Epstein–Barr Virus Hijacks DNA Damage Response Transducers to Orchestrate Its Life Cycle

Abstract
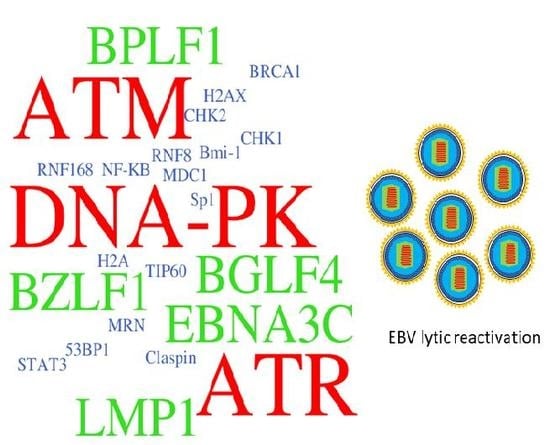
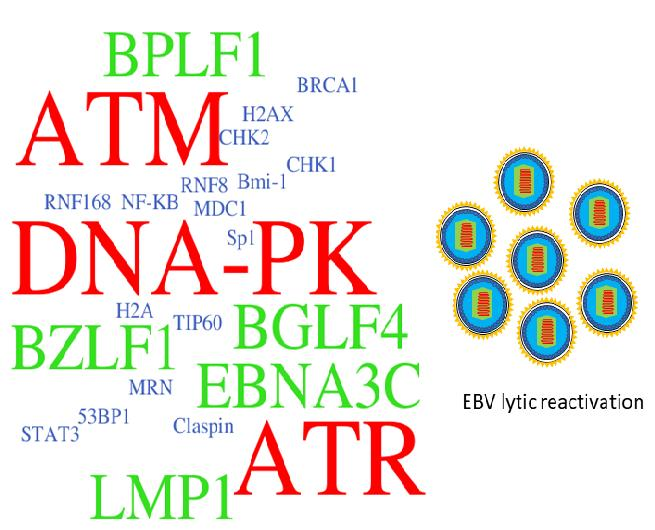{kind=link}
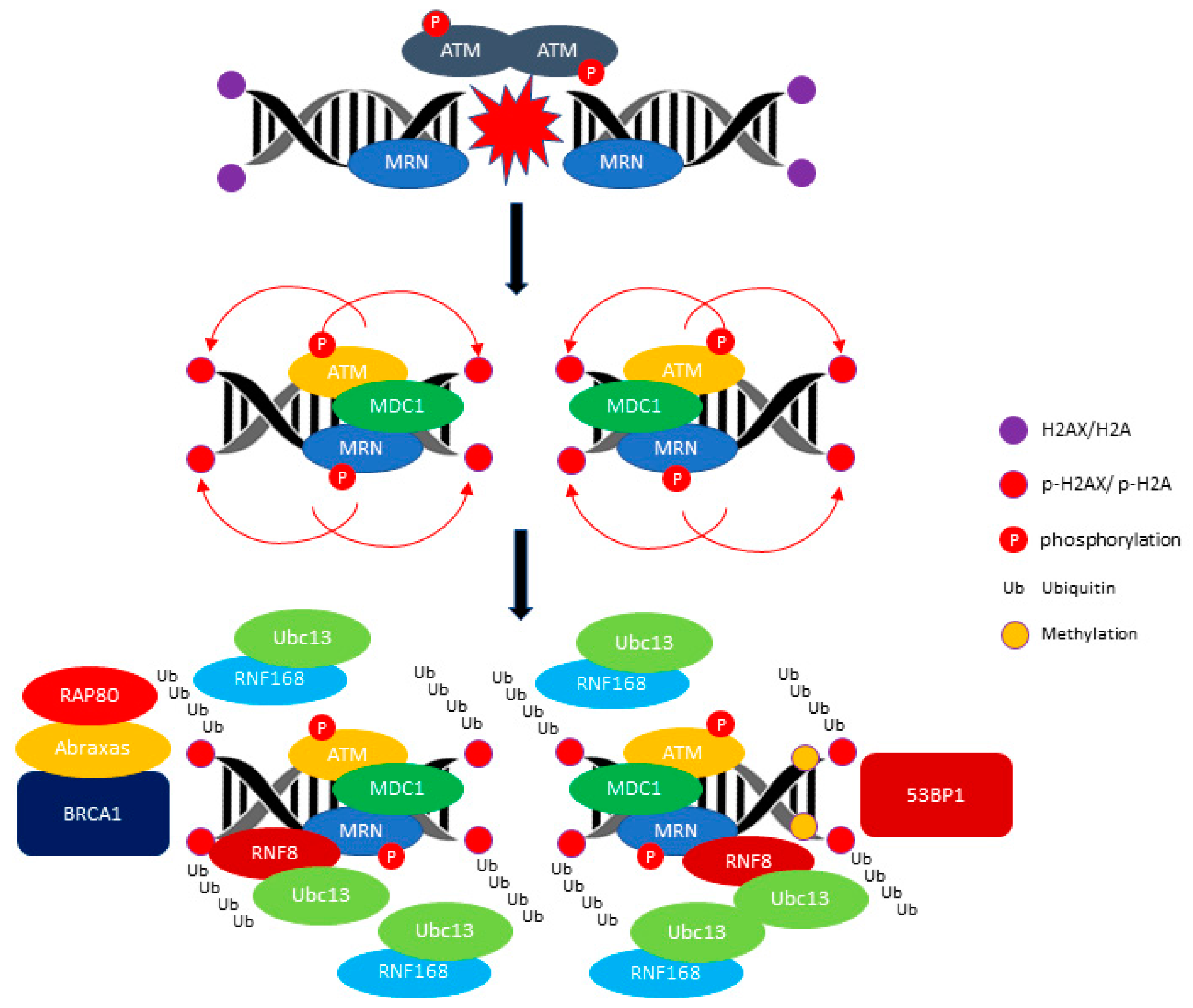{kind=link}
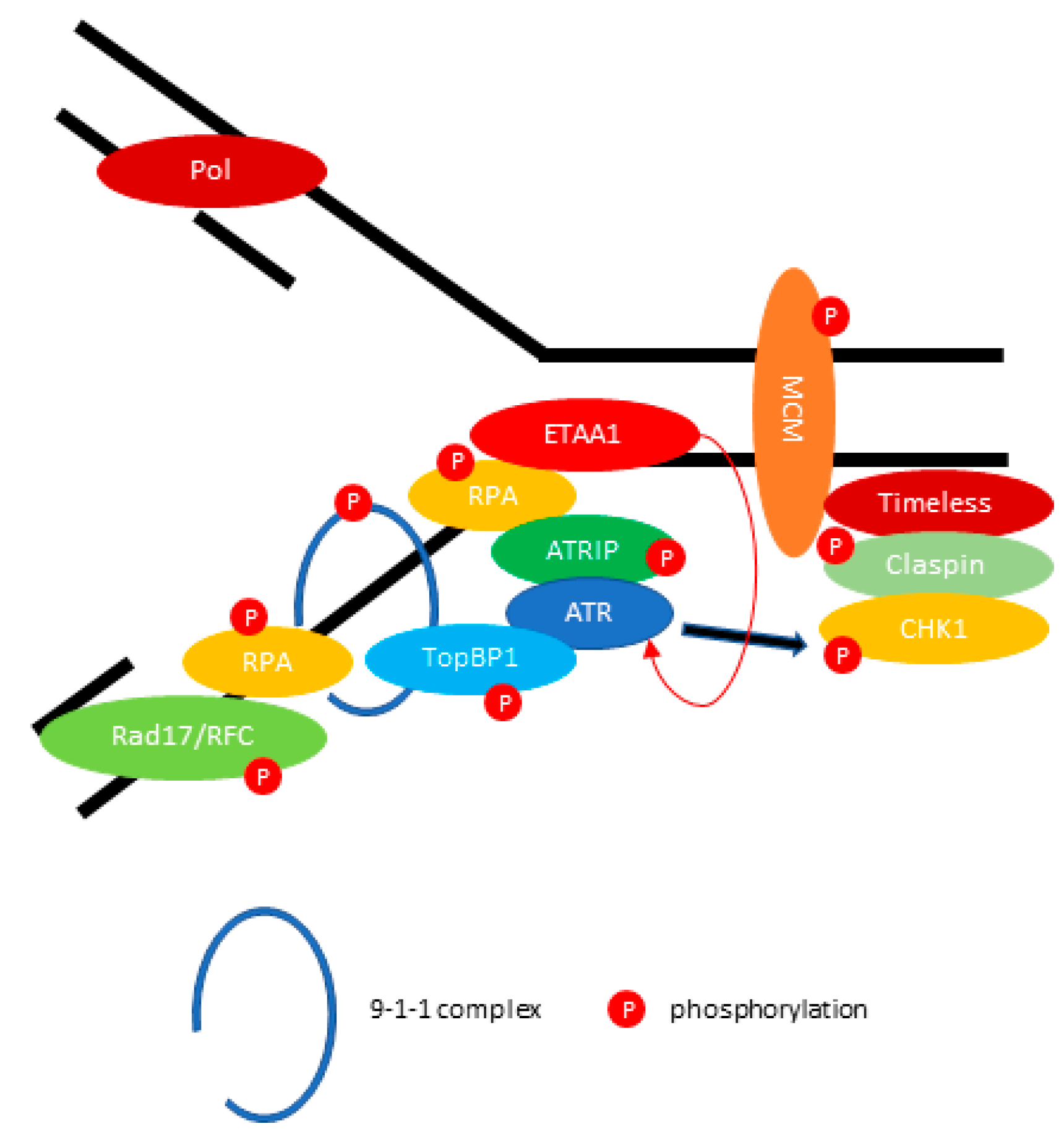{kind=link}
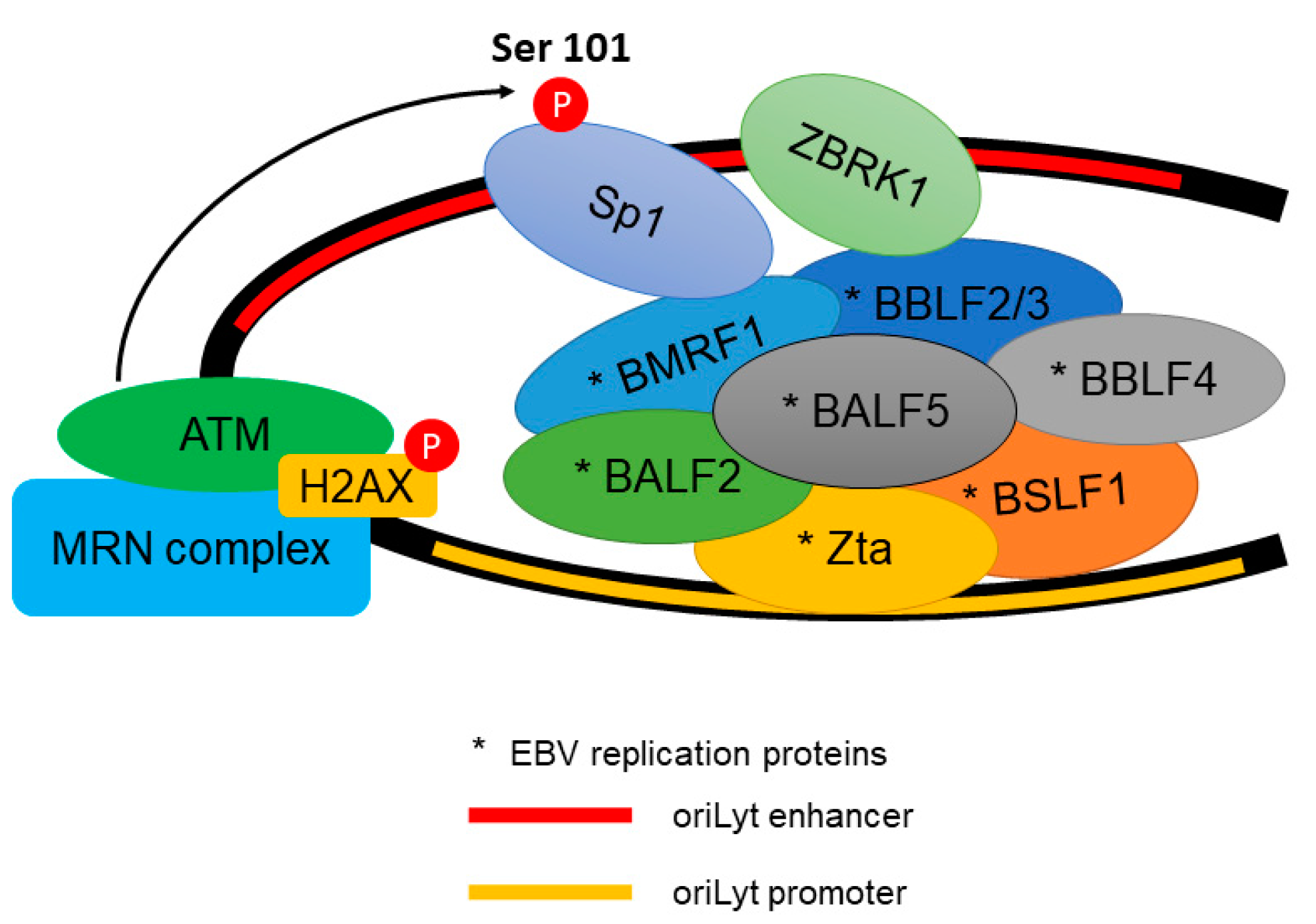{kind=link}
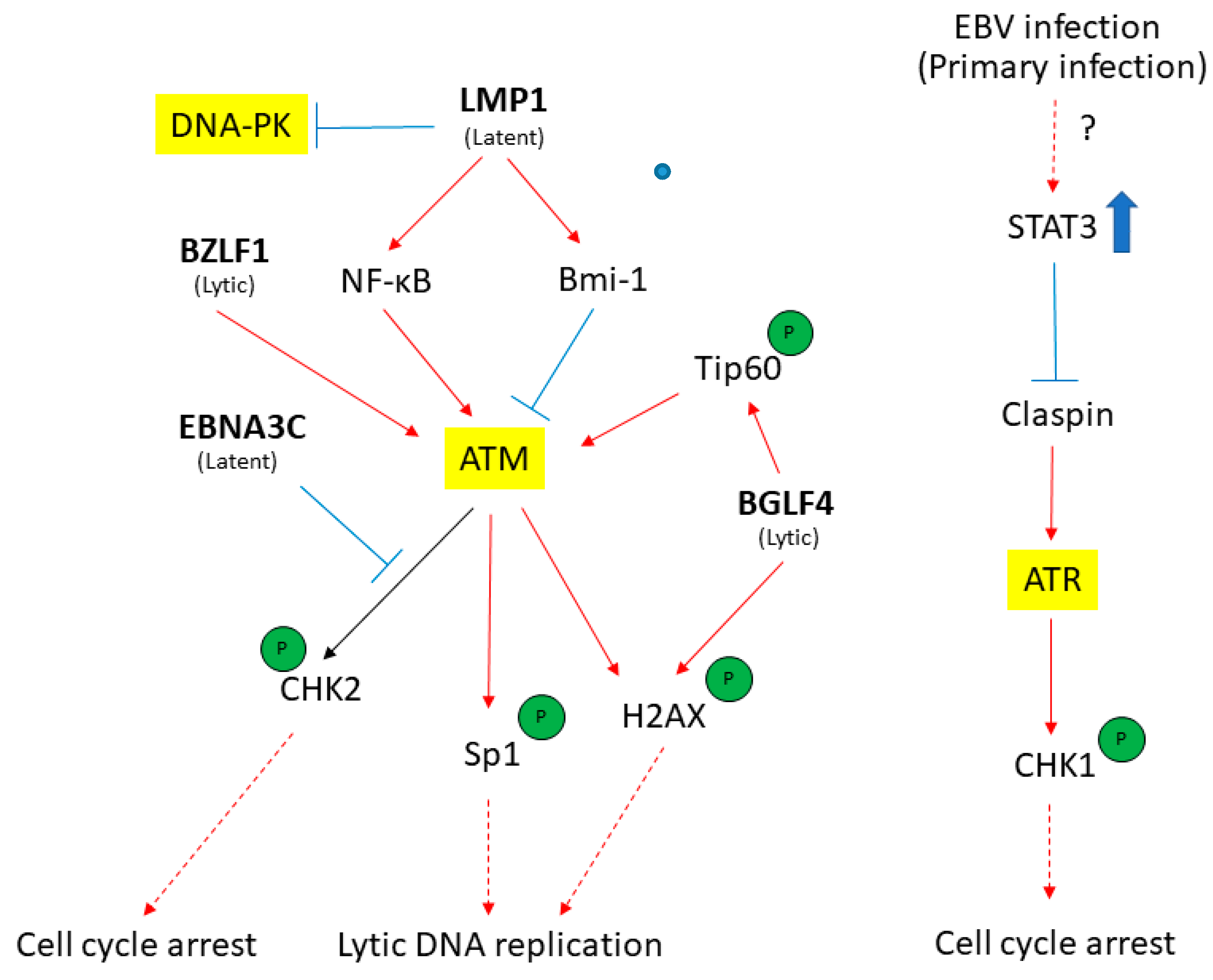{kind=link}
1. Introduction
2. Major Transducers in DNA Damage Response (DDR)
2.1. ATM: A Versatile Protein for Double-Stranded DNA Repair
2.2. ATR: Resolving the Problem of Replication Forks Stalling
2.3. DNA-PK: Another DSB Transducer for NHEJ DNA Repair
3. ATM-Mediated DDR in Response to EBV Infection
4. EBV Disrupts the ATR-Mediated Checkpoint Response
5. DNA-PK: An Emerging DDR Protein to Regulate EBV Infection
6. Other DNA Repair Pathways Affected by EBV Lytic Reactivation
7. DDR Activation in Response to EBV Lytic DNA Replication
8. Summary
Acknowledgments
Conflicts of Interest
References
- Epstein, A. Why and how epstein-barr virus was discovered 50 years ago. Curr. Top. Microbiol. Immunol. 2015, 390, 3–15. [Google Scholar] [PubMed]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Chua, M.L.K.; Wee, J.T.S.; Hui, E.P.; Chan, A.T.C. Nasopharyngeal carcinoma. Lancet 2016, 387, 1012–1024. [Google Scholar] [CrossRef]
- Tsao, S.W.; Yip, Y.L.; Tsang, C.M.; Pang, P.S.; Lau, V.M.; Zhang, G.; Lo, K.W. Etiological factors of nasopharyngeal carcinoma. Oral Oncol. 2014, 50, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Gahn, T.A.; Schildkraut, C.L. The epstein-barr virus origin of plasmid replication, orip, contains both the initiation and termination sites of DNA replication. Cell 1989, 58, 527–535. [Google Scholar] [CrossRef]
- Schepers, A.; Pich, D.; Mankertz, J.; Hammerschmidt, W. Cis-acting elements in the lytic origin of DNA replication of epstein-barr virus. J. Virol. 1993, 67, 4237–4245. [Google Scholar] [PubMed]
- Schepers, A.; Pich, D.; Hammerschmidt, W. Activation of oriLyt, the lytic origin of DNA replication of epstein-barr virus, by BZLF1. Virology 1996, 220, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Fixman, E.D.; Hayward, G.S.; Hayward, S.D. Trans-acting requirements for replication of epstein-barr virus ori-lyt. J. Virol. 1992, 66, 5030–5039. [Google Scholar] [PubMed]
- Cornaby, C.; Jafek, J.L.; Birrell, C.; Mayhew, V.; Syndergaard, L.; Mella, J.; Cheney, W.; Poole, B.D. EBI2 expression in B lymphocytes is controlled by the epstein-barr virus transcription factor, BRRF1 (Na), during viral infection. J. Gen. Virol. 2017, 98, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.K.; Delecluse, H.J.; Gruffat, H.; Morrison, T.E.; Feng, W.H.; Sergeant, A.; Kenney, S.C. The BRRF1 early gene of epstein-barr virus encodes a transcription factor that enhances induction of lytic infection by BRLF1. J. Virol. 2004, 78, 4983–4992. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ragoczy, T.; Heston, L.; Miller, G. The epstein-barr virus Rta protein activates lytic cycle genes and can disrupt latency in B lymphocytes. J. Virol. 1998, 72, 7978–7984. [Google Scholar] [PubMed]
- Feederle, R.; Kost, M.; Baumann, M.; Janz, A.; Drouet, E.; Hammerschmidt, W.; Delecluse, H.J. The epstein-barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 2000, 19, 3080–3089. [Google Scholar] [CrossRef] [PubMed]
