Why do Individuals Differ in Viral Susceptibility? A Story Told by Model Organisms




{kind=link}
{kind=link}
Abstract
1. Introduction
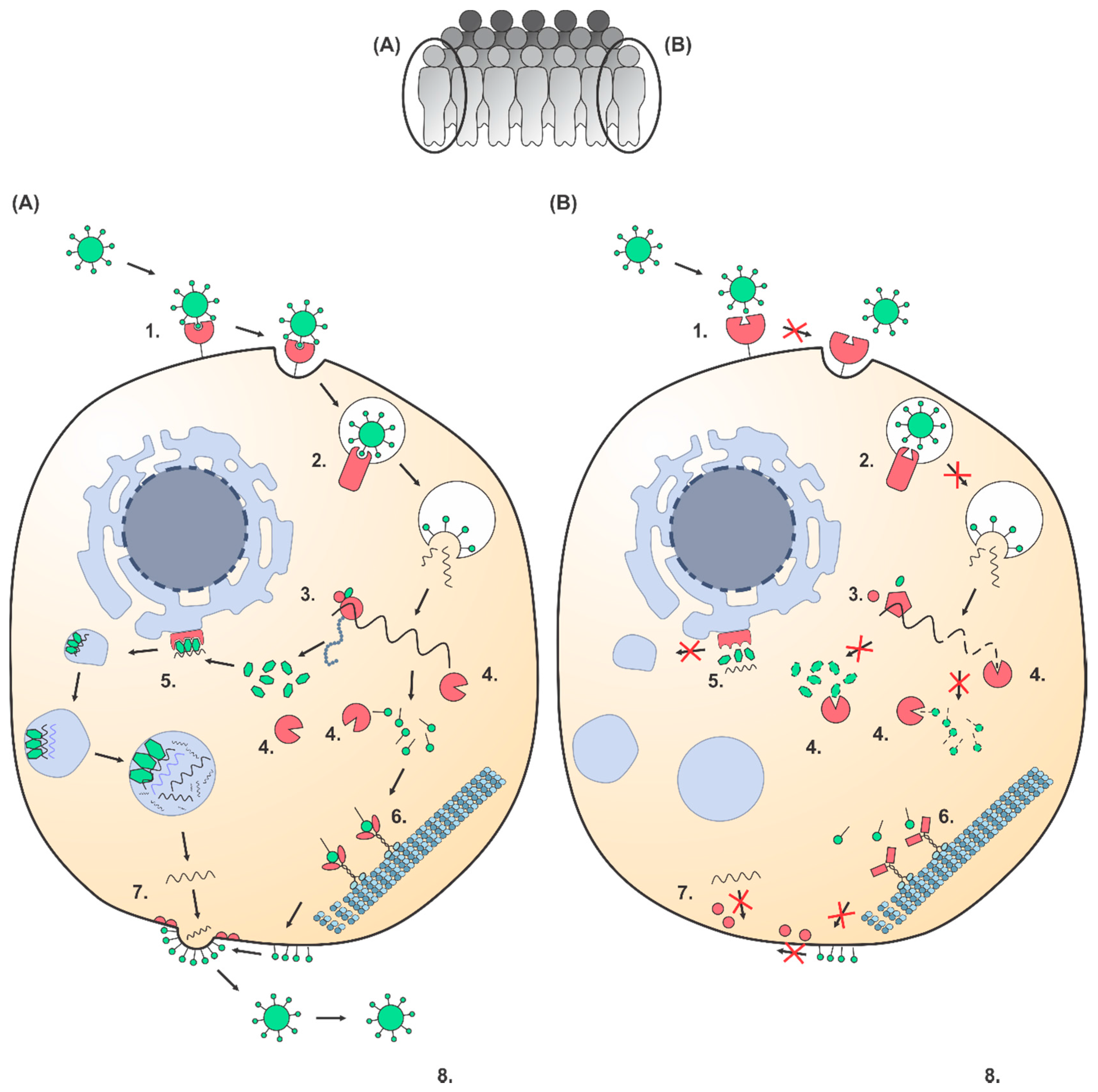
2. Polymorphisms in Host Factors that Interact with Viruses Cause Individual Differences in Viral Susceptibility
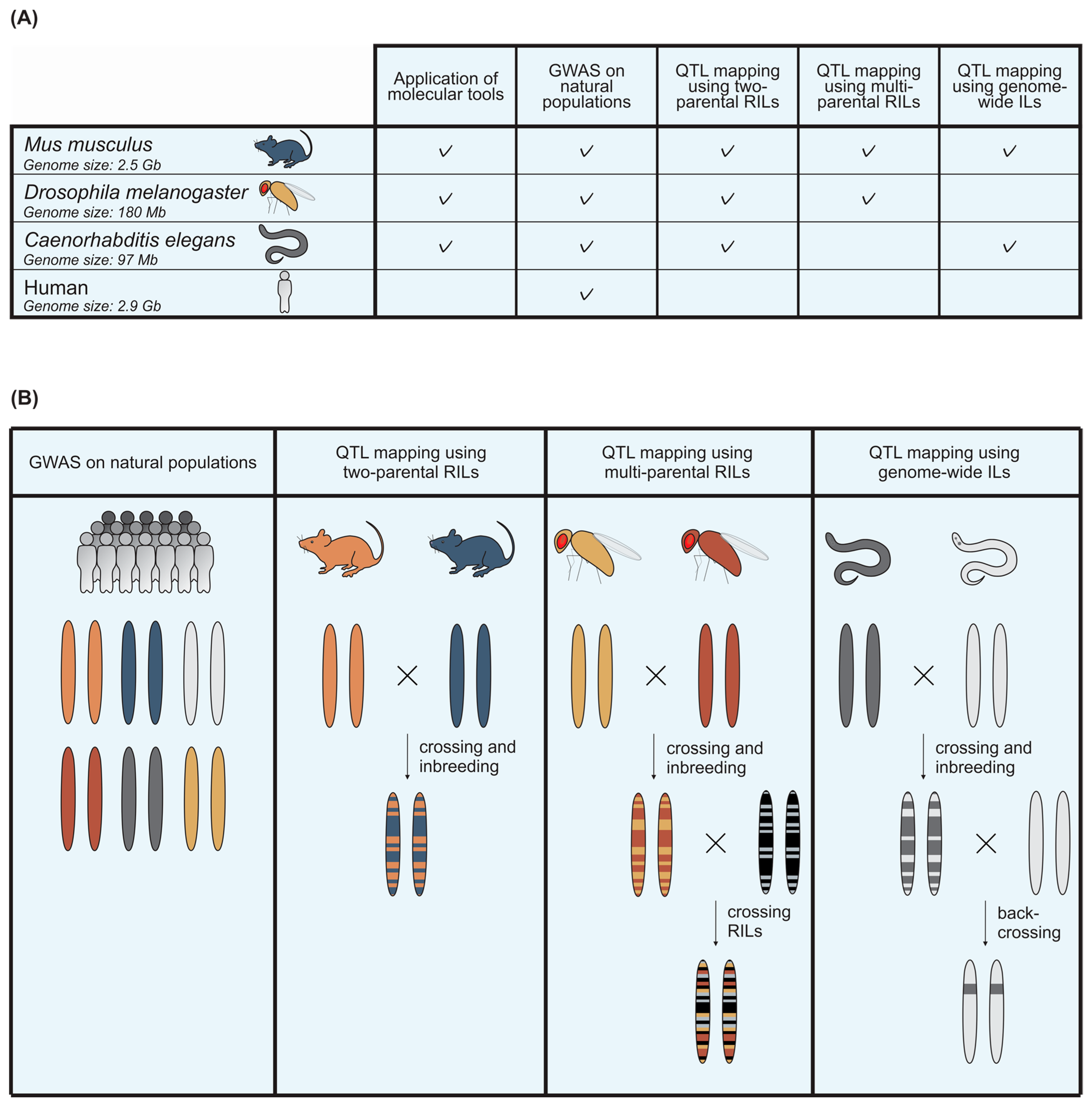
3. Use of Model Organisms to Unravel the Interplay between Host Genetic Variation and Viral Infection
4. Mus musculus
5. Drosophila melanogaster
6. Caenorhabditis elegans
7. Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Gail, M.H.; Weinberg, C.R.; Carroll, R.J.; Chung, C.C.; Wang, Z.; Chanock, S.J.; Fraumeni, J.F.; Chatterjee, N. Distribution of allele frequencies and effect sizes and their interrelationships for common genetic susceptibility variants. Proc. Natl. Acad. Sci. USA 2011, 108, 18026–18031. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.S.; Ehrenreich, I.M.; Loo, W.T.; Lite, T.-L.V.; Kruglyak, L. Finding the sources of missing heritability in a yeast cross. Nature 2013, 494, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.I.; van de Geijn, B.; Raj, A.; Knowles, D.A.; Petti, A.A.; Golan, D.; Gilad, Y.; Pritchard, J.K. RNA splicing is a primary link between genetic variation and disease. Science 2016, 352, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Gasch, A.P.; Payseur, B.A.; Pool, J.E. The Power of Natural Variation for Model Organism Biology. Trends Genet. 2016, 32, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Zignego, A.L.; Wojcik, G.L.; Cacoub, P.; Visentini, M.; Casato, M.; Mangia, A.; Latanich, R.; Charles, E.D.; Gragnani, L.; Terrier, B.; et al. Genome-wide association study of hepatitis C virus- and cryoglobulin-related vasculitis. Genes Immun. 2014, 15, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Al-Qahtani, A.; Khalak, H.G.; Alkuraya, F.S.; Al-Hamoudy, W.; Alswat, K.; Al Balwi, M.A.; Al Abdulkareem, I.; Sanai, F.M.; Abdo, A.A. Genome-wide association study of chronic hepatitis B virus infection reveals a novel candidate risk allele on 11q22.3. J. Med. Genet. 2013, 50, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.N.; Naka, I.; Sa-Ngasang, A.; Anantapreecha, S.; Chanama, S.; Wichukchinda, N.; Sawanpanyalert, P.; Patarapotikul, J.; Tsuchiya, N.; Ohashi, J. A replication study confirms the association of GWAS-identified SNPs at MICB and PLCE1in Thai patients with dengue shock syndrome. BMC Med. Genet. 2014, 15, 58. [Google Scholar] [CrossRef] [PubMed]
- Van Manen, D.; van ‘t Wout, A.B.; Schuitemaker, H. Genome-wide association studies on HIV susceptibility, pathogenesis and pharmacogenomics. Retrovirology 2012, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, Y.; Xu, H.; Tang, K.; Wu, H.; Lu, L.; Wang, Z.; Chen, Z.; Xu, J.; Zhu, Y.; et al. Genome-Wide Association Studies of HIV-1 Host Control in Ethnically Diverse Chinese Populations. Sci. Rep. 2015, 5, 10879. [Google Scholar] [CrossRef] [PubMed]
