Abstract
Glioblastoma, one of the most aggressive primary brain tumors, is characterized by highly immunosuppressive microenvironment. This contributes to glioblastoma resistance to standard treatment modalities and allows tumor growth and recurrence. Several immune-targeted approaches have been recently developed and are currently under preclinical and clinical investigation. Oncolytic viruses, including the autonomous protoparvovirus H-1 (H-1PV), show great promise as novel immunotherapeutic tools. In a first phase I/IIa clinical trial (ParvOryx01), H-1PV was safe and well tolerated when locally or systemically administered to recurrent glioblastoma patients. The virus was able to cross the blood–brain (tumor) barrier after intravenous infusion. Importantly, H-1PV treatment of glioblastoma patients was associated with immunogenic changes in the tumor microenvironment. Tumor infiltration with activated cytotoxic T cells, induction of cathepsin B and inducible nitric oxide (NO) synthase (iNOS) expression in tumor-associated microglia/macrophages (TAM), and accumulation of activated TAM in cluster of differentiation (CD) 40 ligand (CD40L)-positive glioblastoma regions was detected. These are the first-in-human observations of H-1PV capacity to switch the immunosuppressed tumor microenvironment towards immunogenicity. Based on this pilot study, we present a tentative model of H-1PV-mediated modulation of glioblastoma microenvironment and propose a combinatorial therapeutic approach taking advantage of H-1PV-induced microglia/macrophage activation for further (pre)clinical testing.
1. Glioblastoma: Current Clinical State-Of-The-Art
Glioblastoma is the most common and aggressive primary brain tumor. It has a dismal prognosis and is typically characterized by largely inevitable recurrence within six months to one year after initial treatment [1,2]. The current standard of care for newly diagnosed patients includes maximal surgical resection and subsequent concurrent treatment with radiation and temozolomide (TMZ), followed by adjuvant chemotherapy [3]. Due to glioblastoma invasiveness and frequent location in injury-prone brain areas controlling essential motor and cognitive functions, radical resection is only feasible in a subset of patients [1]. Furthermore, at recurrence only a minority of the patients is eligible for second surgery [4], which is so far the only approach firmly associated with improved survival if complete resection of the recurrent tumor is accomplished [5]. In recurrent glioblastoma patients, standard cytotoxic agents, corticosteroids, TMZ rechallenge, carboplatin and irinotecan are applied, among other agents, to palliate symptoms and improve quality of life, but fail to prolong the time to progression [6]. In 2009, the humanized monoclonal antibody bevacizumab targeting tumor angiogenesis (through the vascular endothelial growth factor (VEGF)), was approved for the treatment of recurrent glioblastoma patients on the basis of its ability to achieve superior progression-free survival (PFS), yet in the lamentable absence of meaningful overall survival (OS) improvement [7].
2. Immune Suppression in Glioblastoma Patients
Glioblastoma patients generally show profound local and systemic immune suppression [8]. This tumor-supportive environment is characterized by reduced cluster of differentiation (CD)4+ T lymphocyte counts, increased Forkhead-Box-Protein P3 (FoxP3+) regulatory T cell (Treg) numbers, and elevated circulating myeloid-derived suppressor cells [9,10,11]. Defective receptor expression by monocytes and impaired dendritic cell (DC) function has also been described [12]. Glioblastoma cells and associated microglia/macrophages express various factors that contribute to the establishment of functional immune suppression in the tumor microenvironment. Immunosuppressive and tumor-promoting cytokines, such as interleukin (IL)-1, IL-6, IL-10, transforming growth factor (TGF)-β, colony stimulating factor (CSF) 1, VEGF, and prostaglandin E2 (PGE2), among others, are secreted and impair anti-glioma immune responses. Furthermore, glioblastoma and associated microglia/macrophage cells upregulate several surface molecules (CD95, CD70, programmed cell death ligand 1 (PD-L1)) which inhibit T cell activation and induce T cell apoptosis, thus reducing the numbers of tumor-infiltrating immune effector cells [8,13]. In consequence, the profound immune masking results in tumor evasion from host immune surveillance and leads to subsequent tumor growth and invasion [13]. These observations, together with unavoidable recurrence and resistance of recurrent glioblastoma to conventional treatments, prompted the search for new, immune-targeted therapeutic options to reverse tumor-/tumor microenvironment-driven immune suppression and activate antitumor immune responses.
3. Immune-Targeted Therapeutic Approaches for Glioblastoma
Data from preclinical studies provided convincing evidence that immunotherapy is able to promote efficient anti-glioma immune responses [14,15]. Modulation of human cytomegalovirus (CMV)-specific DCs, application of glioma stem cell-antigens-loaded DC vaccines, cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and PD-1 blockage, etc., are among the numerous extensive works (as reviewed in detail in [14,15]) that have been recently done and allowed for a clinical translation of glioblastoma immune targeting. Several immunotherapy approaches are currently under clinical evaluation, including tumor vaccination, immune checkpoint inhibition and adoptive T cell transfer.
