chronic hepadnavirus infection. Hepatol. Int. 2008, 2, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Marongiu, F.; Doratiotto, S.; Montisci, S.; Pani, P.; Laconi, E. Liver repopulation and carcinogenesis: Two sides of the same coin? Am. J. Pathol. 2008, 172, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.E.; Szmuness, W.; Goodman, A.I.; Weseley, S.A.; Fotino, M. Hepatitis B vaccine: Immune responses in haemodialysis patients. Lancet 1980, 2, 1211–1213. [Google Scholar] [CrossRef]
- Maupas, P.; Goudeau, A.; Coursaget, P.; Drucker, J.; Bagros, P. Immunisation against hepatitis B in man. Lancet 1976, 1, 1367–1370. [Google Scholar] [CrossRef]
- WHO. Global Health Sector Strategy on Viral Hepatitis 2016–2021: Towards Ending Viral Hepatitis; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- WHO. Hepatitis B vaccines: Who position paper—Recommendations. Vaccine 2010, 28, 589–590. [Google Scholar]
- WHO. Immunization Coverage. Available online: http://www.who.int/mediacentre/factsheets/fs378/en/ (accessed on 2 March 2017).
- Hsu, H.M.; Chen, D.S.; Chuang, C.H.; Lu, J.C.; Jwo, D.M.; Lee, C.C.; Lu, H.C.; Cheng, S.H.; Wang, Y.F.; Wang, C.Y.; et al. Efficacy of a mass hepatitis B vaccination program in taiwan. Studies on 3464 infants of hepatitis B surface antigen-carrier mothers. JAMA 1988, 260, 2231–2235. [Google Scholar] [CrossRef] [PubMed]
- Fortuin, M.; Chotard, J.; Jack, A.D.; Maine, N.P.; Mendy, M.; Hall, A.J.; Inskip, H.M.; George, M.O.; Whittle, H.C. Efficacy of hepatitis B vaccine in the Gambian expanded programme on immunisation. Lancet 1993, 341, 1129–1131. [Google Scholar] [CrossRef]
- Whittle, H.C.; Maine, N.; Pilkington, J.; Mendy, M.; Fortuin, M.; Bunn, J.; Allison, L.; Howard, C.; Hall, A. Long-term efficacy of continuing hepatitis B vaccination in infancy in two Gambian villages. Lancet 1995, 345, 1089–1092. [Google Scholar] [CrossRef]
- Peto, T.J.; Mendy, M.E.; Lowe, Y.; Webb, E.L.; Whittle, H.C.; Hall, A.J. Efficacy and effectiveness of infant vaccination against chronic hepatitis B in the Gambia hepatitis intervention study (1986–90) and in the nationwide immunisation program. BMC Infect. Dis. 2014, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Chaouch, H.; Taffon, S.; Villano, U.; Equestre, M.; Bruni, R.; Belhadj, M.; Hannachi, N.; Aouni, M.; Letaief, A.; Ciccaglione, A.R. Naturally occurring surface antigen variants of hepatitis B virus in Tunisian patients. Intervirology 2016, 59, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Tsebe, K.V.; Burnett, R.J.; Hlungwani, N.P.; Sibara, M.M.; Venter, P.A.; Mphahlele, M.J. The first five years of universal hepatitis B vaccination in south africa: Evidence for elimination of HBsAg carriage in under 5-year-olds. Vaccine 2001, 19, 3919–3926. [Google Scholar] [CrossRef]
- Chang, M.H.; You, S.L.; Chen, C.J.; Liu, C.J.; Lai, M.W.; Wu, T.C.; Wu, S.F.; Lee, C.M.; Yang, S.S.; Chu, H.C.; et al. Long-term effects of hepatitis B immunization of infants in preventing liver cancer. Gastroenterology 2016, 151, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, M.; Thursz, M.R. Field battle against hepatitis B infection and HCC in Africa. J. Hepatol. 2016, 66, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Shimakawa, Y.; Bottomley, C.; Njie, R.; Mendy, M. The association between maternal hepatitis B e antigen status, as a proxy for perinatal transmission, and the risk of hepatitis B e antigenaemia in Gambian children. BMC Public Health 2014, 14, 532. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Luo, H.; Wang, F.; Zheng, H.; Gong, X.; Chen, Y.; Wu, Z.; Miao, N.; Kane, M.; Hennessey, K.; et al. Evaluation of policies and practices to prevent mother to child transmission of hepatitis B virus in China: Results from China GAVI project final evaluation. Vaccine 2013, 31 (Suppl. 9), J36–J42. [Google Scholar] [CrossRef] [PubMed]
- Gueye, S.B.; Diop-Ndiaye, H.; Lo, G.; Mintsa, S.; Guindo, I.; Dia, A.; Sow-Sall, A.; Gaye-Diallo, A.; Mboup, S.; Toure-Kane, C. HBV carriage in children born from HIV-seropositive mothers in Senegal: The need of birth-dose HBV vaccination. J. Med. Virol. 2016, 88, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Godbole, G.; Irish, D.; Basarab, M.; Mahungu, T.; Fox-Lewis, A.; Thorne, C.; Jacobs, M.; Dusheiko, G.; Rosenberg, W.M.; Suri, D.; et al. Management of hepatitis B in pregnant women and infants: A multicentre audit from four London hospitals. BMC Pregnancy Childbirth 2013, 13, 222. [Google Scholar] [CrossRef] [PubMed]