- Zúñiga, J.; Buendía-Roldán, I.; Zhao, Y.; Jiménez, L.; Torres, D.; Romo, J.; Ramírez, G.; Cruz, A.; Vargas-Alarcon, G.; Sheu, C.C.; et al. Genetic variants associated with severe pneumonia in A/H1N1 influenza infection. Eur. Respir. J. 2012, 39, 604–610. [Google Scholar] [CrossRef] [PubMed]
- McLaren, P.J.; Coulonges, C.; Bartha, I.; Lenz, T.L.; Deutsch, A.J.; Bashirova, A.; Buchbinder, S.; Carrington, M.N.; Cossarizza, A.; Dalmau, J.; et al. Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc. Natl. Acad. Sci. USA 2015, 112, 14658–14663. [Google Scholar] [CrossRef] [PubMed]
- Van der Sijde, M.R.; Ng, A.; Fu, J. Systems genetics: From GWAS to disease pathways. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 10, 1903–1909. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 Deficiency in Patients with Herpes Simplex Encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, S.; Jouanguy, E.; Al-Hajjar, S.; Fieschi, C.; Al-Mohsen, I.Z.; Al-Jumaah, S.; Yang, K.; Chapgier, A.; Eidenschenk, C.; Eid, P.; et al. Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nat. Genet. 2003, 33, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Chapman, S.J.; Hill, A.V.S. Human genetic susceptibility to infectious disease. Nat. Rev. Genet. 2012, 13, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Brodin, P.; Jojic, V.; Gao, T.; Bhattacharya, S.; Angel, C.J.L.; Furman, D.; Shen-Orr, S.; Dekker, C.L.; Swan, G.E.; Butte, A.J.; et al. Variation in the human immune system is largely driven by non-heritable influences. Cell 2015, 160, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Skalka, A.M.; Flint, S.J.; Enquiest, L.W.; Krug, R.M.; Racaniello, V.R. Principles of Virology, 4th ed.; ASM Press: Washington, DC, USA, 2009; ISBN 9781555819514. [Google Scholar]
- Liu, R.; Paxton, W.A.; Choe, S.; Ceradini, D.; Martin, S.R.; Horuk, R.; MacDonald, M.E.; Stuhlmann, H.; Koup, R.A.; Landau, N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.-M.; Saragosti, S.; Lapouméroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Carrington, M.; Winkler, C.; Huttley, G.A.; Smith, M.W.; Allikmets, R.; Goedert, J.J.; Buchbinder, S.P.; Vittinghoff, E.; Gomperts, E.; et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 1996, 273, 1856–1862. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Hancock, D.B.; Gaddis, N.C.; Levy, J.L.; Bierut, L.J.; Kral, A.H.; Johnson, E.O. Associations of common variants in the BST2 region with HIV-1 acquisition in African American and European American people who inject drugs. Aids 2015, 29, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Laplana, M.; Caruz, A.; Pineda, J.A.; Puig, T.; Fibla, J. Association of BST-2 gene variants with HIV disease progression underscores the role of