3.1. Tumor Vaccination
In newly diagnosed glioblastoma patients, Rindopepimut—an epidermal growth factor receptor variant III (EGFR vIII)-specific peptide vaccine—reached phase III clinical testing but disappointingly, when combined with TMZ, failed to add significant OS benefit compared to TMZ alone [16]. This was ascribed to the relatively low percentage of newly diagnosed glioblastomas which express this EGFR mutant [17]. Indeed, the vast heterogeneity of tumor-specific antigen expression in glioblastoma is a major factor limiting the tumor vaccination approach. Hope in that regard has been raised by the identification of ten novel glioblastoma-associated tumor antigens [18] whose curative potential as a multi-peptide vaccine, using CD8+ T cell epitopes, has been tested in phase I/II clinical trials [14]. A phase III clinical assessment of a DC-based vaccine is currently active and results are awaited.
3.2. Immune Checkpoint Inhibition
The first Food and Drug Administration (FDA)-approved immune checkpoint inhibitor was the humanized anti-CTLA-4 antibody ipilimumab. Although an improved OS was achieved in a phase III melanoma clinical trial [19], ipilimumab showed severe immune-related adverse effects [20] and a clinical utility limited to only a small subset of glioblastoma patients [14]. Other immune checkpoint inhibitors are presently under clinical evaluation in glioblastoma patients, in particular anti-PD-1 (nivolumab, pembrolizumab) and anti-PD-L1 (MEDI4736) antibodies.
3.3. Adoptive T Cell Transfer
One clinical trial, in which autologous T cells specific for glioblastoma-expressed CMV antigens were adoptively transferred to recurrent glioblastoma patients, reported significant OS prolongation [21]. Another trial of autologous therapy with CMV-specific T cells is ongoing. Also ongoing are clinical studies using adoptive transfer of autologous T cells with chimeric antigen receptor (CAR) targeting different glioblastoma-associated antigens [14,15].
Despite the impressive research progress recently made in cancer/glioblastoma immunotherapy, it has to be noted that some of the approaches were associated with immune-related adverse effects, imposing the need for concerned risk–benefit assessment. Some studies have reported gastrointestinal, dermatological and endocrine toxicities attributable to immune checkpoint blockade [22]. Furthermore, life-threatening neurotoxicity and other severe complications, including on-target off-tumor toxicities, were documented after CAR T cell infusion [23].
4. Need for Novel Immunotherapeutic Strategies: Oncolytic Viruses
As apparent from above, an improved arsenal of immunotherapeutic agents is still needed. Recently, cancer immunotherapy using oncolytic viruses (OVs) has gained much attention and raised considerable hopes [24,25,26,27]. These agents possess the ability of infecting and killing malignant cells without causing harm to normal tissues. This oncoselectivity is a complex phenomenon and is largely due to the dependence of OV life cycle on various tumor cell-specific factors. OV-induced tumor cell toxicity coupled with host immune system stimulation warrant OV clinical development as targeted multimodal cancer therapeutics.
5. Oncolytic H-1 Parvovirus
An emerging candidate is the rodent H-1 protoparvovirus (H-1PV), the smallest among all OVs. H-1PV is endowed with natural anticancer activity and is nonpathogenic for humans. These properties, together with the lack of pre-existing immunity in the human population [28], the potential of H-1PV for application via multiple routes (intratumoral, intravenous, intranasal), and its capacity to cross the blood–brain barrier, make this virus a suitable tool for oncolytic virotherapy of several tumor entities, including those of the central nervous system [29,30,31,32]. In addition, H-1PV oncolysis-associated proinflammatory host immune responses observed in preclinical studies raise hopes that parvovirotherapy may offer a safe alternative to current immune-targeted approaches [33].
6. Immunotherapeutic Potential of Oncolytic H-1PV: Preclinical Evidence
6.1. In Tumor Models Other than Glioma
The ability of H-1PV-infected tumor cells to activate antitumor immune responses has been demonstrated in several preclinical cancer models, in particular hepatoma [34], melanoma [35], pancreatic [36], colorectal [37,38] and nasopharyngeal [39] carcinomas. In these models, H-1PV-infected tumor cell lysates induced DC maturation, proinflammatory cytokine secretion, tumor-associated antigen cross-presentation, and T/Natural Killer (NK) cell stimulation.
6.2. In Glioma Models
In contrast to pancreatic cancer and melanoma, for which extensive preclinical work demonstrated enhanced immunogenicity of H-1PV-infected versus non-infected tumors, considerably less is known about H-1PV-infected glioma interactions with the host immune system. In glioma-bearing immunocompetent animals, spleno- and lymphadenomegaly are observed upon infection with minute virus of mice (an autonomous protoparvovirus closely related to H-1PV), together with increased interferon-gamma (IFN-γ) production in spleen and tumor-draining lymph nodes. Proinflammatory cytokine release and upregulated CD80/83/86 expression were detected in antigen-presenting cells, including microglia [40]. In regard to H-1PV, the only preclinical evidence gathered so far comes from an orthotopic rat glioma model, in which H-1PV treatment was able to eradicate even advanced tumors [41]. This therapeutic effect was however present only if the host immune system was intact: T cell depletion impaired H-1PV-induced tumor regression. Importantly, the sole presence of T cells, in the absence of H-1PV treatment, was not sufficient to cause glioma suppression. These observations argue for a role of host T cell responses in H-1PV-promoted oncosuppression [32].