- Ragoczy, T.; Miller, G. Role of the epstein-barr virus Rta protein in activation of distinct classes of viral lytic cycle genes. J. Virol. 1999, 73, 9858–9866. [Google Scholar] [PubMed]
- Hammerschmidt, W.; Sugden, B. Identification and characterization of oriLyt, a lytic origin of DNA replication of epstein-barr virus. Cell 1988, 55, 427–433. [Google Scholar] [CrossRef]
- Cho, M.S.; Tran, V.M. A concatenated form of epstein-barr viral DNA in lymphoblastoid cell lines induced by transfection with BZLF1. Virology 1993, 194, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, W.; Sugden, B. Replication of epstein-barr viral DNA. Cold Spring Harb. Perspect. Biol. 2013, 5, a013029. [Google Scholar] [CrossRef] [PubMed]
- El-Guindy, A.; Ghiassi-Nejad, M.; Golden, S.; Delecluse, H.J.; Miller, G. Essential role of Rta in lytic DNA replication of epstein-barr virus. J. Virol. 2013, 87, 208–223. [Google Scholar] [CrossRef] [PubMed]
- Hadinoto, V.; Shapiro, M.; Sun, C.C.; Thorley-Lawson, D.A. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog. 2009, 5, e1000496. [Google Scholar] [CrossRef] [PubMed]
- Temple, R.M.; Zhu, J.; Budgeon, L.; Christensen, N.D.; Meyers, C.; Sample, C.E. Efficient replication of epstein-barr virus in stratified epithelium in vitro. Proc. Natl. Acad. Sci. USA 2014, 111, 16544–16549. [Google Scholar] [CrossRef] [PubMed]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.M.; Yip, Y.L.; Lo, K.W.; Deng, W.; To, K.F.; Hau, P.M.; Lau, V.M.; Takada, K.; Lui, V.W.; Lung, M.L.; et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E3473–E3482. [Google Scholar] [CrossRef] [PubMed]
- Kenney, S.C.; Mertz, J.E. Regulation of the latent-lytic switch in epstein-barr virus. Semin. Cancer Biol. 2014, 26, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, R.; Grand, R.J. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses 2015, 7, 2542–2591. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, P.A.; Luftig, M.A. At a crossroads: Human DNA tumor viruses and the host DNA damage response. Future Virol. 2011, 6, 813–830. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Tourigny, J.P.; Forte, E.; Salinas, R.E.; Dave, S.S.; Luftig, M.A. Analysis of epstein-barr virus-regulated host gene expression changes through primary B-cell outgrowth reveals delayed kinetics of latent membrane protein 1-mediated NF-κB activation. J. Virol. 2012, 86, 11096–11106. [Google Scholar] [CrossRef] [PubMed]
- O’Nions, J.; Turner, A.; Craig, R.; Allday, M.J. Epstein-barr virus selectively deregulates DNA damage responses in normal B cells but has no detectable effect on regulation of the tumor suppressor p53. J. Virol. 2006, 80, 12408–12413. [Google Scholar] [CrossRef] [PubMed]
- Gruhne, B.; Sompallae, R.; Marescotti, D.; Kamranvar, S.A.; Gastaldello, S.; Masucci, M.G. The epstein-barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2009, 106, 2313–2318. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, P.A.; Yan, C.M.; Forte, E.; Bocedi, A.; Tourigny, J.P.; White, R.E.; Allday, M.J.; Patel, A.; Dave, S.S.; Kim, W.; et al. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses epstein-barr virus transformation of primary human B cells. Cell Host Microbe 2010, 8, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Robison, J.G.; Lu, L.; Dixon, K.; Bissler, J.J. DNA lesion-specific co-localization of the Mre11/Rad50/Nbs1 (MRN) complex and replication protein A (RPA) to repair foci. J. Biol. Chem. 2005, 280, 12927–12934. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, M.; Bree, R.T.; Lowndes, N.F. The MRN complex: Coordinating and mediating the response to broken chromosomes. EMBO Rep. 2003, 4, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- So, S.; Davis, A.J.; Chen, D.J. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J. Cell Biol. 2009, 187, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T. Mechanisms of ATM activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xu, Y.; Roy, K.; Price, B.D. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell. Biol. 2007, 27, 8502–8509. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.M.; Li, J.J. ATM-NF-κB connection as a target for tumor radiosensitization. Curr. Cancer Drug Targets 2007, 7, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Cremona, C.A.; Behrens, A. ATM signalling and cancer. Oncogene 2014, 33, 3351–3360. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Lukas, C.; Melander, F.; Stucki, M.; Falck, J.; Bekker-Jensen, S.; Goldberg, M.; Lerenthal, Y.; Jackson, S.P.; Bartek, J.; Lukas, J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J. 2004, 23, 2674–2683. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. Mdc1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. Rnf168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. Rnf8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the Rnf8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. Rnf8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S.; et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Elledge, S.J. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl. Acad. Sci. USA 2007, 104, 20759–20763. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Sung, P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol. Cell. Biol. 2014, 34, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Minton, K. DNA damage response: ATR prevents premature apoptosis. Nat. Rev. Mol. Cell Biol. 2015, 16, 640. [Google Scholar] [CrossRef] [PubMed]
- Zou, L. Single- and double-stranded DNA: Building a trigger of ATR-mediated DNA damage response. Genes Dev. 2007, 21, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Bomgarden, R.D.; Yean, D.; Yee, M.C.; Cimprich, K.A. A novel protein activity mediates DNA binding of an ATR-atrip complex. J. Biol. Chem. 2004, 279, 13346–13353. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Elledge, S.J. Sensing DNA damage through atrip recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, Z.; Liu, Y.; Zou, Y. Preferential localization of hyperphosphorylated replication protein a to double-strand break repair and checkpoint complexes upon DNA damage. Biochem. J. 2005, 391, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Delacroix, S.; Wagner, J.M.; Kobayashi, M.; Yamamoto, K.; Karnitz, L.M. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007, 21, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kumagai, A.; Dunphy, W.G. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J. Biol. Chem. 2007, 282, 28036–28044. [Google Scholar] [CrossRef] [PubMed]