BST-2 in HIV type 1 infection. J. Infect. Dis. 2013, 207, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Ovsyannikova, I.G.; Ryan, J.E.; Vierkant, R.A.; Pankratz, V.S.; Jacobson, R.M.; Poland, G.A. Immunologic significance of HLA class I genes in measles virus-specific IFN-γ and IL-4 cytokine immune responses. Immunogenetics 2005, 57, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Salek-Ardakani, S.; Arrand, J.R.; Mackett, M. Epstein–Barr Virus Encoded Interleukin-10 Inhibits HLA-Class I, ICAM-1, and B7 Expression on Human Monocytes: Implications for immune evasion by EBV. Virology 2002, 304, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Pittman, K.J.; Glover, L.C.; Wang, L.; Ko, D.C. The Legacy of Past Pandemics: Common human mutations that protect against infectious disease. PLoS Pathog. 2016, 12, e1005680. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.S.; Schmidt, A.; Dittmann Chevillotte, M.; Wisskirchen, C.; Hellmuth, J.; Willms, S.; Gilmore, R.H.; Glas, J.; Folwaczny, M.; Müller, T.; et al. Polymorphisms in melanoma differentiation-associated gene 5 link protein function to clearance of hepatitis C virus. Hepatology 2015, 61, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.L.; Thio, C.L.; Martin, M.P.; Qi, Y.; Ge, D.; O’hUigin, C.; Kidd, J.; Kidd, K.; Khakoo, S.I.; Alexander, G.; et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009, 461, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Ndungo, E.; Herbert, A.S.; Raaben, M.; Obernosterer, G.; Biswas, R.; Miller, E.H.; Wirchnianski, A.S.; Carette, J.E.; Brummelkamp, T.R.; Whelan, S.P.; et al. A Single Residue in Ebola Virus Receptor NPC1 Influences Cellular Host Range in Reptiles. mSphere 2016, 1, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Ndungo, E.; Kaczmarek, M.E.; Herbert, A.S.; Binger, T.; Kuehne, A.I.; Jangra, R.K.; Hawkins, J.A.; Gifford, R.J.; Biswas, R.; et al. Filovirus receptor NPC1 contributes to species-specific patterns of ebolavirus susceptibility in bats. Elife 2015, 4, e11785. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Muhsin, M.; Atienza, G.A.; Kwak, D.-Y.; Kim, S.-M.; De Leon, T.B.; Angeles, E.R.; Coloquio, E.; Kondoh, H.; Satoh, K.; et al. Single nucleotide polymorphisms in a gene for translation initiation factor (eIF4G) of rice (Oryza sativa) associated with resistance to Rice tungro spherical virus. Mol. Plant Microbe Interact. 2010, 23, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.; Manolio, T. How to Interpret a Genome-wide Association Study. JAMA 2008, 11, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Cogni, R.; Cao, C.; Day, J.P.; Bridson, C.; Jiggins, F.M. The genetic architecture of resistance to virus infection in Drosophila. Mol. Ecol. 2016, 25, 5228–5241. [Google Scholar] [CrossRef] [PubMed]
