
MimiLook: A Phylogenetic Workflow for Detection of Gene Acquisition in Major Orthologous Groups of Megavirales




Abstract
:1. Introduction
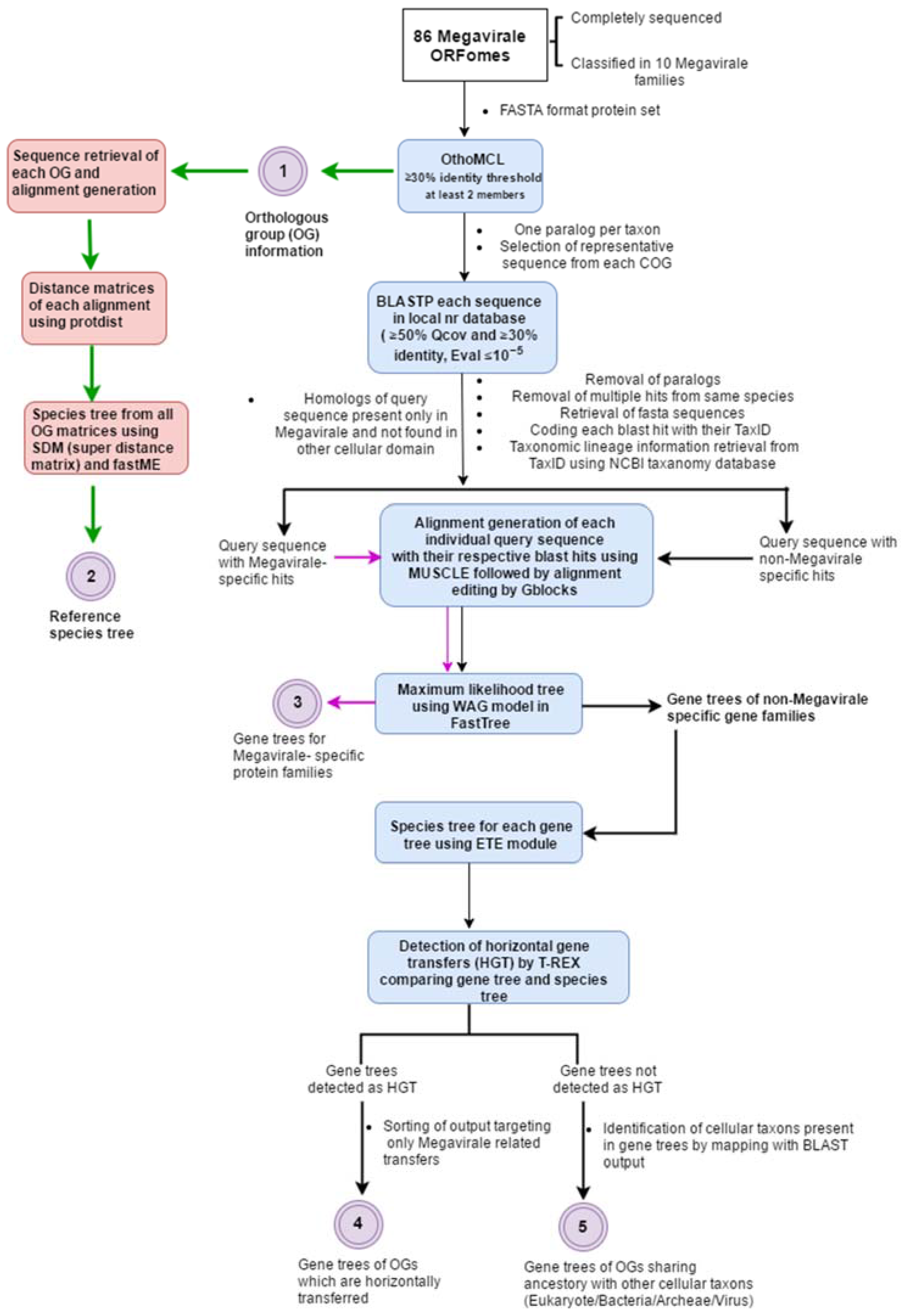
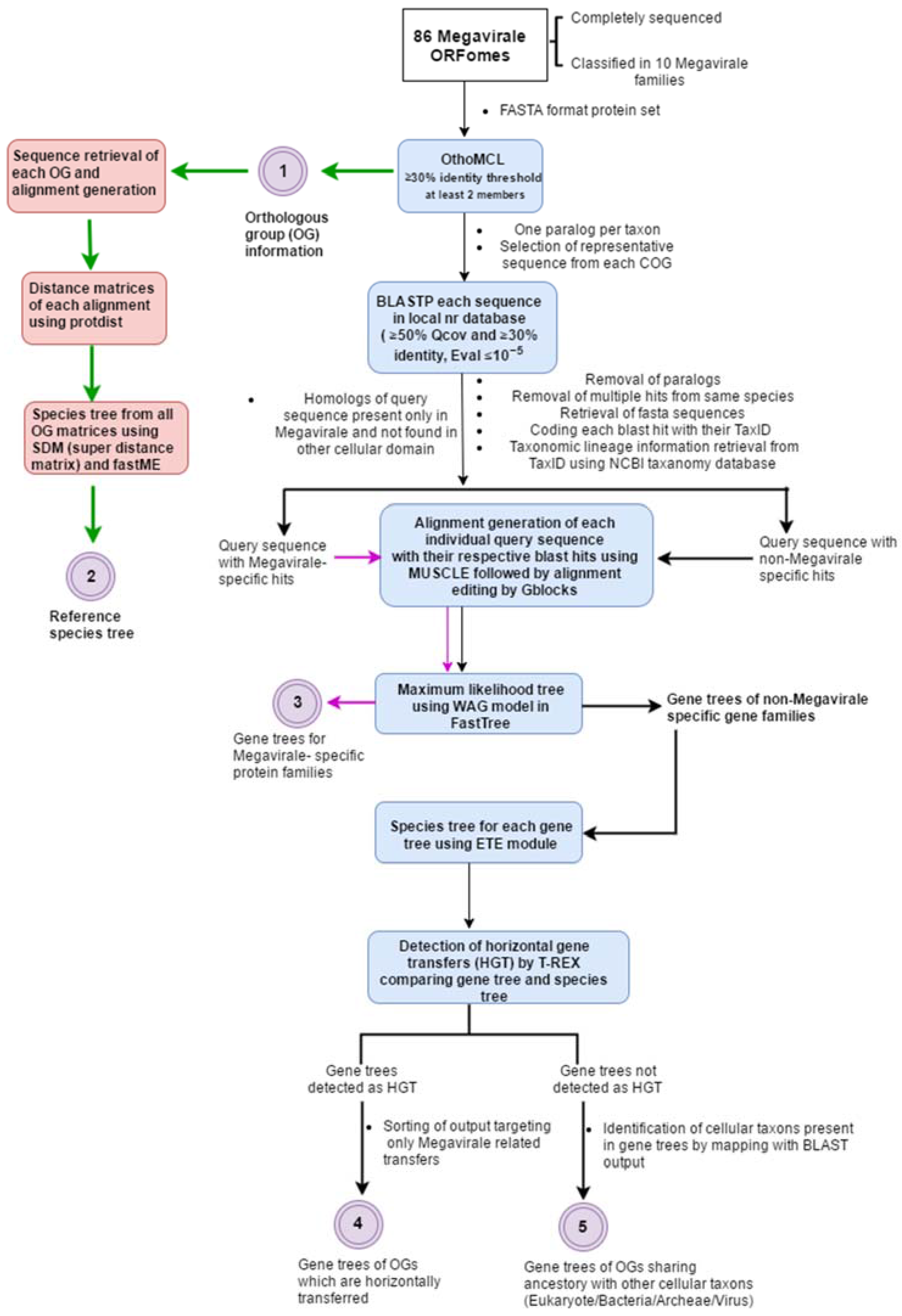
2. A General Overview of Workflow
2.1. Complete ORFomes
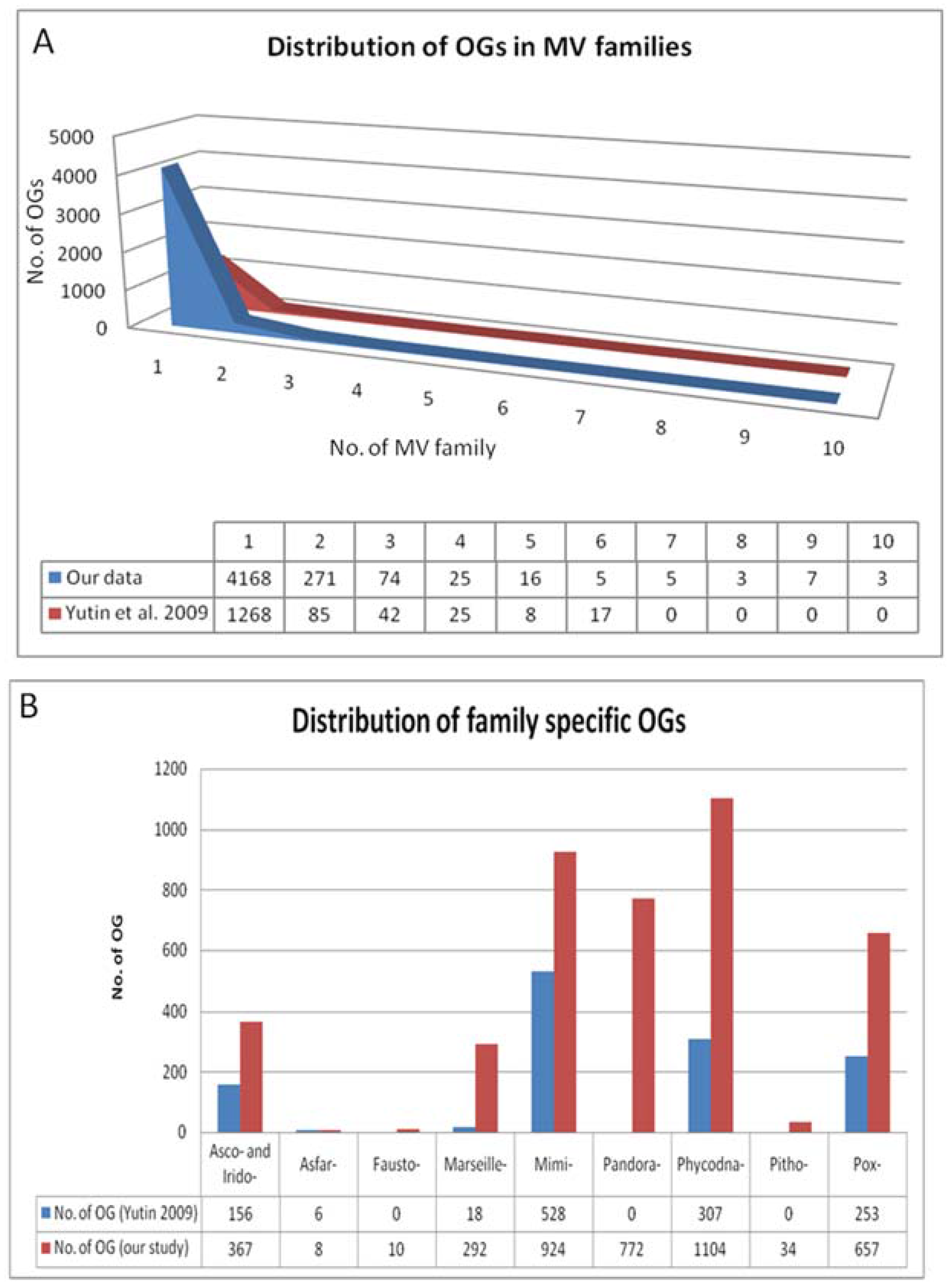
2.2. Detection of Orthologous Groups
2.3. Search for Homologs and Curation of Blast Output
2.4. Alignment Generation, Alignment Editing and Tree Preparation
2.5. Detection of Horizontal Gene Transfer Event
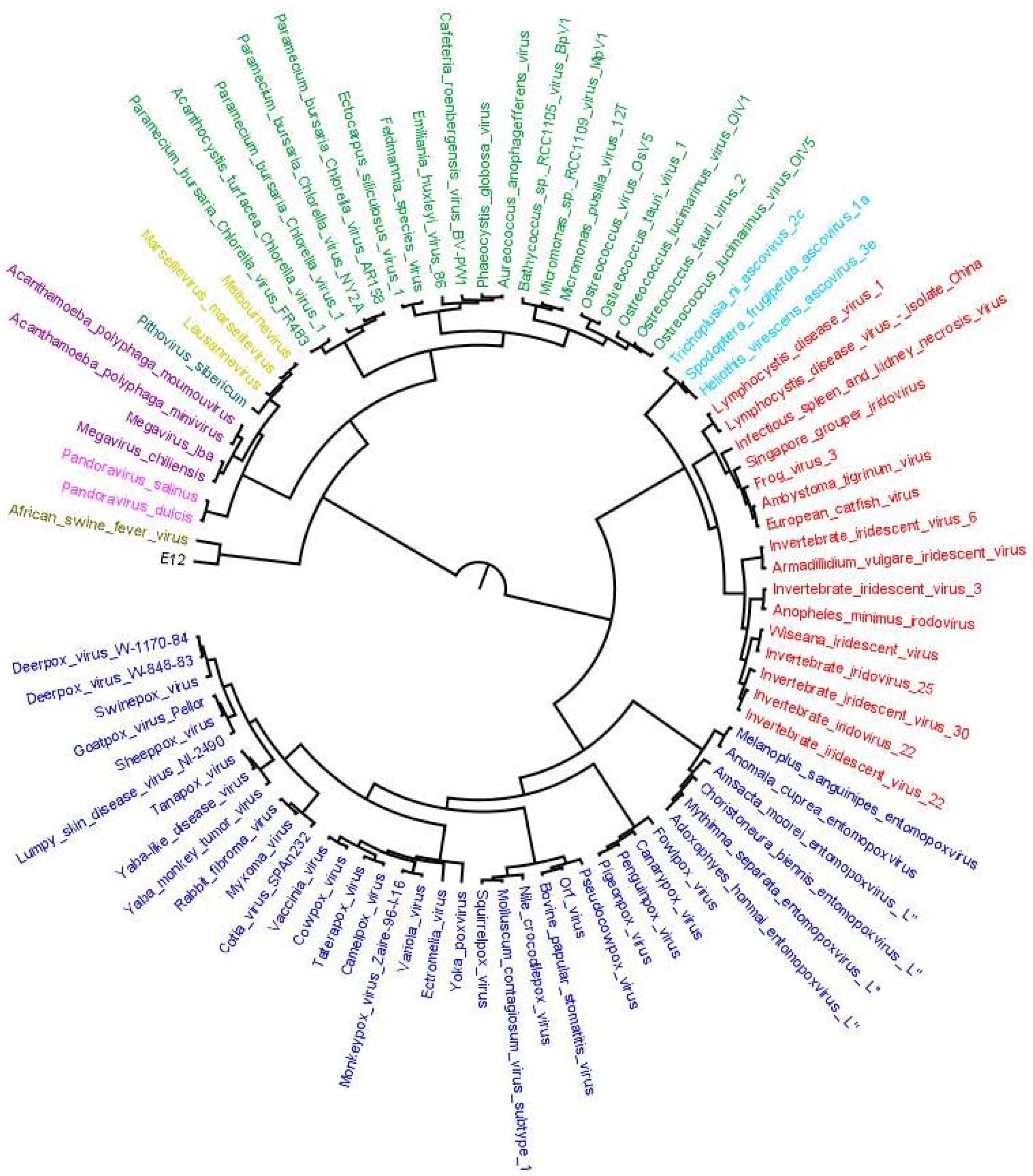
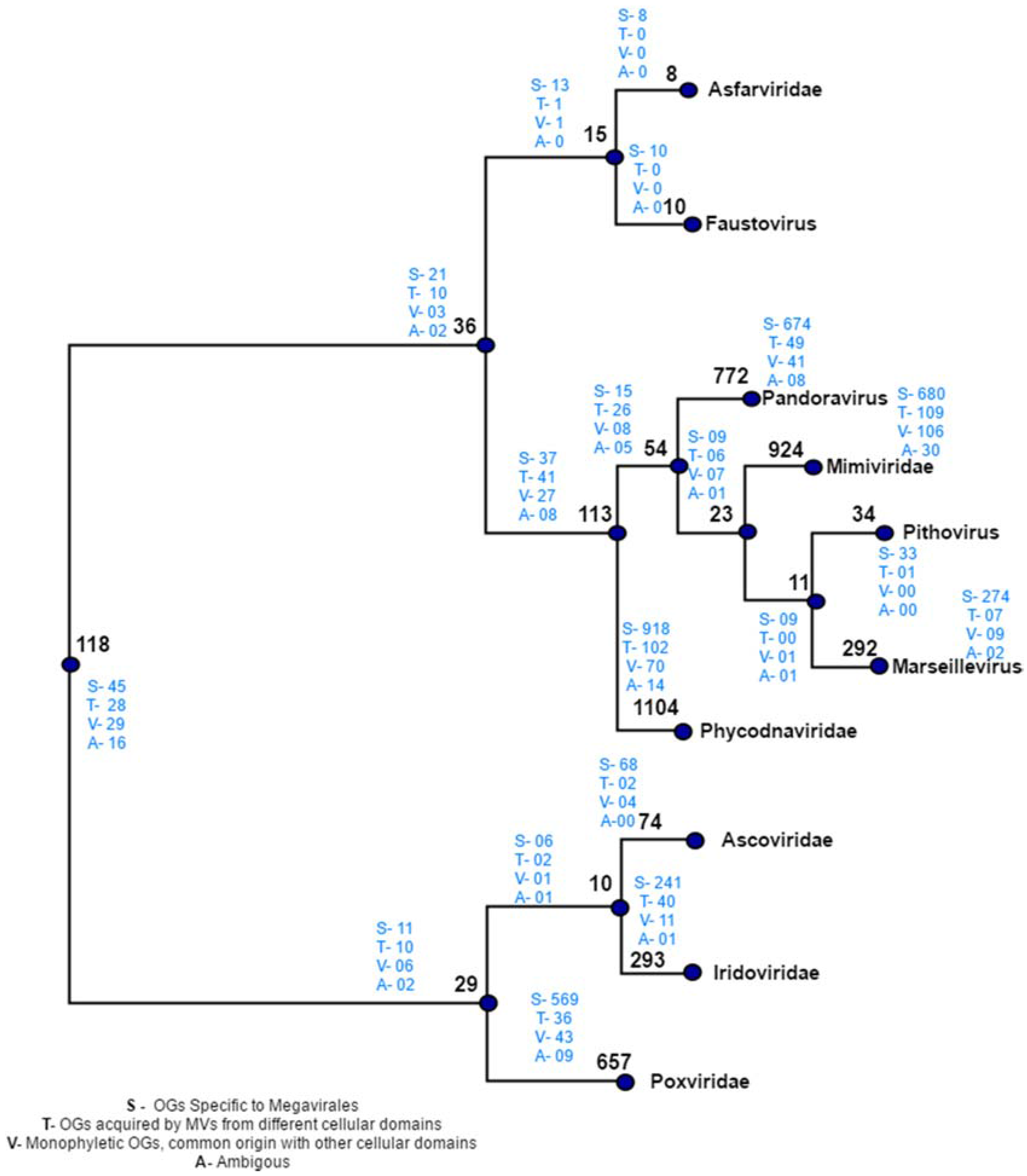
2.6. Plotting OGs and Evolutionary Scenarios on Reference Megavirale Family Tree
3. Implementation
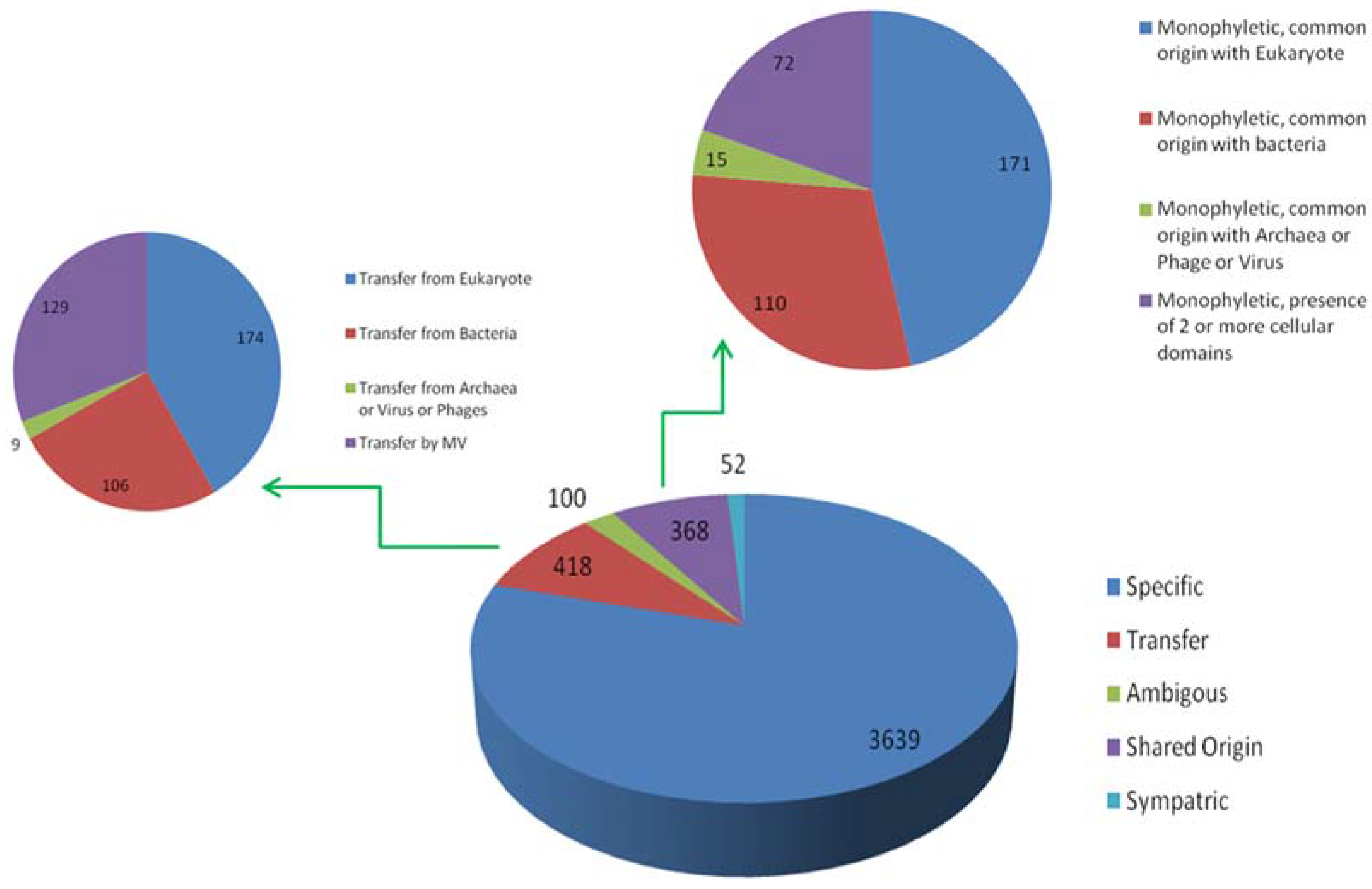
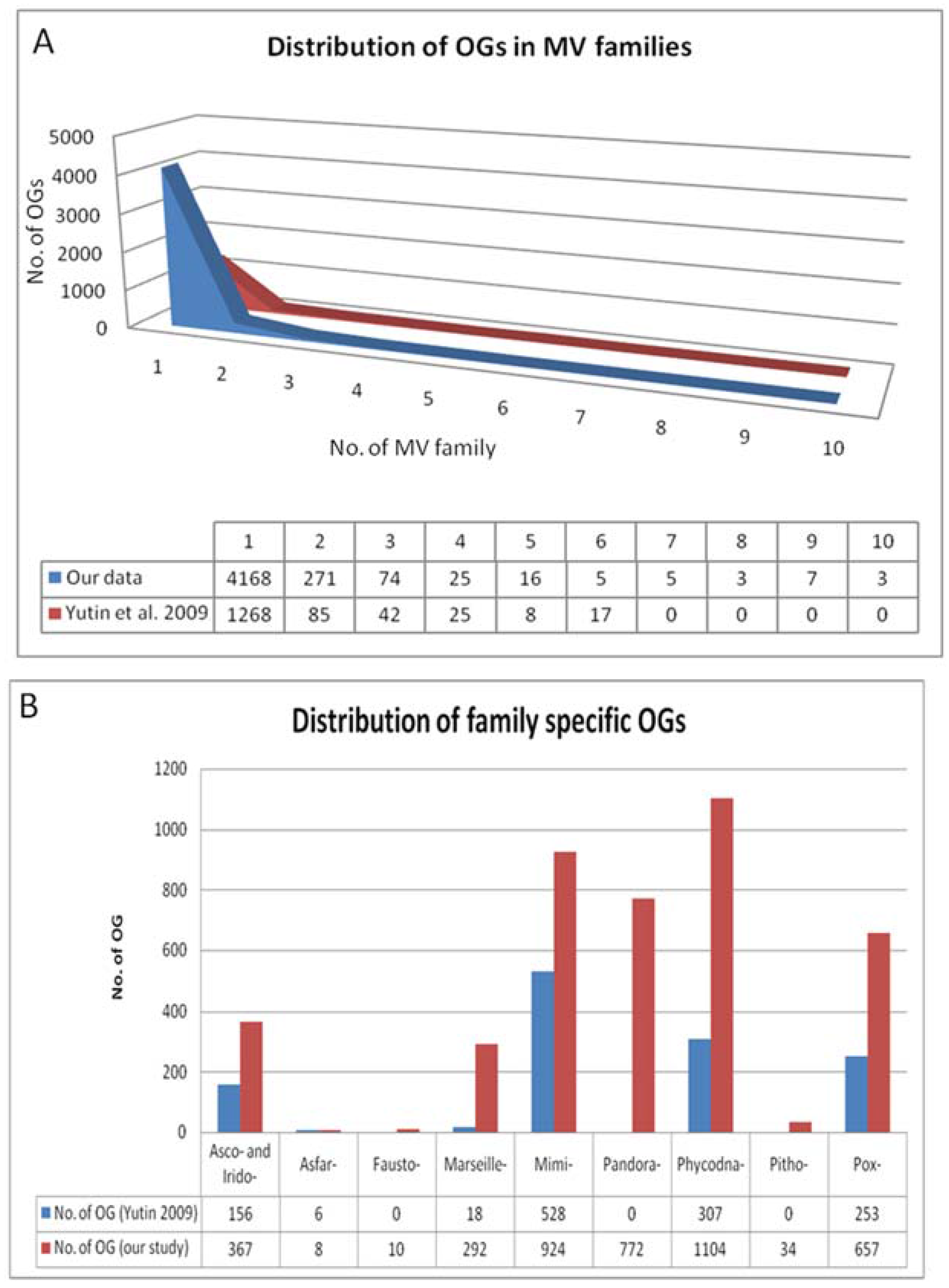
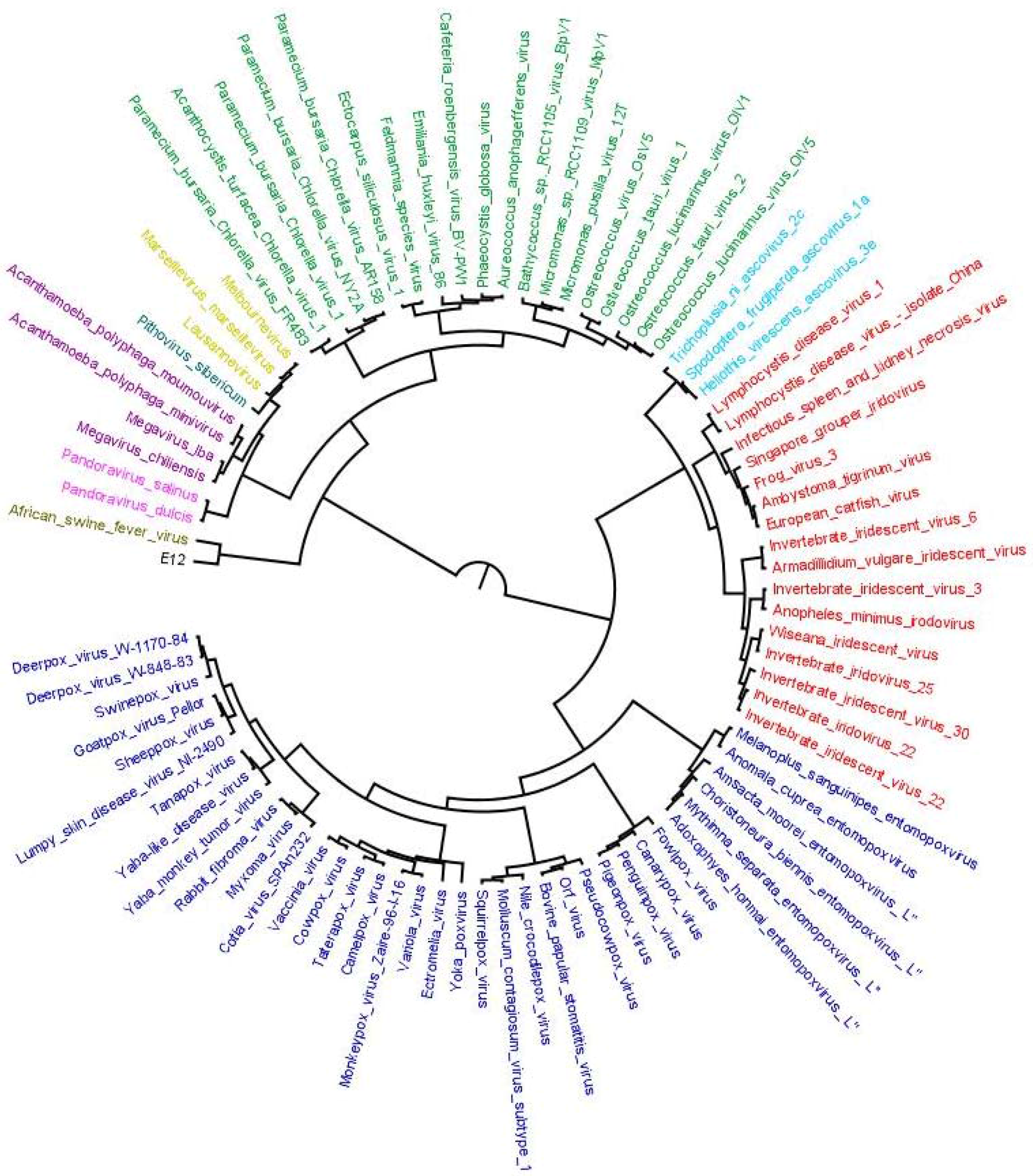
4. Results of MimiLook Analysis in the Proposed Order Megavirales
5. Discussion and Future Perspective
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- La Scola, B.; Audic, S.; Robert, C.; Jungang, L.; de Lambellerie, X.; Drancourt, M.; Birtles, R.; Claverie, J.M.; Raoult, D. A giant virus in amoebae. Science 2003, 299, 2033. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; La Scola, B.; Suzan, M.; Claverie, J.M. The 1.2-megabase genome sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Boyer, M.; Yutin, N.; Pagnier, I.; Barrassi, L.; Fournous, G.; Espinosa, L.; Robert, C.; Azza, S.; Sun, S.; Rossmann, M.G.; et al. Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc. Natl. Acad. Sci. USA 2009, 106, 21848–21853. [Google Scholar] [CrossRef] [PubMed]