- Keeble, S.; Quested, J.; Barker, D.; Varadarajan, A.; Shankar, A.G. Immunization of babies born to HBsAg positive mothers: An audit on the delivery and completeness of follow up in Norfolk and Suffolk, United Kingdom. Hum. Vaccin. Immunother. 2015, 11, 1153–1156. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.Q.; Duan, Z.P.; Bhamidimarri, K.R.; Zou, H.B.; Liang, X.F.; Li, J.; Tong, M.J. An algorithm for risk assessment and intervention of mother to child transmission of hepatitis B virus. Clin. Gastroenterol. Hepatol. 2012, 10, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, C.; Jia, Z.F.; Wu, X.; Wen, S.M.; Kong, F.; Hu, K.Q.; Li, J.; Jiang, J.; Niu, J.Q. Protective effect of an improved immunization practice of mother-to-infant transmission of hepatitis B virus and risk factors associated with immunoprophylaxis failure. Medicine 2016, 95, e4390. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.Y.; Chang, M.H.; Ni, Y.H.; Chiang, C.L.; Chen, H.L.; Wu, J.F.; Chen, P.J. No increase in prevalence of hepatitis B surface antigen mutant in a population of children and adolescents who were fully covered by universal infant immunization. J. Infect. Dis. 2010, 201, 1192–2000. [Google Scholar] [CrossRef] [PubMed]
- Suwannakarn, K.; Tangkijvanich, P.; Thawornsuk, N.; Theamboonlers, A.; Tharmaphornpilas, P.; Yoocharoen, P.; Chongsrisawat, V.; Poovorawan, Y. Molecular epidemiological study of hepatitis B virus in Thailand based on the analysis of pre-S and S genes. Hepatol. Res. 2008, 38, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Lee le, Y.; Aw, M.; Rauff, M.; Loh, K.S.; Lim, S.G.; Lee, G.H. Hepatitis B immunoprophylaxis failure and the presence of hepatitis B surface gene mutants in the affected children. J. Med. Virol. 2015, 87, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Liu, S.L.; Zhai, X.J.; Zhu, F.C.; Pan, H.; Yu, J.X.; Chen, Y.Z.; Xie, Y.R.; Zhang, X.Y.; Zhang, H.M.; et al. A serological and molecular survey of hepatitis B in children 15 years after inception of the national hepatitis B vaccination program in eastern China. J. Med. Virol. 2009, 81, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.Q.; Duan, Z.; Dai, E.; Zhang, S.; Han, G.; Wang, Y.; Zhang, H.; Zou, H.; Zhu, B.; Zhao, W.; et al. Tenofovir to prevent hepatitis B transmission in mothers with high viral load. NEJM 2016, 374, 2324–2334. [Google Scholar] [CrossRef] [PubMed]
- Kramvis, A.; Clements, C.J. Implementing a birth dose of hepatitis B vaccine for home deliveries in Africa—Too soon? Vaccine 2010, 28, 6408–6410. [Google Scholar] [CrossRef] [PubMed]
- Hodges, M.; Sanders, E.; Aitken, C. Seroprevalence of hepatitis markers; HAV, HBV, HCV and HEV amongst primary school children in Freetown, Sierra Leone. West. Afr. J. Med. 1998, 17, 36–37. [Google Scholar] [PubMed]
- Wurie, I.M.; Wurie, A.T.; Gevao, S.M. Sero-prevalence of hepatitis B virus among middle to high socio-economic antenatal population in Sierra Leone. West. Afr. J. Med. 2005, 24, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Frew, P.M.; Alhanti, B.; Vo-Green, L.; Zhang, S.; Liu, C.; Nguyen, T.; Schamel, J.; Saint-Victor, D.S.; Nguyen, M.L. Multilevel factors influencing hepatitis B screening and vaccination among Vietnamese Americans in Atlanta, Georgia. Yale J. Biol. Med. 2014, 87, 455–471. [Google Scholar] [PubMed]
- Vedio, A.; Liu, E.Z.; Lee, A.C.; Salway, S. Improving access to health care for chronic hepatitis B among migrant Chinese populations: A systematic mixed methods review of barriers and enablers. J. Viral Hepat. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kunoee, A.; Nielsen, J.; Cowan, S. Hepatitis B vaccination coverage and risk factors associated with incomplete vaccination of children born to hepatitis B surface antigen-positive mothers, Denmark, 2006 to 2010. Euro Surveill. 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Inui, A. Chronic hepatitis B in children in the United States and Canada: International origins place the disease burden on children even in the era of universal vaccination. Transl. Pediatr. 2016, 5, 1–4. [Google Scholar]
- Nayagam, S.; Thursz, M.; Sicuri, E.; Conteh, L.; Wiktor, S.; Low-Beer, D.; Hallett, T.B. Requirements for global elimination of hepatitis B: A modelling study. Lancet Infect. Dis. 2016, 12, 1399–1408. [Google Scholar]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kennedy, P.T.F.; Litwin, S.; Dolman, G.E.; Bertoletti, A.; Mason, W.S. Immune Tolerant Chronic Hepatitis B: The Unrecognized Risks. Viruses 2017, 9, 96. https://doi.org/10.3390/v9050096
Kennedy PTF, Litwin S, Dolman GE, Bertoletti A, Mason WS. Immune Tolerant Chronic Hepatitis B: The Unrecognized Risks. Viruses. 2017; 9(5):96. https://doi.org/10.3390/v9050096
Chicago/Turabian StyleKennedy, Patrick T. F., Samuel Litwin, Grace E. Dolman, Antonio Bertoletti, and William S. Mason. 2017. "Immune Tolerant Chronic Hepatitis B: The Unrecognized Risks" Viruses 9, no. 5: 96. https://doi.org/10.3390/v9050096
APA StyleKennedy, P. T. F., Litwin, S., Dolman, G. E., Bertoletti, A., & Mason, W. S. (2017). Immune Tolerant Chronic Hepatitis B: The Unrecognized Risks. Viruses, 9(5), 96. https://doi.org/10.3390/v9050096