- Navadgi-Patil, V.M.; Burgers, P.M. Yeast DNA replication protein Dpb11 activates the Mec1/ATR checkpoint kinase. J. Biol. Chem. 2008, 283, 35853–35859. [Google Scholar] [CrossRef] [PubMed]
- Bass, T.E.; Luzwick, J.W.; Kavanaugh, G.; Carroll, C.; Dungrawala, H.; Glick, G.G.; Feldkamp, M.D.; Putney, R.; Chazin, W.J.; Cortez, D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat. Cell Biol. 2016, 18, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Zhao, Y.; Xu, Y.; Ning, S.; Huo, W.; Hou, M.; Gao, G.; Ji, J.; Guo, R.; Xu, D. Ewing tumor-associated antigen 1 interacts with replication protein a to promote restart of stalled replication forks. J. Biol. Chem. 2016, 291, 21956–21962. [Google Scholar] [CrossRef] [PubMed]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Raschle, M.; Mailand, N. Activation of the ATR kinase by the RPA-binding protein etaa1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Zhou, Q.; Chen, J.; Yuan, J. RPA-binding protein etaa1 is an ATR activator involved in DNA replication stress response. Curr. Biol. 2016, 26, 3257–3268. [Google Scholar] [CrossRef] [PubMed]
- Kemp, M.G.; Akan, Z.; Yilmaz, S.; Grillo, M.; Smith-Roe, S.L.; Kang, T.H.; Cordeiro-Stone, M.; Kaufmann, W.K.; Abraham, R.T.; Sancar, A.; et al. Tipin-replication protein a interaction mediates CHK1 phosphorylation by ATR in response to genotoxic stress. J. Biol. Chem. 2010, 285, 16562–16571. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Kim, S.M.; Dunphy, W.G. Claspin and the activated form of ATR-atrip collaborate in the activation of CHK1. J. Biol. Chem. 2004, 279, 49599–49608. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa-Sugata, N.; Masai, H. Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J. Biol. Chem. 2007, 282, 2729–2740. [Google Scholar] [CrossRef] [PubMed]
- Decottignies, A. Alternative end-joining mechanisms: A historical perspective. Front. Genet. 2013, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Seol, J.H.; Shim, E.Y.; Lee, S.E. Microhomology-mediated end joining: Good, bad and ugly. Mutat. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Shrivastav, M.; de Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, B.P.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair (Amst.) 2014, 17, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Dutton, A.; Woodman, C.B.; Chukwuma, M.B.; Last, J.I.; Wei, W.; Vockerodt, M.; Baumforth, K.R.; Flavell, J.R.; Rowe, M.; Taylor, A.M.; et al. Bmi-1 is induced by the epstein-barr virus oncogene LMP1 and regulates the expression of viral target genes in hodgkin lymphoma cells. Blood 2007, 109, 2597–2603. [Google Scholar] [CrossRef] [PubMed]
- Gruhne, B.; Sompallae, R.; Masucci, M.G. Three epstein-barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene 2009, 28, 3997–4008. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Yap, L.F.; Fung, M.; Starzcynski, J.; Saleh, A.; Morgan, S.; Dawson, C.; Chukwuma, M.B.; Maina, E.; Buettner, M.; et al. The ATM tumour suppressor gene is down-regulated in EBV-associated nasopharyngeal carcinoma. J. Pathol. 2009, 217, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yang, L.; Xiao, L.; Tang, M.; Liu, L.; Li, Z.; Deng, M.; Sun, L.; Cao, Y. Down-regulation of EBV-LMP1 radio-sensitizes nasal pharyngeal carcinoma cells via NF-κB regulated ATM expression. PLoS ONE 2011, 6, e24647. [Google Scholar] [CrossRef] [PubMed]