- Philippe, N.; Legendre, M.; Doutre, G.; Couté, Y.; Poirot, O.; Lescot, M.; Arslan, D.; Seltzer, V.; Bertaux, L.; Bruley, C.; et al. Pandoraviruses: Amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 2013, 341, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Bartoli, J.; Shmakova, L.; Jeudy, S.; Labadie, K.; Adrait, A.; Lescot, M.; Poirot, O.; Bertaux, L.; Bruley, C.; et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc. Natl. Acad. Sci. USA. 2014, 111, 4274–4279. [Google Scholar] [CrossRef] [PubMed]
- Reteno, D.G.; Benamar, S.; Khalil, J.B.; Andreani, J.; Armstrong, N.; Klose, T.; Rossmann, M.; Colson, P.; Raoult, D.; La Scola, B. Faustovirus, an asfarvirus-related new lineage of giant viruses infecting amoebae. J. Virol. 2015, 89, 6585–6594. [Google Scholar] [CrossRef] [PubMed]
- Legendre, M.; Lartigue, A.; Bertaux, L.; Jeudy, S.; Bartoli, J.; Lescot, M.; Alempic, J.M.; Ramus, C.; Bruley, C.; Labadie, K.; et al. In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba. Proc. Natl. Acad. Sci. USA 2015, 112, 5327–5335. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Aravind, L.; Koonin, E.V. Common origin of four diverse families of large eukaryotic DNA viruses. J. Virol. 2001, 75, 11720–11734. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Wolf, Y.I.; Raoult, D.; Koonin, E.V. Eukaryotic large nucleo-cytoplasmic DNA viruses: Clusters of orthologous genes and reconstruction of viral genome evolution. Virol. J. 2009, 6, 223. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; de Lamballerie, X.; Yutin, N.; Asgari, S.; Bigot, Y.; Bideshi, D.K.; Cheng, X.W.; Federici, B.A.; Van Etten, J.L.; Koonin, E.V.; et al. “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch. Virol. 2013, 158, 2517–2521. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.M. Viruses take center stage in cellular evolution. Genome Biol. 2006, 7, 110. [Google Scholar] [CrossRef] [PubMed]