7. Immunotherapeutic Potential of H-1PV in Glioblastoma Patients: First Evidence of Immunogenic Tumor Microenvironment Conversion
Several OVs have been tested in glioma clinical trials and hold considerable promise as novel targeted brain tumor therapeutics. The most advanced are the clinical studies using herpes simplex virus (HSV), Newcastle disease virus (NDV), as well as adeno-, reo-, vaccinia, measles, polio-, and vesicular stomatitis virus (VSV) [42,43]. ParvOryx01 (NCT01301430) was the pilot clinical trial of an oncolytic parvovirus, H-1PV, in patients with recurrent glioblastoma [44]. This trial investigated the application of escalating parvovirus doses via either intravenous or intracerebral routes. In addition to assessing H-1PV safety, tolerability, pharmacokinetics, shedding and maximum tolerated dose, tumor tissue samples were acquired at resection and allowed the analysis of glioblastoma cells and glioblastoma microenvironment nine days after parvovirotherapy. The results of the ParvOryx01 clinical trial have been recently published [45]. Together with the promising clinical observations (reliable safety, good tolerability, well predictable pharmacokinetics, induction of neutralizing antibodies in a virus dose-dependent manner while providing a window for booster H-1PV reapplications, and extended median survival in comparison with recent meta-analyses), several intriguing findings arose from the in-situ analyses of resected parvovirus-treated tumors.
7.1. Glioblastoma Infiltration with Immune Cells
In comparison with non-parvovirus-treated (historical) controls, increased tumor infiltration with CD45+/CD3+/CD8+ T lymphocytes was observed in some ParvOryx01 patients. Tumor-infiltrating CD4+ and CD25+ cells were also present, but importantly, FoxP3+ Treg cells were only a minor subfraction of the tumor immune cell infiltrate [45]. Of note, tumor-infiltrating cytotoxic T cells (CTLs) were found to be PD-1-negative.
7.2. Activation Status of Glioblastoma-Infiltrating Immune Cells
The activated state of tumor-infiltrating CTLs was demonstrated by the abundant expression of granzyme B and the presence of perforin [45]. The CD4+ helper T cell activation marker CD 40 ligand (CD40L) was also expressed in parvovirus-treated glioblastomas. Surprisingly, CD40L expression was also detected in non-lymphocyte-infiltrated glioblastomas, hinting at CD40L cellular source other than CD4+ T cells.
7.3. CD40L Expression by Glioblastoma Cells
In line with the above, CD40L expression was detected in non-macrophage, EGFR-positive, i.e., most likely glioblastoma cells. In contrast, CD40 expression was not observed, either on tumor or on tumor-associated microglia/macrophage (TAM) cells. Interestingly, CD40L expression in glioblastoma cells was first reported in 2015 and found to be a positive prognostic factor, while co-expression of both CD40 and CD40L in glioblastoma cells correlated with negative prognosis [46]. It is noteworthy that in the ParvOryx01 study, CD40+ cells with a non-macrophage phenotype were observed in the tumor blood vessel lumen in patients who received H-1PV by intravenous infusion (Figure 1). With the limitation imposed by the unavailability of tumor samples from time points later than nine days after treatment, we hypothesize that CD40-expressing peripheral blood DCs or monocytes may be recruited to H-1PV-infected tumors and interact with CD40L+ glioblastoma cells.
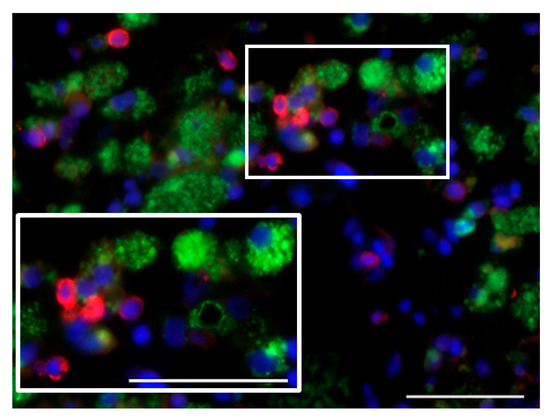
Figure 1.
Intra- and perivascularly located cluster of differentiation (CD)40+ cells lacking CD68 expression infiltrate the tumor of an intravenously parvovirus-treated glioblastoma patient. CD40, red; CD68, green; DAPI, blue. Scale bars: 50 μm.
7.4. Proinflammatory Cytokine Production in Glioblastoma Microenvironment
Two proinflammatory cytokines, IFN-γ and IL-2, were detected in the glioblastoma microenvironment of ParvOryx01 patients who also showed increased tumor infiltration with lymphocytes [45]. IL-12 was not found at detectable levels, at least at the single time point at which tissue material was collected. With this limitation imposed by the trial’s risk-minimizing design, the kinetics of proinflammatory cytokine production in parvovirus-treated glioblastomas could not be followed, and a transient, short lasting expression may be missed. The cellular source of IFN-γ and IL-2 was not identified within the frame of the trial, but two types of cells (tumor-infiltrating CD4+ T cells and/or activated TAM, see below) appear as the most likely candidates.