- Le Clorennec, C.; Ouk, T.S.; Youlyouz-Marfak, I.; Panteix, S.; Martin, C.C.; Rastelli, J.; Adriaenssens, E.; Zimber-Strobl, U.; Coll, J.; Feuillard, J.; et al. Molecular basis of cytotoxicity of epstein-barr virus (EBV) latent membrane protein 1 (LMP1) in EBV latency III B cells: LMP1 induces type II ligand-independent autoactivation of CD95/Fas with caspase 8-mediated apoptosis. J. Virol. 2008, 82, 6721–6733. [Google Scholar] [CrossRef] [PubMed]
- Dirmeier, U.; Hoffmann, R.; Kilger, E.; Schultheiss, U.; Briseno, C.; Gires, O.; Kieser, A.; Eick, D.; Sugden, B.; Hammerschmidt, W. Latent membrane protein 1 of epstein-barr virus coordinately regulates proliferation with control of apoptosis. Oncogene 2005, 24, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, T.; Verma, S.C.; Lan, K.; Murakami, M.; Robertson, E.S. The ATM/ATR signaling effector CHK2 is targeted by epstein-barr virus nuclear antigen 3C to release the G2/M cell cycle block. J. Virol. 2007, 81, 6718–6730. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed]
- Deming, P.B.; Flores, K.G.; Downes, C.S.; Paules, R.S.; Kaufmann, W.K. ATR enforces the topoisomerase II-dependent G2 checkpoint through inhibition of PLK1 kinase. J. Biol. Chem. 2002, 277, 36832–36838. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.J.; Dutta, A. ATR pathway is the primary pathway for activating G2/M checkpoint induction after re-replication. J. Biol. Chem. 2007, 282, 30357–30362. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bhaduri-McIntosh, S. A central role for STAT3 in Gammaherpesvirus-life cycle and -diseases. Front. Microbiol. 2016, 7, 1052. [Google Scholar] [CrossRef] [PubMed]
- Buettner, M.; Heussinger, N.; Niedobitek, G. Expression of epstein-barr virus (EBV)-encoded latent membrane proteins and STAT3 activation in nasopharyngeal carcinoma. Virchows Arch. 2006, 449, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Kawanishi, M.; Hiraku, Y.; Murata, M.; Huang, G.W.; Huang, Y.; Luo, D.Z.; Mo, W.G.; Fukui, Y.; Kawanishi, S. Reactive nitrogen species-dependent DNA damage in EBV-associated nasopharyngeal carcinoma: The relation to STAT3 activation and EGFR expression. Int. J. Cancer 2008, 122, 2517–2525. [Google Scholar] [CrossRef] [PubMed]
- Shair, K.H.; Bendt, K.M.; Edwards, R.H.; Bedford, E.C.; Nielsen, J.N.; Raab-Traub, N. EBV latent membrane protein 1 activates AKT, NF-κB, and STAT3 in B cell lymphomas. PLoS Pathog. 2007, 3, e166. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.P.; Townsend, P.A.; Knight, R.A.; Scarabelli, T.M.; Latchman, D.S.; Stephanou, A. STAT3 modulates the DNA damage response pathway. Int. J. Exp. Pathol. 2010, 91, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Shields, B.J.; Hauser, C.; Bukczynska, P.E.; Court, N.W.; Tiganis, T. DNA replication stalling attenuates tyrosine kinase signaling to suppress S Phase progression. Cancer Cell 2008, 14, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Shields, B.J.; Tiganis, T. Replication checkpoint control by a PTK/STAT3/Cyclin D1 axis. Cell Cycle 2009, 8, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Koganti, S.; Hui-Yuen, J.; McAllister, S.; Gardner, B.; Grasser, F.; Palendira, U.; Tangye, S.G.; Freeman, A.F.; Bhaduri-McIntosh, S. STAT3 interrupts ATR-CHK1 signaling to allow oncovirus-mediated cell proliferation. Proc. Natl. Acad. Sci. USA 2014, 111, 4946–4951. [Google Scholar] [CrossRef] [PubMed]