- Arslan, D.; Legendre, M.; Seltzer, V.; Abergel, C.; Claverie, J.M. Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc. Natl. Acad. Sci. USA 2011, 108, 17486–17491. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.; López-García, P. Comment on “The 1.2-megabase genome sequence of Mimivirus”. Science 2005, 308, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Balaji, S.; Koonin, E.V.; Aravind, L. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 2006, 117, 156–184. [Google Scholar] [CrossRef] [PubMed]
- Filée, J.; Siguier, P.; Chandler, M. I am what I eat and I eat what I am: Acquisition of bacterial genes by giant viruses. Trends Genet. 2007, 23, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Filée, J.; Pouget, N.; Chandler, M. Phylogenetic evidence for extensive lateral acquisition of cellular genes by Nucleocytoplasmic large DNA viruses. BMC Evol. Biol. 2008, 8, 320. [Google Scholar] [CrossRef]
- Moreira, D.; Brochier-Armanet, C. Giant viruses, giant chimeras: The multiple evolutionary histories of Mimivirus genes. BMC Evol. Biol. 2008, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Colson, P.; Raoult, D.; Koonin, E.V. Mimiviridae: Clusters of orthologous genes, reconstruction of gene repertoire evolution and proposed expansion of the giant virus family. Virol. J. 2013, 10, 106. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Wolf, Y.I.; Koonin, E.V. Origin of giant viruses from smaller DNA viruses not from a fourth domain of cellular life. Virology 2014, 466–467, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Suhre, K.J. Gene and genome duplication in Acanthamoeba polyphaga Mimivirus. J. Virol. 2005, 79, 14095–14101. [Google Scholar] [CrossRef] [PubMed]
- Filée, J.; Chandler, M. Convergent mechanisms of genome evolution of large and giant DNA viruses. Res. Microbiol. 2008, 159, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Desnues, C.; la Scola, B.; Yutin, N.; Fournous, G.; Robert, C.; Azza, S.; Jardot, P.; Monteil, S.; Campocasso, A.; Koonin, E.V.; et al. Provirophages and transpovirons as the diverse mobilome of giant viruses. Proc. Natl. Acad. Sci. USA 2012, 109, 18078–18083. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Koonin, E.V. Polintons: A hotbed of eukaryotic virus, transposon and plasmid evolution. Nat. Rev. Microbiol. 2015, 13, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Hooper, S.D.; Berg, O.G. Detection of genes with atypical nucleotide sequence in microbial genomes. J. Mol. Evol. 2002, 54, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Deschavanne, P.; Giron, A.; Vilain, J.; Dufraigne, C.; Fertil, B. Genomic signature is preserved in short DNA fragments. In Proceedings of the 13th IEEE International Conference on BioInformatics and BioEngineering, Arlilngton, VA, USA, 8–10 November 2000; pp. 161–167. [Google Scholar]
- Dufraigne, C.; Fertil, B.; Lespinats, S.; Giron, A.; Deschavanne, P. Detection and characterization of horizontal transfers in prokaryotes using genomic signature. Nucleic Acids Res. 2005, 33, e6. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Zhaxybayeva, O.; Gogarten, J.P.; Charlebois, R.L.; Doolittle, W.F.; Papke, R.T. Phylogenetic analyses of cyanobacterial genomes: Quantification of horizontal gene transfer events. Genome Res. 2006, 16, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Snel, B.; Bork, P.; Huynen, M.A. Genomes in flux: The evolution of archaeal and proteobacterial gene content. Genome Res. 2002, 12, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Hallett, M.; Lagergren, J. Efficient algorithms for lateral gene transfer problems. In Proceedings of the Fifth Annual International Conference on Research in Computational Biology; El-Mabrouk, N., Lengauer, T., Sankoff, D., Eds.; ACM Press: New York, NY, USA, 2001; pp. 149–156. [Google Scholar]