7.5. Induction of Cathepsin B Expression
In agreement with preclinical data demonstrating cathepsin B induction and cytosolic translocation in H-1PV-infected human glioma cells [47], the expression of this lysosomal protease was significantly increased in ParvOryx01 patients, when compared with historical recurrent glioblastoma cases. Contrary to initial expectations, cathepsin B expression was observed not only in tumor cells, but mostly in TAM [45]. As increased cathepsin B production correlates with microglial activation [48], and induction of glioma apoptosis by microglia-derived cathepsin B has been demonstrated in vitro [49], first steps were undertaken within the frame of the ParvOryx01 study, to assess glioblastoma-associated microglia/macrophage activation state. Expression of inducible nitric oxide (NO) synthase (iNOS), a marker of classical (proinflammatory, macrophage phenotype M1) microglia/macrophage activation, was assessed in four ParvOryx01 patients, selected on the basis of strong CD68 positivity. While in one patient iNOS expression was not revealed, iNOS-positive cells were detected in the other three patients’ tumors at day nine after H-1PV application. The availability of resection material from the primary tumor and a second recurrence (respectively preceding and following first recurrence subjected to H-1PV treatment) provided the unique opportunity to analyze glioblastoma microenvironment in a non-parvovirus-exposed tumor, as well as at a later time point after virotherapy. Compared with the control primary glioblastoma, the first recurrence (resected at day nine post-treatment) showed a clear induction of both cathepsin B [45] and iNOS expression in TAM cells, which persisted and even increased in the second recurrence at two months after virus application (Figure 2 and Figure 3). These data should be put together with the in vitro demonstration of human macrophage activation as a result of their abortive infection with H-1PV [36], and with the constitutive NO and oxygen species production in human promonocytic cell clones surviving H-1PV-induced apoptosis [50]. It should also be stated that in a recent study of iNOS expression in grade IV glioblastoma patients, the iNOS levels detected failed to positively correlate with survival [51]. Altogether, the above data provide first hints on H-1PV ability to trigger M1 microglia/macrophage polarization in the glioblastoma microenvironment. This concept is still speculative but would be worth corroborating through further (pre)clinical studies, as it is of major relevance for future combinatorial approaches using H-1PV and other immunotherapeutic agents.
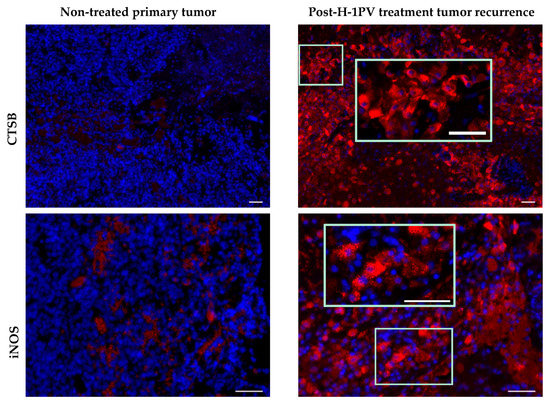
Figure 2.
Comparative analysis of cathepsin B (CTSB) and inducible nitric oxide (NO) synthase (iNOS) expression in non-H-1 parvovirus (H-1PV)-treated primary tumor and in the second tumor recurrence (two months after H-1PV treatment of the first recurrence). CTSB, red; iNOS, red; DAPI, blue. Scale bars: 50 μm.
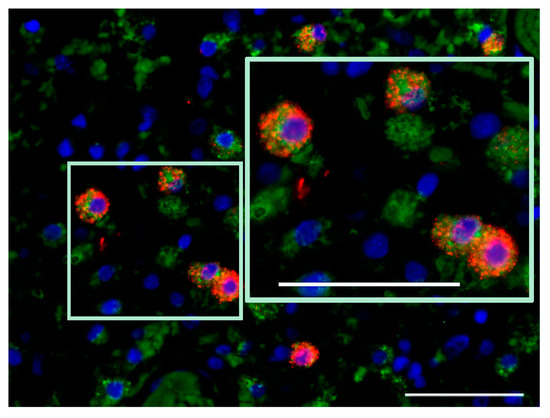
Figure 3.
Microglia/macrophage (CD68+) phenotype of iNOS-expressing cells in the second tumor recurrence (two months after H-1PV treatment of the first recurrence). CD68, green; iNOS, red; DAPI, blue. Scale bars: 50 μm.
8. Other Oncolytic Viruses and Tumor Microenvironment Modulation
In a recent editorial, de Vries et al. emphasized that the most successful OV-based strategy will be the one taking advantage of these viruses’ capacity for modifying the tumor microenvironment [52]. Indeed, several OVs were reported to exert positive effects in regard to tumor microenvironment modulation.