- Mordasini, V.; Ueda, S.; Aslandogmus, R.; Berger, C.; Gysin, C.; Huhn, D.; Sartori, A.A.; Bernasconi, M.; Nadal, D. Activation of ATR-CHK1 pathway facilitates EBV-mediated transformation of primary tonsillar B-cells. Oncotarget 2017, 8, 6461–6474. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hafez, A.Y.; Messinger, J.E.; McFadden, K.; Fenyofalvi, G.; Shepard, C.N.; Lenzi, G.M.; Kim, B.; Luftig, M.A. Limited nucleotide pools restrict epstein-barr virus-mediated B-cell immortalization. Oncogenesis 2017, 6, e349. [Google Scholar] [CrossRef] [PubMed]
- Lees-Miller, S.P.; Long, M.C.; Kilvert, M.A.; Lam, V.; Rice, S.A.; Spencer, C.A. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol. 1996, 70, 7471–7477. [Google Scholar] [PubMed]
- Wang, Y.; Li, H.; Tang, Q.; Maul, G.G.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus ori-lyt-dependent DNA replication: Involvement of host cellular factors. J. Virol. 2008, 82, 2867–2882. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Lim, C.; Lee, J.Y.; Song, Y.J.; Park, J.; Choe, J.; Seo, T. DNA-PK/KU complex binds to latency-associated nuclear antigen and negatively regulates kaposi’s sarcoma-associated herpesvirus latent replication. Biochem. Biophys. Res. Commun. 2010, 394, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLIFE 2012, 1, e00047. [Google Scholar] [CrossRef] [PubMed]
- Han, I.; Harada, S.; Weaver, D.; Xue, Y.; Lane, W.; Orstavik, S.; Skalhegg, B.; Kieff, E. EBNA-LP associates with cellular proteins including DNA-PK and HA95. J. Virol. 2001, 75, 2475–2481. [Google Scholar] [CrossRef] [PubMed]
- Kempkes, B.; Ling, P.D. EBNA2 and its coactivator EBNA-LP. Curr. Top. Microbiol. Immunol. 2015, 391, 35–59. [Google Scholar] [PubMed]
- Lu, J.; Tang, M.; Li, H.; Xu, Z.; Weng, X.; Li, J.; Yu, X.; Zhao, L.; Liu, H.; Hu, Y.; et al. EBV-LMP1 suppresses the DNA damage response through DNA-PK/AMPK signaling to promote radioresistance in nasopharyngeal carcinoma. Cancer Lett. 2016, 380, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Bertolin, A.P.; Mansilla, S.F.; Gottifredi, V. The identification of translesion DNA synthesis regulators: Inhibitors in the spotlight. DNA Repair (Amst) 2015, 32, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Gibbs-Seymour, I.; Bekker-Jensen, S. Regulation of PCNA-protein interactions for genome stability. Nat. Rev. Mol. Cell Biol. 2013, 14, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, A.; Kanda, T.; Yamashita, Y.; Murata, T.; Saito, S.; Kawashima, D.; Isomura, H.; Nishiyama, Y.; Tsurumi, T. Spatiotemporally different DNA repair systems participate in epstein-barr virus genome maturation. J. Virol. 2011, 85, 6127–6135. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schmaus, S.; Wolf, H.; Schwarzmann, F. The reading frame BPLF1 of epstein-barr virus: A homologue of herpes simplex virus protein VP16. Virus Genes 2004, 29, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, C.B.; Vaziri, C.; Shackelford, J.; Pagano, J.S. Epstein-barr virus BPLF1 deubiquitinates PCNA and attenuates polymerase η recruitment to DNA damage sites. J. Virol. 2012, 86, 8097–8106. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Whitehurst, C.B.; Pagano, J.S. The RAD6/18 ubiquitin complex interacts with the epstein-barr virus deubiquitinating enzyme, BPLF1, and contributes to virus infectivity. J. Virol. 2014, 88, 6411–6422. [Google Scholar] [CrossRef] [PubMed]