- MacLeod, D.; Charlebois, R.L.; Doolittle, F.; Bapteste, E. Deduction of probable events of lateral gene transfer through comparison of phylogenetic trees by recursive consolidation and rearrangement. BMC Evol. Biol. 2005, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Beiko, R.G.; Hamilton, N. Phylogenetic identification of lateral genetic transfer events. BMC Evol. Biol. 2006, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Than, C.; Ruths, D.; Nakhleh, L. PhyloNet: A software package for analyzing and reconstructing reticulate evolutionary relationships. BMC Bioinf. 2008, 9, 322. [Google Scholar] [CrossRef] [PubMed]
- Boc, A.; Diallo, A.B.; Makarenkov, V. T-REX: A web server for inferring, validating and visualizing phylogenetic trees and networks. Nucleic Acids Res. 2012, 40, W573–W579. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; Raoult, D. Megavirales composing a fourth domain of life: Mimiviridae and Marseilleviridae. In Viruses: Essential Agents of Life; Witzany, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 217–244. [Google Scholar]
- Pagnier, I.; Reteno, D.G.; Saadi, H.; Boughalmi, M.; Gaia, M.; Slimani, M.; Ngounga, T.; Bekliz, M.; Colson, P.; Raoult, D.; et al. A decade of improvements in Mimiviridae and Marseilleviridae isolation from amoeba. Intervirology 2013, 56, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; La Scola, B.; Birtles, R. The discovery and characterization of Mimivirus, the largest known virus and putative pneumonia agent. Clin. Infect. Dis. 2007, 45, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; La Scola, B.; Raoult, D. Giant viruses of amoebae as human pathogens. Intervirology 2013, 56, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Mueller, L.; Baud, D.; Bertelli, C.; Greub, G. Lausannevirus seroprevalence among asymptomatic young adults. Intervirology 2013, 56, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Filée, J. Genomic comparison of closely related giant viruses supports an accordion-like model of evolution. Front. Microbiol. 