8.1. Preclinical Studies
Zamarin et al. observed marked infiltration of distant tumors with effector, but not Treg cells, in melanoma-bearing mice treated with Newcastle disease virus [53]. Intratumoral administration of an oncolytic adenovirus decreased tumor-infiltrating Treg cell numbers and stimulated IFN-γ-producing CD8+ T cells in a mouse glioblastoma model [54]. Recently, the immunotherapeutic potential of a recombinant polio/rhinovirus chimera was demonstrated in vitro in high-grade glioblastoma [55]. Other preclinical studies using bladder, colon, and breast cancer animal models also pointed to recombinant poxviruses’ positive effects on the immunological components of the tumor microenvironment, as summarized in [52].
8.2. Clinical Studies
In a clinical setting, positive effects (dense tumor infiltration with effector T cells, decreased Treg and myeloid-derived suppressor cell numbers, and increased IFN-γ production by CD4+ and CD8+ tumor-infiltrating lymphocytes) of recombinant vaccinia and HSV, and of measles virus on the tumor microenvironment were demonstrated in melanoma and cutaneous T cell lymphoma patients [56,57,58,59]. Remarkably, responses were observed in both injected and non-injected melanoma lesions, in patients who were intratumorally treated with a granulocyte-macrophage colony-stimulating factor-encoding second-generation HSV [60]. The added value of poxvirus-mediated intratumoral expression of specific tumor-associated antigens was supported by clinical trials in patients with prostate and locally advanced pancreatic cancer [52]. However, clinical data on antitumor immune responses and tumor microenvironment modulation in OV-treated patients remain presently limited [61].
9. Tumor Microenvironment Modulation by H-1PV: Current Hypothesis and Perspective
Glioblastoma microenvironment is marked by profound immunosuppression [8]. This tumor-supportive microenvironment [51,62] is established by glioblastoma stem cells, tumor and TAM cells. The pilot ParvOryx01 study gathered initial evidence of tumor microenvironment immunogenic conversion in recurrent glioblastoma patients who underwent local or systemic H-1 parvovirotherapy (Table 1).

Table 1.
Main characteristics of glioblastoma microenvironment as described in the literature or observed in H-1 parvovirus-treated glioblastoma patients.
Based on both preclinical experience and the observations from ParvOryx01 patients, we believe that H-1PV treatment may lead to glioblastoma-associated microglia/macrophage activation. This is supported by the increased expression of CD68, cathepsin B and iNOS in tumors of H-1PV-treated patients, in comparison with historical non-parvovirus-treated controls. As a direct result of their abortive parvovirus infection and/or indirectly, via proinflammatory mediators released by infected tumor cells, microglia/macrophages get activated and may exert toxic effects on neighboring glioblastoma cells through cathepsin B and NO release (Figure 4). Another likely scenario, supported by evidence from animal models [40], is that microglia/macrophage capacity for expressing immune costimulatory molecules (usually strongly compromised in glioblastoma) may also be increased upon H-1PV-triggered activation. Tumor tissue collection in this first trial, primarily designed to assess safety, did not allow performing such studies, but in future H-1PV glioblastoma trials, tumor infiltration with DCs, the CD40/CD40L axis, and costimulatory molecule expression on TAM and DCs will be worth investigating.
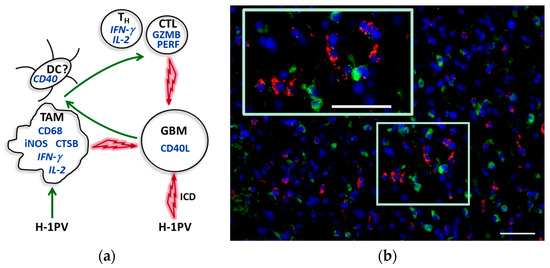
Figure 4.
Central role of TAM in the immunogenic conversion of glioblastoma microenvironment upon H-1PV infection. (a) Tentative model based on existing (pre)clinical data. Interacting cells are presented with corresponding proinflammatory products (in italics if cellular source unproved). The identification of CD40+ myeloid cells as being denditric cells (DCs) is putative. Green and red arrows indicate activating and cytotoxic interactions, respectively. For details, see main text. TH, helper T cell; CTL, cytotoxic T lymphocyte; GZMB, granzyme B; PERF, perforin; GBM, glioblastoma; ICD, immunogenic cell death; (b) Activated TAM (CD68, green) in CD40L-positive (red) tumor region. Scale bars: 50 μm.
In conclusion, H-1PV—an oncolytic parvovirus which showed excellent safety and tolerability upon local and systemic application in glioblastoma patients—may represent a novel tool for therapeutic TAM activation (Figure 4). Immunogenic conversion of TAM is the subject of intensive investigations, as exemplified by the recent preclinical demonstration of the antifungal agent’s amphotericin B capacity for restoring the suppressed ability of glioblastoma-associated microglia/macrophages to target brain tumor-initiating cells [65]. We suggest that in future attempts at prolonging time to glioblastoma recurrence, H-1PV (alone or with other treatments targeting TAM) may be combined with other immunotherapeutic modalities, in particular CAR T cell-based strategies and immune checkpoint blockade, to achieve synergistic antiglioblastoma effects.