- Dyson, O.F.; Pagano, J.S.; Whitehurst, C.B. The translesion polymerase Pol η is required for efficient epstein-barr virus infectivity and is regulated by the viral deubiquitinating enzyme BPLF1. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Kenney, S.C. Reactivation and lytic replication of EBV. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, A.; Fujita, M.; Zhang, L.; Shirata, N.; Daikoku, T.; Sugaya, Y.; Isomura, H.; Nishiyama, Y.; Tsurumi, T. Epstein-barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J. Biol. Chem. 2005, 280, 8156–8163. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; Lo, Y.C.; Chua, H.H.; Chiu, H.Y.; Tsai, S.C.; Chen, J.Y.; Lo, K.W.; Tsai, C.H. Critical role of p53 in histone deacetylase inhibitor-induced epstein-barr virus Zta expression. J. Virol. 2008, 82, 7745–7751. [Google Scholar] [CrossRef] [PubMed]
- Chua, H.H.; Chiu, H.Y.; Lin, S.J.; Weng, P.L.; Lin, J.H.; Wu, S.W.; Tsai, S.C.; Tsai, C.H. P53 and Sp1 cooperate to regulate the expression of epstein-barr viral Zta protein. J. Med. Virol. 2012, 84, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Hagemeier, S.R.; Barlow, E.A.; Meng, Q.; Kenney, S.C. The cellular ataxia telangiectasia-mutated kinase promotes epstein-barr virus lytic reactivation in response to multiple different types of lytic reactivation-inducing stimuli. J. Virol. 2012, 86, 13360–13370. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Shirata, N.; Murata, T.; Nakasu, S.; Kudoh, A.; Iwahori, S.; Nakayama, S.; Chiba, S.; Isomura, H.; Kanda, T.; et al. Transient increases in p53-responsible gene expression at early stages of epstein-barr virus productive replication. Cell Cycle 2010, 9, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kamura, T.; Shirata, N.; Murata, T.; Kudoh, A.; Iwahori, S.; Nakayama, S.; Isomura, H.; Nishiyama, Y.; Tsurumi, T. Degradation of phosphorylated p53 by viral protein-ECS E3 ligase complex. PLoS Pathog. 2009, 5, e1000530. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Shirata, N.; Kudoh, A.; Iwahori, S.; Nakayama, S.; Murata, T.; Isomura, H.; Nishiyama, Y.; Tsurumi, T. Expression of epstein-barr virus BZLF1 immediate-early protein induces p53 degradation independent of MDM2, leading to repression of p53-mediated transcription. Virology 2009, 388, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhu, J.; Xie, Z.; Liao, G.; Liu, J.; Chen, M.R.; Hu, S.; Woodard, C.; Lin, J.; Taverna, S.D.; et al. Conserved herpesvirus kinases target the DNA damage response pathway and Tip60 histone acetyltransferase to promote virus replication. Cell Host Microbe 2011, 10, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Wang’ondu, R.; Teal, S.; Park, R.; Heston, L.; Delecluse, H.; Miller, G. DNA damage signaling is induced in the absence of epstein-barr virus (EBV) lytic DNA replication and in response to expression of Zebra. PLoS ONE 2015, 10, e0126088. [Google Scholar] [CrossRef] [PubMed]