2015, 6, 593. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, J.; Christian, J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, S. Graph Clustering by Flow Simulation. Ph.D. Thesis, University of Utrecht, Utrecht, The Netherlands, 2000. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. PHYLIP: Phylogenetic inference package, version 3.2. Cladistics 1989, 19, 164–166. [Google Scholar]
- Criscuolo, A.; Berry, V.; Douzery, E.J.; Gascuel, O. SDM: A fast distance-based approach for (super) tree building in phylogenomics. Syst. Biol. 2006, 55, 740–755. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Paramvir, S.D.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Linder, C.R.; Warnow, T. RAxML and FastTree: Comparing two methods for large-scale maximumlLikelihood phylogeny estimation. PLoS ONE 2011, 16, e27731. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE3: Reconstruction, analysis and visualization of phylogenomic data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Ragan, M.A. On surrogate methods for detecting lateral gene transfer. FEMS Microbiol. Lett. 2001, 201, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Palmer, J.D. Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 2008, 9, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Filee, J. Multiple occurrences of giant virus core genes acquired by eukaryotic genomes: The visible part of the iceberg? Virology 2014, 466–467, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Maumus, F.; Epert, A.; Nogue, F.; Blanc, G. Plant genomes enclose footprints of past infections by giant virus relatives. Nat. Commun. 2014, 5, 42–68. [Google Scholar] [CrossRef] [PubMed]