- Magwire, M.M.; Fabian, D.K.; Schweyen, H.; Cao, C.; Longdon, B.; Bayer, F.; Jiggins, F.M. Genome-Wide Association Studies Reveal a Simple Genetic Basis of Resistance to Naturally Coevolving Viruses in Drosophila melanogaster. PLoS Genet. 2012, 8, e1003057. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, T.; Lucas, M.; Simon-Chazottes, D.; Frenkiel, M.-P.; Montagutelli, X.; Ceccaldi, P.-E.; Deubel, V.; Guenet, J.-L.; Despres, P. A nonsense mutation in the gene encoding 2′-5′-oligoadenylate synthetase/L1 isoform is associated with West Nile virus susceptibility in laboratory mice. Proc. Natl. Acad. Sci. USA 2002, 99, 11311–11316. [Google Scholar] [CrossRef] [PubMed]
- Welton, A.R.; Chesler, E.J.; Sturkie, C.; Anne, U.; Hirsch, G.N.; Spindler, K.R.; Jackson, A.U. Identification of Quantitative Trait Loci for Susceptibility to Mouse Adenovirus Type 1. J. Virol. 2005, 79, 11517–11522. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, A.L.; Okumura, A.; Ferris, M.T.; Green, R.; Feldmann, F.; Kelly, S.M.; Scott, D.P.; Safronetz, D.; Haddock, E.; LaCasse, R.; et al. Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Science 2014, 346, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Ashe, A.; Bélicard, T.; Le Pen, J.; Sarkies, P.; Frézal, L.; Lehrbach, N.J.; Félix, M.A.; Miska, E.A. A deletion polymorphism in the Caenorhabditis elegans RIG-I homolog disables viral RNA dicing and antiviral immunity. Elife 2013, 2, e00994. [Google Scholar] [CrossRef] [PubMed]
- King, E.G.; Merkes, C.M.; Mcneil, C.L.; Hoofer, S.R.; Sen, S.; Broman, K.W.; Long, A.D.; Macdonald, S.J. Genetic dissection of a model complex trait using the Drosophila Synthetic Population Resource. Genome Res. 2012, 22, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Iraqi, F.A.; Mahajne, M.; Salaymah, Y.; Sandovski, H.; Tayem, H.; Vered, K.; Balmer, L.; Hall, M.; Manship, G.; Morahan, G.; et al. The genome architecture of the collaborative cross mouse genetic reference population. Genetics 2012, 190, 389–401. [Google Scholar]
- Chinwalla, A.T.; Cook, L.L.; Delehaunty, K.D.; Fewell, G.A.; Fulton, L.A.; Fulton, R.S.; Graves, T.A.; Hillier, L.W.; Mardis, E.R.; McPherson, J.D.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef] [PubMed]
- Gardner, J.; Anraku, I.; Le, T.T.; Larcher, T.; Major, L.; Roques, P.; Schroder, W.A.; Higgs, S.; Suhrbier, A. Chikungunya Virus Arthritis in Adult Wild-Type Mice. J. Virol. 2010, 84, 8021–8032. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, R.R.; Bouvier, N.M. Animal models for influenza virus pathogenesis, transmission, and immunology. J. Immunol. Methods 2014, 410, 60–79. [Google Scholar] [CrossRef] [PubMed]