Acknowledgments
We thank Alexandra Just for excellent technical assistance, and are deeply indebted to Anne Régnier-Vigouroux (Johannes Gutenberg University, Mainz) and Volker Daniel (Institute of Immunology, University Hospital, Heidelberg) for critical reading of the manuscript. We are grateful to the ParvOryx01 trial team [44,45] for making the tumor samples described in this report available with all necessary approvals. The ParvOryx01 clinical study and the accompanying research program were sponsored by ORYX GmbH & Co. KG (Baldham, Germany).
Author Contributions
A.L.A. conceived and designed the immunological analysis of tumor microenvironment; M.B. performed in situ immunofluorescent staining experiments; K.G. and A.U. conducted patients’ treatment and post-treatment tumor tissue collection; A.L.A. and J.R. wrote the paper.
Conflicts of Interest
K.G. and J.R. are co-inventors in patents/patent applications relating to the cooperation between parvoviruses and the immune system.
References
- Wilson, T.A.; Karajanis, M.A.; Harter, D.H. Glioblastoma multiforme: State of the art and future therapeutics. Surg. Neurol. Int. 2014, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Mirimanoff, R.O. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology: Central Nervous System Cancers, Version 1.2015. 2015. Available online: https://www.nccn.org/professionals/physician_gls/pdf/cns.pdf (accessed on 17 October 2017).
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro Oncol. 2013, 15, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Suchorska, B.; Weller, M.; Tabatabai, G.; Senft, C.; Hau, P.; Sabel, M.C.; Herrlinger, U.; Ketter, R.; Schlegel, U.; Marosi, C.; et al. Complete resection of contrast-enhancing tumor volume is associated with improved survival in recurrent glioblastoma—Results from the DIRECTOR trial. Neuro Oncol. 2016, 18, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, E.; Tosoni, A.; Bartolini, S.; Mazzocchi, V.; Fioravanti, A.; Brandes, A.A. Treatment options for recurrent glioblastoma: Pittfalls and future trends. Expert Rev. Anticancer Ther. 2009, 9, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Razavi, S.-M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune evasion strategies of glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, M.P.; Lin, Y.; New, K.C.; Bulur, P.A.; O’Neill, B.P.; Gastineau, D.A.; Dietz, A.B. Systemic immune suppression in glioblastoma: The interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. 2010, 12, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E.; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased regulatory T cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006, 66, 3294–3302. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.C.; Gonzalez, G.C.; Zhang, L.; Ibrahim, G.; Kelly, J.J.; Gustafson, M.P.; Lin, Y.; Dietz, A.B.; Forsyth, P.A.; Wee Yong, V.; et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol. 2010, 12, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Ogden, A.T.; Horgan, D.; Waziri, A.; Anderson, D.; Louca, J.; McKhann, G.M.; Sisti, M.B.; Parsa, A.T.; Bruce, J.N. Defective receptor expression and dendritic cell differentiation of monocytes in glioblastomas. Neurosurgery 2006, 59, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 2015, 17, vii9–vii14. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Zhang, H.; Gu, L.; Ye, B.; Jian, Z.; Stary, C.; Xiong, X. Advances in immunotherapy for glioblastoma multiforme. J. Immunol. Res. 2017, 2017, 3597613. [Google Scholar] [CrossRef] [PubMed]
- Weiss, T.; Weller, M.; Roth, P. Immunotherapy for glioblastoma: Concepts and challenges. Curr. Opin. Neurol. 2015, 28, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Marucci, A. Rindopeptimut Misses OS Endpoint in Phase III Glioblastoma Trial; OncLive: Cranbury, NJ, USA, 2016; Available online: http://www.onclive.com/web-exclusives/rindopepimut-misses-os-endpoint-in-phase-iii-glioblastoma-trial (accessed on 20 October 2017).