- Forterre, P. Giant viruses: Conflicts in revisiting the virus concept. Intervirology 2010, 53, 362–378. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
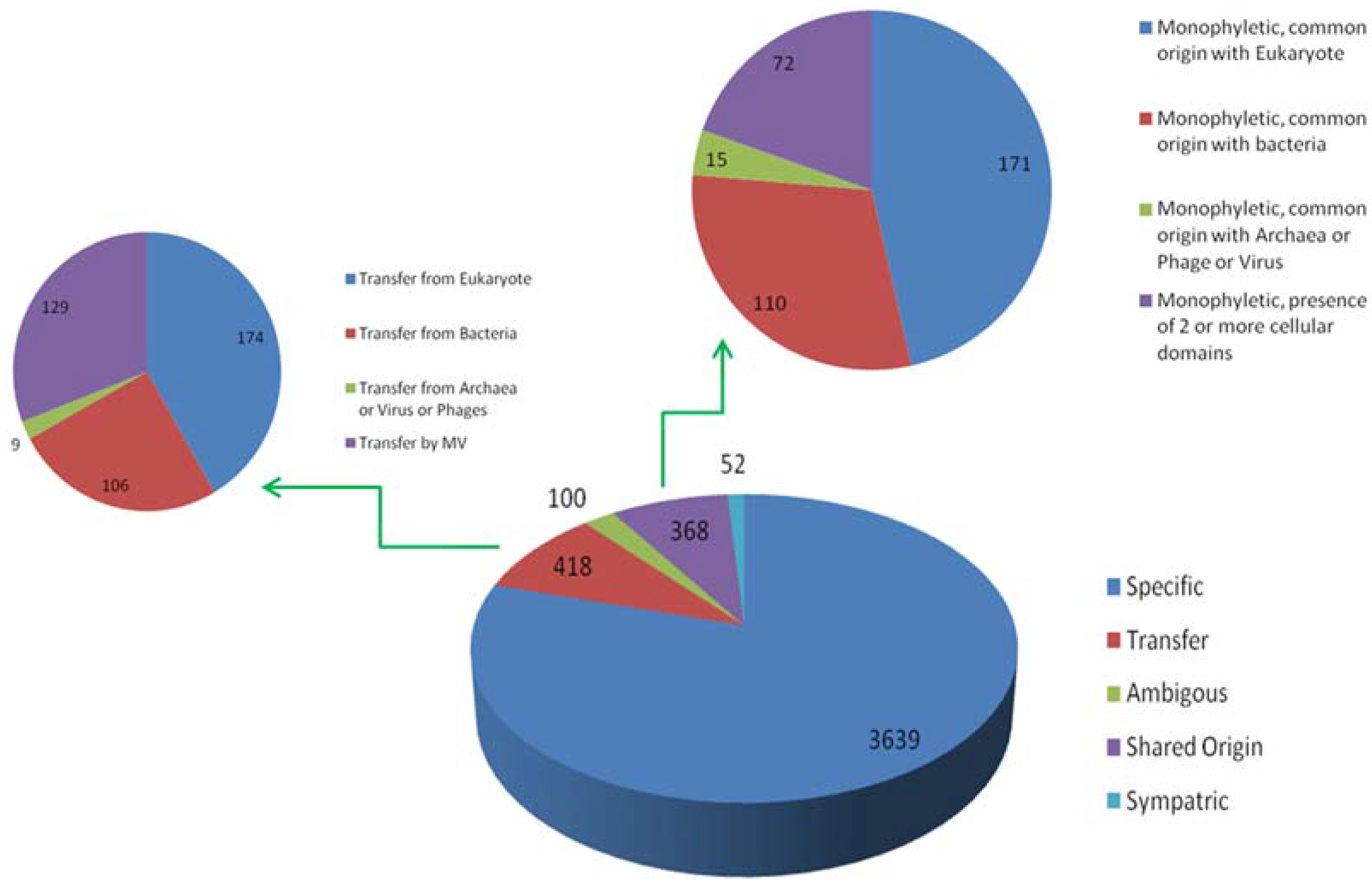{kind=link}
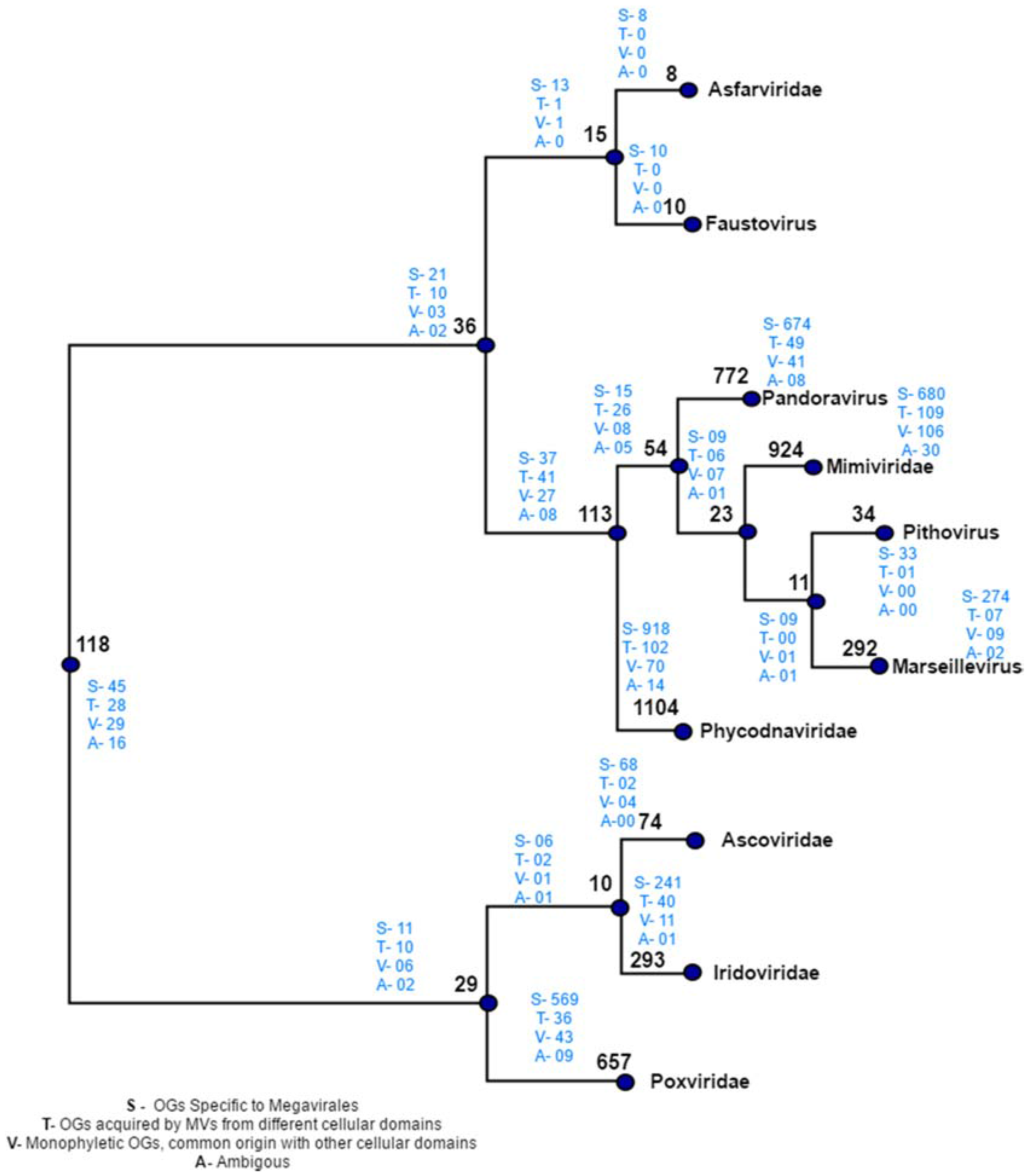{kind=link}
| ORF Name | Source of Transfer [40] | OG ID | Evolutionary Scenario Detected | Putative Donor/Receptor |
|---|---|---|---|---|
| OtV1_33/OsV5_33 | Eukaryote | OG_02476 | Transfer from eukaryote | Magnoliophyta |
| OtV1_113/OsV5_131 | Eukaryote | OG_04266 | Transfer from eukaryote | Pyrenomonadales |
| OsV5_125 | Virus | OG_00455 | Specific to Megavirales | - |
| OsV5_218 | Eukaryote | OG_00469 | Specific to Megavirales | - |
| OtV2_3 | Bacteria | OG_00262 | Shared origin with bacteria | - |
| OtV2_29 | Bacteria | OG_04066 | Transfer from bacteria | Clostridiales |
| OtV2_30 | Bacteria | OG_00775 | Transfer from bacteria | Alphaproteobacteria |
| OtV2_40 | Ambiguous | OG_02248 | Transfer from phages | Phages (Myoviridae) |
| OtV2_78/OlV1_89 | Bacteria | OG_00556 | Transfer from bacteria | Helicobacter (Proteobacteria) |
| OtV2_158/OlV1_172 | Eukaryote | OG_02251 | Shared origin with eukaryote | - |
| OtV2_165 | Bacteria | OG_02131 | Specific to Megavirales | MV specific |
| OtV2_167 | Ambiguous | OG_02177 | Shared origin with bacteria | - |
| OtV2_179 | Virus | OG_04069 | Shared origin with phages | - |
| OtV2_193/OlV1_206 | Eukaryote | OG_02255 | Specific to Megavirales | MV specific |
| OtV2_201/OlV1_213 | Host | OG_02256 | Transfer from eukaryote | Saccharomycetes |
| OtV2_202/OlV1_214 | Ambiguous | OG_02257 | Probable transfer | Probable transfer from Rickettsiaceae (bacteria) |
| OtV2_222/OlV1_234 | Host | OG_01016 | Transfer from eukaryote | Mamiellales |
| OlV1_190 | Eukaryote | OG_02481 | Sympatric transfer | Transfer from Bacteria; Gene with eukaryotes |
| OlV1_236 | Host | OG_01062 | Specific to Megavirales | MV-specific |