- Victor Garcia, J. Humanized mice for HIV and AIDS research. Curr. Opin. Virol. 2016, 19, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Guabiraba, R.; Ryffel, B. Dengue virus infection: Current concepts in immune mechanisms and lessons from murine models. Immunology 2014, 141, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Brehm, M.A.; Garcia, J.V.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Mestas, J.; Hughes, C.C.W. Of Mice and Not Men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Buchner, D.A.; Nadeau, J.H. Contrasting genetic architectures in different mouse reference populations used for studying complex traits. Genome Res. 2015, 25, 775–791. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Lee, S.-J.; Kim, K.E.; Lee, H.S.; Oh, N.; Park, I.; Ko, E.; Oh, S.J.; Lee, Y.-S.; Kim, D.; et al. Amelioration of sepsis by TIE2 activation-induced vascular protection. Sci. Transl. Med. 2016, 8, 335. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.C.; David, S.; Zhang, R.; Berghelli, A.; Milam, K.; Higgins, S.J.; Hunter, J.; Mukherjee, A.; Wei, Y.; Tran, M.; et al. Gene control of tyrosine kinase TIE2 and vascular manifestations of infections. Proc. Natl. Acad. Sci. USA 2016, 113, 2472–2477. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, J.H.; Singer, J.B.; Matin, A.; Lander, E.S. Analysing complex genetic traits with chromosome substitution strains. Nat. Genet. 2000, 24, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Deshmukh, H.; Johnson, N.; Cowell, L.G.; Rude, T.H.; Scott, W.K.; Nelson, C.L.; Zaas, A.K.; Marchuk, D.A.; Keum, S.; et al. Two genes on A/J chromosome 18 are associated with susceptibility to Staphylococcus aureus infection by combined microarray and QTL analyses. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Thach, D.C.; Kleeberger, S.R.; Tucker, P.C.; Griffin, D.E. Genetic Control of Neuroadapted Sindbis Virus Replication in Female Mice Maps to Chromosome 2 and Associates with Paralysis and Mortality. J. Virol. 2001, 75, 8674–8680. [Google Scholar] [CrossRef] [PubMed]
- Nedelko, T.; Kollmus, H.; Klawonn, F.; Spijker, S.; Lu, L.; Heßman, M.; Alberts, R.; Williams, R.W.; Schughart, K. Distinct gene loci control the host response to influenza H1N1 virus infection in a time-dependent manner. BMC Genom. 2012, 13, 411. [Google Scholar] [CrossRef] [PubMed]
- Boon, A.C.M.; deBeauchamp, J.; Hollmann, A.; Luke, J.; Kotb, M.; Rowe, S.; Finkelstein, D.; Neale, G.; Lu, L.; Williams, R.W.; et al. Host Genetic Variation Affects Resistance to Infection with a Highly Pathogenic H5N1 Influenza A Virus in Mice. J. Virol. 2009, 83, 10417–10426. [Google Scholar] [CrossRef] [PubMed]