- Pelloski, C.E.; Ballman, K.V.; Furth, A.F.; Zhang, L.; Lin, E.; Sulman, E.P.; Bhat, K.; McDonald, J.M.; Yung, W.K.; Colman, H.; et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J. Clin. Oncol. 2007, 25, 2288–2294. [Google Scholar] [CrossRef] [PubMed]
- Dutoit, V.; Herold-Mende, C.; Hilf, N.; Schoor, O.; Beckhove, P.; Bucher, J.; Dorsch, K.; Flohr, S.; Fritsche, J.; Lewandrowski, P.; et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain 2012, 135, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermot, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimimab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Kefford, R.; Marshall, M.A. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Schuessler, A.; Smith, C.; Beagley, L.; Boyle, G.M.; Rehan, S.; Matthews, K.; Jones, L.; Crough, T.; Dasari, V.; Klein, K.; et al. Autologous T-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res. 2014, 74, 3466–3476. [Google Scholar] [CrossRef] [PubMed]
- Gelao, L.; Criscitiello, C.; Esposito, A.; Goldhirsch, A.; Curigliano, G. Immune checkpoint blockade in cancer treatment: A double-edged sword cross-targeting the host as an “innocent bystander”. Toxins 2014, 6, 914–933. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Schüßler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T cells—Challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Lawler, S.E.; Chiocca, E.A. Oncolytic virus-mediated immunotherapy: A combinatorial approach for cancer treatment. J. Clin. Oncol. 2015, 33, 2812–2814. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Cancer Immunother. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Harrington, K.J.; Pandha, H.S.; Vile, R.G.; Melcher, A.A.; Errington, F. Oncolytic viruses: A novel form of immunotherapy. Expert Rev. Anticancer Ther. 2008, 8, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Newman, S.J.; McCallin, P.F.; Sever, J.L. Attempts to isolate H-1 virus from spontaneous human abortions: A negative report. Teratology 1970, 3, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Rommelaere, L.; Geletneky, K.; Angelova, A.L.; Daeffler, L.; Dinsart, C.; Kiprianova, I.; Schlehofer, J.R.; Raykov, Z. Oncolytic parvoviruses as cancer therapeutics. Cytokine Growth Factor Rev. 2010, 21, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Bonifati, S.; Scott, E.M.; Angelova, A.L.; Rommelaere, J. Oncolytic parvoviruses: From basic virology to clinical applications. Virol. J. 2015, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.L.; Geletneky, K.; Nüesch, J.P.F.; Rommelaere, J. Tumor selectivity of oncolytic parvoviruses: From in vitro and animal models to cancer patients. Front. Bioeng. Biotechnol. 2015, 3, 55. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Nüesch, J.P.F.; Angelova, A.; Kiprianova, I.; Rommelaere, J. Double-faceted mechanism of parvoviral oncosuppression. Curr. Opin. Virol. 2015, 13, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.; Goepfert, K.; Heinrich, B.; Breitbach, C.J.; Delic, M.; Galle, P.R.; Rommelaere, J. Oncolytic virotherapy as emerging immunotherapeutic modality: Potential of parvovirus H-1. Front. Oncol. 2014, 4, 92. [Google Scholar] [CrossRef] [PubMed]
- Raykov, Z.; Grekova, S.; Galabov, A.S.; Balboni, G.; Koch, U.; Aprahamian, M.; Rommelaere, J. Combined oncolytic and vaccination activities of parvovirus H-1 in a metastatic tumor model. Oncol. Rep. 2007, 17, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.H.; Zeidler, M.; Wilsberg, V.; Cornelis, J.J.; Woelfel, T.; Rommelaere, J.; Galle, P.R.; Heike, M. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum. Gene Ther. 2005, 16, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Grekova, S.; Aprahamian, M.; Giese, N.; Schmitt, S.; Giese, T.; Falk, C.S.; Daeffler, L.; Cziepluch, C.; Rommelaere, J.; Raykov, Z. Immune cells participate in the oncosuppressive activity of parvovirus H-1PV and are activated as a result of their abortive infection. Cancer Biol. Ther. 2010, 10, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, B.; Goepfert, K.; Delic, M.; Galle, P.R.; Moehler, M. Influence of the oncolytic parvovirus H-1, CTLA-4 antibody tremelimumab and cytostatic drugs on the human immune system in a human in vitro model of colorectal cancer cells. Onco Targets Ther. 2013, 6, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Dempe, S.; Dinsart, C.; Rommelaere, J. Enhancement of NK cell antitumor responses using an oncolytic parvovirus. Int. J. Cancer 2011, 128, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Moralés, O.; Richard, A.; Martin, N.; Mrizak, D.; Sénéchal, M.; Miroux, C.; Pancré, V.; Rommelaere, J.; Caillet-Fauquet, P.; de Lanoit, Y.; et al. Activation of a helper but not regulatory human CD4+ T cell response by oncolytic H-1 parvovirus. PLoS ONE 2012, 7, e32197. [Google Scholar] [CrossRef] [PubMed]
- Grekova, S.P.; Raykov, Z.; Zawatzky, R.; Rommelaere, J.; Koch, U. Activation of a glioma-specific immune response by oncolytic parvovirus Minute Virus of Mice infection. Cancer Gene Ther. 2012, 19, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Kiprianova, I.; Ayache, A.; Koch, R.; Herrero Y Calle, M.; Deleu, L.; Sommer, C.; Thomas, N.; Rommelaere, J.; Schlehofer, J. Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro Oncol. 2010, 12, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Lamfers, M.L. Clinical trials of oncolytic viruses for gliomas. In Current Clinical Oncology: High-Grade Gliomas: Diagnosis and Treatment; Barnett, G.H., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2007; pp. 391–403. ISBN 978-1-59745-185-7. [Google Scholar]
