
Quantification of Lyssavirus-Neutralizing Antibodies Using Vesicular Stomatitis Virus Pseudotype Particles


Abstract
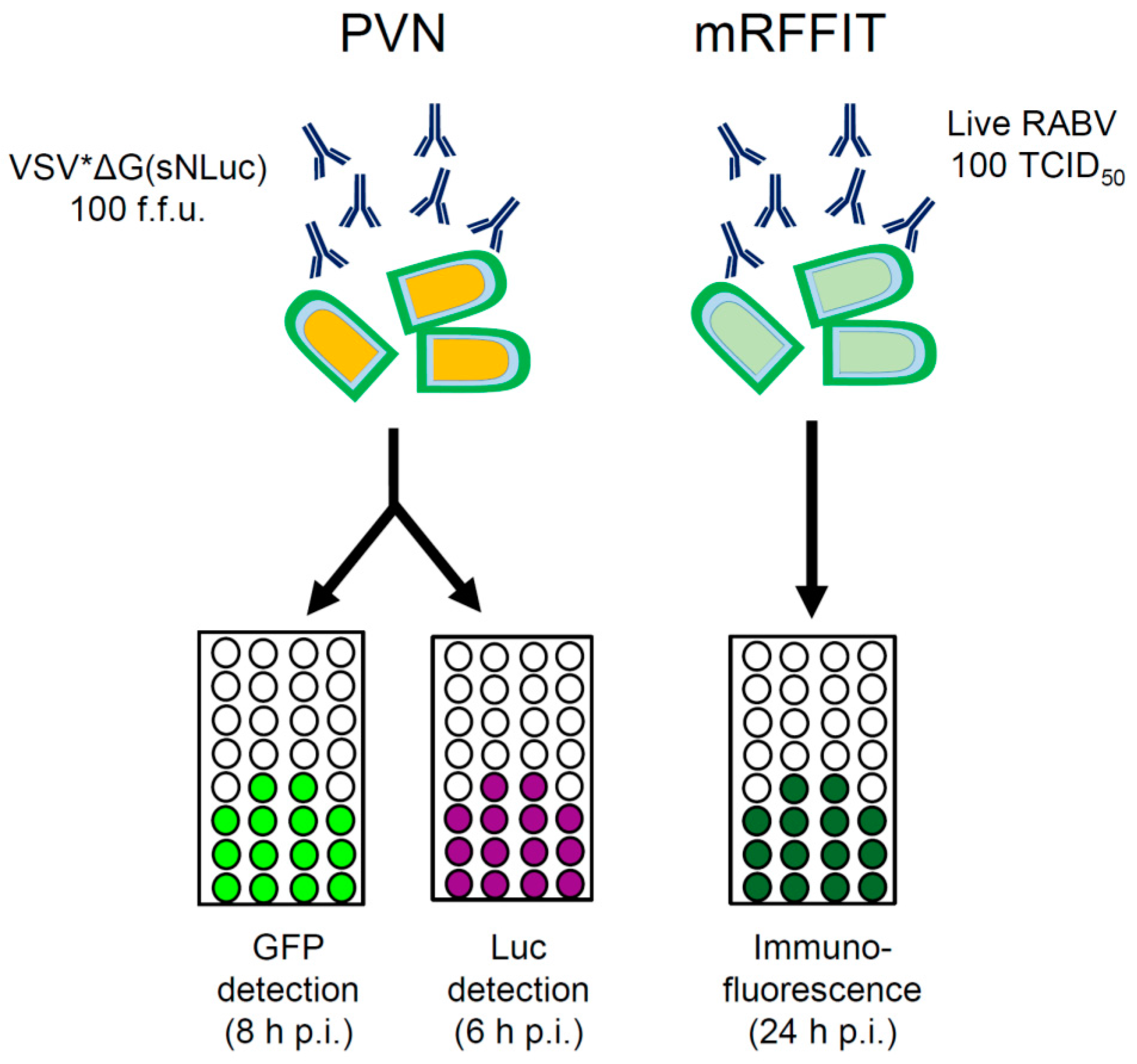
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Generation of Helper Cell Lines
2.3. Antibodies and Immune Sera
2.4. Immunofluorescence Analysis
2.5. Cell Surface Biotinylation
2.6. Generation of Pseudotype Virus
2.7. Virus Neutralization Tests
2.8. Statistical Analysis