- Horby, P.; Sudoyo, H.; Viprakasit, V.; Fox, A.; Thai, P.Q.; Yu, H.; Davila, S.; Hibberd, M.; Dustan, S.J.; Monteerarat, Y.; et al. What is the evidence of a role for host genetics in susceptibility to influenza A/H5N1? Epidemiol. Infect. 2010, 138, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Ferris, M.T.; Aylor, D.L.; Bottomly, D.; Whitmore, A.C.; Aicher, L.D.; Bell, T.A.; Bradel-Tretheway, B.; Bryan, J.T.; Buus, R.J.; Gralinski, L.E.; et al. Modeling Host Genetic Regulation of Influenza Pathogenesis in the Collaborative Cross. PLoS Pathog. 2013, 9, e1003196. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-L.; Hatesuer, B.; Bergmann, S.; Nedelko, T.; Schughart, K. Protection from Severe Influenza Virus Infections in Mice Carrying the Mx1 Influenza Virus Resistance Gene Strongly Depends on Genetic Background. J. Virol. 2015, 89, 9998–10009. [Google Scholar] [CrossRef] [PubMed]
- Turan, K.; Mibayashi, M.; Sugiyama, K.; Saito, S.; Numajiri, A.; Nagata, K. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res. 2004, 32, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Killip, M.J.; Staeheli, P.; Randall, R.E.; Jackson, D. The Human Interferon-Induced MxA Protein Inhibits Early Stages of Influenza A Virus Infection by Retaining the Incoming Viral Genome in the Cytoplasm. J. Virol. 2013, 87, 13053–13058. [Google Scholar] [CrossRef] [PubMed]
- Ciancanelli, M.J.; Abel, L.; Zhang, S.Y.; Casanova, J.L. Host genetics of severe influenza: From mouse Mx1 to human IRF7. Curr. Opin. Immunol. 2016, 38, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Perelygin, A.A.; Scherbik, S.V.; Zhulin, I.B.; Stockman, B.M.; Li, Y.; Brinton, M.A. Positional cloning of the murine flavivirus resistance gene. Proc. Natl. Acad. Sci. USA 2002, 99, 9322–9327. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.K.; Lisco, A.; McDermott, D.H.; Huynh, L.; Ward, J.M.; Johnson, B.; Johnson, H.; Pape, J.; Foster, G.A.; Krysztof, D.; et al. Genetic variation in OAS1 is a risk factor for initial infection with West Nile virus in man. PLoS Pathog. 2009, 5, e1000321. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.L.; Waldron, F.M.; Robertson, S.; Crowson, D.; Ferrari, G.; Quintana, J.F.; Brouqui, J.M.; Bayne, E.H.; Longdon, B.; Buck, A.H.; et al. The discovery, distribution, and evolution of viruses associated with drosophila melanogaster. PLoS Biol. 2015, 13, e1002210. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Massouras, A.; Inoue, Y.; Peiffer, J.; Ràmia, M.; Tarone, A.M.; Turlapati, L.; Zichner, T.; Zhu, D.; Lyman, R.F.; et al. Natural variation in genome architecture among 205 Drosophila melanogaster Genetic Reference Panel lines. Genome Res. 2014, 24, 1193–1208. [Google Scholar] [CrossRef] [PubMed]
- Mackay, T.F.C.; Richards, S.; Stone, E.A.; Barbadilla, A.; Ayroles, J.F.; Zhu, D.; Casillas, S.; Han, Y.; Magwire, M.M.; Cridland, J.M.; et al. The Drosophila melanogaster Genetic Reference Panel. Nature 2012, 482, 173–178. [Google Scholar] [CrossRef] [PubMed]
- King, E.G.; Macdonald, S.J.; Long, A.D. Properties and power of the Drosophila synthetic population resource for the routine dissection of complex traits. Genetics 2012, 191, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Magwire, M.M.; Bayer, F.; Jiggins, F.M. A Polymorphism in the Processing Body Component Ge-1 Controls Resistance to a Naturally Occurring Rhabdovirus in Drosophila. PLoS Pathog. 2016, 12, e1005387. [Google Scholar]