- Hau, P.M.; Deng, W.; Jia, L.; Yang, J.; Tsurumi, T.; Chiang, A.K.; Huen, M.S.; Tsao, S.W. Role of ATM in the formation of the replication compartment during lytic replication of epstein-barr virus in nasopharyngeal epithelial cells. J. Virol. 2015, 89, 652–668. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Feederle, R.; Kremmer, E.; Hammerschmidt, W. Cellular transcription factors recruit viral replication proteins to activate the epstein-barr virus origin of lytic DNA replication, oriLyt. EMBO J. 1999, 18, 6095–6105. [Google Scholar] [CrossRef] [PubMed]
- Iwahori, S.; Yasui, Y.; Kudoh, A.; Sato, Y.; Nakayama, S.; Murata, T.; Isomura, H.; Tsurumi, T. Identification of phosphorylation sites on transcription factor Sp1 in response to DNA damage and its accumulation at damaged sites. Cell. Signal. 2008, 20, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, B.A.; Kelly, C.M.; Kim, J.; Hornsby, S.M.; Azizkhan-Clifford, J. Phosphorylation of Sp1 in response to DNA damage by ataxia telangiectasia-mutated kinase. Mol. Cancer Res. 2007, 5, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Beishline, K.; Kelly, C.M.; Olofsson, B.A.; Koduri, S.; Emrich, J.; Greenberg, R.A.; Azizkhan-Clifford, J. Sp1 facilitates DNA double-strand break repair through a nontranscriptional mechanism. Mol. Cell. Biol. 2012, 32, 3790–3799. [Google Scholar] [CrossRef] [PubMed]
- Iwahori, S.; Shirata, N.; Kawaguchi, Y.; Weller, S.K.; Sato, Y.; Kudoh, A.; Nakayama, S.; Isomura, H.; Tsurumi, T. Enhanced phosphorylation of transcription factor Sp1 in response to herpes simplex virus type 1 infection is dependent on the ataxia telangiectasia-mutated protein. J. Virol. 2007, 81, 9653–9664. [Google Scholar] [CrossRef] [PubMed]
- Daikoku, T.; Kudoh, A.; Sugaya, Y.; Iwahori, S.; Shirata, N.; Isomura, H.; Tsurumi, T. Postreplicative mismatch repair factors are recruited to epstein-barr virus replication compartments. J. Biol. Chem. 2006, 281, 11422–11430. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.G.; Verrall, E.; Schelcher, C.; Rhie, A.; Doherty, A.J.; Sinclair, A.J. Functional interaction between epstein-barr virus replication protein Zta and host DNA damage response protein 53BP1. J. Virol. 2009, 83, 11116–11122. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Deng, W.; Hau, P.M.; Liu, J.; Lau, V.M.; Cheung, A.L.; Huen, M.S.; Tsao, S.W. Epstein-barr virus BZLF1 protein impairs accumulation of host DNA damage proteins at damage sites in response to DNA damage. Lab. Investig. 2015, 95, 937–950. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hau, P.M.; Tsao, S.W. Epstein–Barr Virus Hijacks DNA Damage Response Transducers to Orchestrate Its Life Cycle. Viruses 2017, 9, 341. https://doi.org/10.3390/v9110341
Hau PM, Tsao SW. Epstein–Barr Virus Hijacks DNA Damage Response Transducers to Orchestrate Its Life Cycle. Viruses. 2017; 9(11):341. https://doi.org/10.3390/v9110341
Chicago/Turabian StyleHau, Pok Man, and Sai Wah Tsao. 2017. "Epstein–Barr Virus Hijacks DNA Damage Response Transducers to Orchestrate Its Life Cycle" Viruses 9, no. 11: 341. https://doi.org/10.3390/v9110341
APA StyleHau, P. M., & Tsao, S. W. (2017). Epstein–Barr Virus Hijacks DNA Damage Response Transducers to Orchestrate Its Life Cycle. Viruses, 9(11), 341. https://doi.org/10.3390/v9110341