| MpV1_8 | Host | OG_04280 | Shared origin with eukaryote | - |
| MpV1_9 | Host | OG_04281 | Transfer from eukaryote | Mamiellales |
| MpV1_201 | Bacteria | OG_00262 | Transfer from bacteria | Enterobacteriales (independent acquisition) |
| MpV1_203 | Virus | OG_00202 | Transfer from Megavirales | Insectomime virus and Port-Miou virus |
| Ny2A_137/AR158_126 | Ambiguous | OG_00853 | Sympatric transfer | Transfer from Chlorella variabilis; Outside bacteria |
| Ny2A_359 | Ambiguous | OG_00554 | Ambiguous | Ambiguous (mix of bacteria and bacteriophages) |
| Ny2A_543/AR158_487 | Ambiguous | OG_00554 | Ambiguous | Ambiguous (mix of bacteria and bacteriophages) |
| Ny2A_542/AR158_486 | Virus | OG_03082 | Shared origin with phages | - |
| Chiliensis_647 | Ambiguous | OG_01369 | Transfer from Megavirales | Brucellaceae (bacteria) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, S.; Panda, A.; Colson, P.; Raoult, D.; Pontarotti, P. MimiLook: A Phylogenetic Workflow for Detection of Gene Acquisition in Major Orthologous Groups of Megavirales. Viruses 2017, 9, 72. https://doi.org/10.3390/v9040072
Jain S, Panda A, Colson P, Raoult D, Pontarotti P. MimiLook: A Phylogenetic Workflow for Detection of Gene Acquisition in Major Orthologous Groups of Megavirales. Viruses. 2017; 9(4):72. https://doi.org/10.3390/v9040072
Chicago/Turabian StyleJain, Sourabh, Arup Panda, Philippe Colson, Didier Raoult, and Pierre Pontarotti. 2017. "MimiLook: A Phylogenetic Workflow for Detection of Gene Acquisition in Major Orthologous Groups of Megavirales" Viruses 9, no. 4: 72. https://doi.org/10.3390/v9040072
APA StyleJain, S., Panda, A., Colson, P., Raoult, D., & Pontarotti, P. (2017). MimiLook: A Phylogenetic Workflow for Detection of Gene Acquisition in Major Orthologous Groups of Megavirales. Viruses, 9(4), 72. https://doi.org/10.3390/v9040072