- Mussabekova, A.; Daeffler, L.; Imler, J.L. Innate and intrinsic antiviral immunity in Drosophila. Cell. Mol. Life Sci. 2017, 74, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wilfert, L.; Jiggins, F.M. Disease association mapping in Drosophila can be replicated in the wild. Biol. Lett. 2010, 6, 666–668. [Google Scholar] [CrossRef] [PubMed]
- Bangham, J.; Obbard, D.J.; Kim, K.-W.; Haddrill, P.R.; Jiggins, F.M. The age and evolution of an antiviral resistance mutation in Drosophila melanogaster. Proc. R. Soc. B 2007, 274, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Ciot, H.L.; Ciota, T.; Jia, Y.; Puig-Basagoiti, F.; Kramer, L.D.; Shi, P.-Y.; Glaser, R.L. West Nile Virus Infection of Drosophila melanogaster Induces a Protective RNAi Response. Virology 2008, 377, 197–206. [Google Scholar]
- Haddad, R.I.; Clark, J.R.; Wein, R.O.; Grillone, G.A. Broad RNA interference-mediated antiviral immunity and virus- specific inducible responses in Drosophila. J. Immunol. 2013, 190, 650–658. [Google Scholar]
- Sabin, L.R.; Zhou, R.; Gruber, J.J.; Lukinova, N.; Bambina, S.; Lau, C.; Thompson, C.B.; Cherry, S. Ars2 regulates both miRNA- and siRNA-dependent silencing and suppresses RNA virus infection in Drosophila. Cell 2009, 138, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.; Pettersson, J.; Higgs, S.; Charrel, R.; de Lamballerie, X. Emerging arboviruses: Why today? One Health 2017, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, B.E.; Phillips, P.C. Caenorhabditis elegans as a platform for molecular quantitative genetics and the systems biology of natural variation. Genet. Res. 2010, 92, 331–348. [Google Scholar] [CrossRef] [PubMed]
- Andersen, E.C.; Gerke, J.P.; Shapiro, J.A.; Crissman, J.R.; Ghosh, R.; Bloom, J.S.; Félix, M.-A.; Kruglyak, L. Chromosome-scale selective sweeps shape Caenorhabditis elegans genomic diversity. Nat. Genet. 2012, 44, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.E.; Zdraljevic, S.; Roberts, J.P.; Andersen, E.C. CeNDR, the Caenorhabditis elegans natural diversity resource. Nucleic Acids Res. 2017, 45, D650–D657. [Google Scholar] [CrossRef] [PubMed]
- Rockman, M.V.; Kruglyak, L. Recombinational landscape and population genomics of caenorhabditis elegans. PLoS Genet. 2009, 5, e1000419. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Álvarez, O.A.; Gutteling, E.W.; Tijsterman, M.; Fu, J.; Riksen, J.A.G.; Hazendonk, E.; Prins, P.; Plasterk, R.H.A.; Jansen, R.C.; et al. Mapping determinants of gene expression plasticity by genetical genomics in C. elegans. PLoS Genet. 2006, 2, 2155–2161. [Google Scholar] [CrossRef] [PubMed]