- Duffy, M.R.; Fisher, K.; Seymour, L.W. Making oncolytic virotherapy a clinical reality: The European contribution. Hum. Gene Ther. 2017, 28, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Huesing, J.; Rommelaere, J.; Schlehofer, J.R.; Leuchs, B.; Dahm, M.; Krebs, O.; von Knebel Doeberitz, M.; Huber, B.; Hajda, J. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer 2012, 12, 99. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.; Neumann, J.-O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 parvovirus shows safety and signs of immunogenic activity in a first phase I/IIa glioblastoma trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [PubMed]
- Chonan, M.; Saito, R.; Shoji, T.; Shibahara, I.; Kanamori, M.; Sonoda, Y.; Watanabe, M.; Kikushi, T.; Ishii, N.; Tominaga, T. CD40/CD40L expression correlates with the survival of patients with glioblastomas and an augmentation in CD40 signaling enhances the efficacy of vaccinations against glioma models. Neuro Oncol. 2015, 17, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Di Piazza, M.; Mader, C.; Geletneky, K.; Herrero Y Calle, M.; Weber, E.; Schlehofer, J.; Deleu, L.; Rommelaere, J. Cytosolic activation of cathepsins mediates parvovirus H-1-induced killing of cisplatin and TRAIL-resistant glioma cells. J. Virol. 2007, 81, 4186–4198. [Google Scholar] [CrossRef] [PubMed]
- Kingham, P.J.; Pocock, J.M. Microglial secreted cathepsin B induces neuronal apoptosis. J. Neurochem. 2001, 76, 1475–1484. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.Y.; Yoo, B.C.; Jung, J.W.; Oh, E.S.; Hwang, J.S.; Shin, J.A.; Kim, S.Y.; Cha, S.H.; Han, I.O. Induction of glioma apoptosis by microglia-secreted molecules: The role of nitric oxide and cathepsin B. Biochim. Biophys. Acta 2009, 1793, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Rayet, B.; Lopez-Guerrero, J.-A.; Rommelaere, J.; Dinsart, C. Induction of programmed cell death by parvovirus H-1 in U937 cells: Connection with the tumor necrosis factor α signalling pathway. J. Virol. 1998, 72, 8893–8903. [Google Scholar] [PubMed]
- Lisi, L.; Ciotti, G.M.; Braun, D.; Kalinin, S.; Currò, D.; Dello Russo, C.; Coli, A.; Mangiola, A.; Anile, C.; Feinstein, D.L.; et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci. Lett. 2017, 645, 106–112. [Google Scholar] [CrossRef] [PubMed]
- De Vries, C.R.; Kaufman, H.L.; Lattime, E.C. Oncolytic viruses: Focusing on the tumor microenvironment. Cancer Gene Ther. 2015, 22, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 22ra32. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Dey, M.; Chang, A.L.; Kim, J.W.; Miska, J.; LIng, A.; Nettlebeck, D.M.; Han, Y.; Zhang, L.; Lesniak, M.S. Intratumoral oncolytic adenoviral treatment modulates the glioma microenvironment and facilitates systemic tumor-antigen-specific T cell therapy. Oncoimmunology 2015, 4, e1022302. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.C.; Holl, E.K.; Boczkowski, D.; Dobrikova, E.; Mosaheb, M.; Chandramohan, V.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci. Transl. Med. 2017, 9, eaan4220. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, M.J.; Maguire, H.C., Jr.; Eisenlohr, L.C.; Laughlin, C.E.; Monken, C.E.; McCue, P.A.; Kovatich, A.J.; Lattime, E.C. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999, 6, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; DeRaffele, G.; Divito, J.; Hörig, H.; Lee, D.; Panicali, D.; Voulo, M. A phase I trial of intralesional rV-Tricom vaccine in the tratment of malignant melanoma. Hum. Gene Ther. 2001, 12, 1459–1480. [Google Scholar] [PubMed]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Heinzerling, L.; Künzi, V.; Oberholzer, P.A.; Kündig, T.; Naim, H.; Dummer, R. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood 2005, 106, 2287–2294. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef] [PubMed]
- Aitken, A.S.; Roy, D.G.; Bourgeois-Daigneault, M.-C. Taking a stab at cancer; oncolytic virus-mediated anti-cancer vaccination strategies. Biomedicines 2017, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Kees, T.; Lohr, J.; Noack, J.; Mora, R.; Gdinya, G.; Tödt, G.; Ernst, A.; Radlwimmer, B.; Falk, C.S.; Herold-Mende, C.; et al. Microglia isolated from patients with glioma gain antitumor activities on poly (I:C) stimulation. Neuro Oncol. 2012, 14, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, W.C.; Kong, J.; Gao, J.; Gutman, D.A.; Cooper, L.A.D.; Appin, C.; Park, Y.; Scarpace, L.; Mikkelsen, T.; Cohen, M.L.; et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin. Cancer Res. 2013, 19, 4951–4960. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, A.; Lesniak, M.S. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro Oncol. 2006, 8, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Saskar, S.; Döring, A.; Zemp, F.J.; Silva, C.; Lun, X.; Wang, Y.; Kelly, J.; Hader, W.; Hamilton, M.; Mercier, P.; et al. Therapeutic activation of macrophages and microglia to suppress brain tumor-initiating cells. Nat. Neurosci. 2014, 17, 46–55. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).