- Doroszuk, A.; Snoek, L.B.; Fradin, E.; Riksen, J.; Kammenga, J. A genome-wide library of CB4856/N2 introgression lines of CB4856/N2 introgression lines of Caenorhabditis elegans. Nucleic Acids Res. 2009, 37, e110. [Google Scholar] [CrossRef] [PubMed]
- Andersen, E.C.; Shimko, T.C.; Crissman, J.R.; Ghosh, R.; Bloom, J.S.; Seidel, H.S.; Gerke, J.P.; Kruglyak, L. A Powerful New Quantitative Genetics Platform, Combining Caenorhabditis elegans High-Throughput Fitness Assays with a Large Collection of Recombinant Strains. G3 Genes Genomes Genet. 2015, 5, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, M.R.; Rockman, M.V. Fine-Scale Crossover Rate Variation on the Caenorhabditis elegans X Chromosome. G3 Genes Genomes Genet. 2016, 6, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Harry, B.L.; Zhou, Q.; Skeen-Gaar, R.R.; Ge, X.; Lee, E.S.; Mitani, S.; Xue, D. Hepatitis B virus X protein targets the Bcl-2 protein CED-9 to induce intracellular Ca2+ increase and cell death in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2012, 109, 18465–18470. [Google Scholar] [CrossRef] [PubMed]
- Gammon, D.B.; Ishidate, T.; Li, L.; Gu, W.; Silverman, N.; Mello, C.C. The Antiviral RNA Interference Response Provides Resistance to Lethal Arbovirus Infection and Vertical Transmission in Caenorhabditis elegans. Curr. Biol. 2017, 27, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Félix, M.A.; Ashe, A.; Piffaretti, J.; Wu, G.; Nuez, I.; Bélicard, T.; Jiang, Y.; Zhao, G.; Franz, C.J.; Goldstein, L.D.; et al. Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biol. 2011, 9, e1000586. [Google Scholar] [CrossRef] [PubMed]
- Sterken, M.G.; Snoek, L.B.; Bosman, K.J.; Daamen, J.; Riksen, J.A.G.; Bakker, J.; Pijlman, G.P.; Kammenga, J.E. A heritable antiviral RNAi response limits orsay virus infection in Caenorhabditis elegans N2. PLoS ONE 2014, 9, e89760. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M. Master sensors of pathogenic RNA—RIG-I like receptors. Immunobiology 2013, 218, 1322–1335. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Sluijs, L.; Pijlman, G.P.; Kammenga, J.E. Why do Individuals Differ in Viral Susceptibility? A Story Told by Model Organisms. Viruses 2017, 9, 284. https://doi.org/10.3390/v9100284
Van Sluijs L, Pijlman GP, Kammenga JE. Why do Individuals Differ in Viral Susceptibility? A Story Told by Model Organisms. Viruses. 2017; 9(10):284. https://doi.org/10.3390/v9100284
Chicago/Turabian StyleVan Sluijs, Lisa, Gorben P. Pijlman, and Jan E. Kammenga. 2017. "Why do Individuals Differ in Viral Susceptibility? A Story Told by Model Organisms" Viruses 9, no. 10: 284. https://doi.org/10.3390/v9100284
APA StyleVan Sluijs, L., Pijlman, G. P., & Kammenga, J. E. (2017). Why do Individuals Differ in Viral Susceptibility? A Story Told by Model Organisms. Viruses, 9(10), 284. https://doi.org/10.3390/v9100284