3. Results
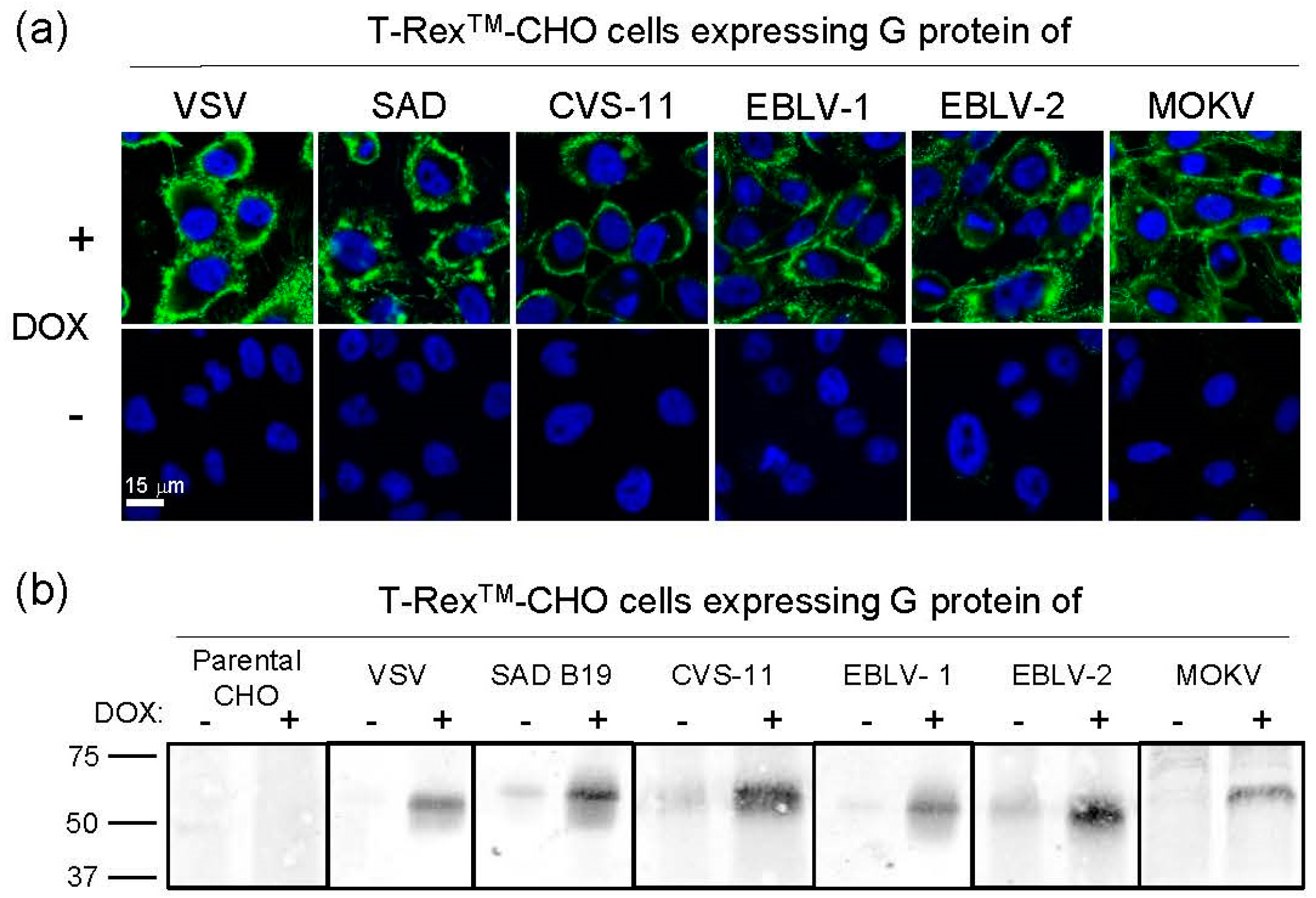
3.1. Generation of Transgenic Helper Cell Lines
3.2. Generation of VSV Pseudotypes
3.3. Neutralization of Pseudotype Virus
3.4. Analysis of Pseudotype Viruses Bearing Other Lyssavirus Glycoproteins
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rupprecht, C.E.; Hanlon, C.A.; Hemachudha, T. Rabies re-examined. Lancet Infect. Dis. 2002, 2, 327–343. [Google Scholar] [CrossRef]
- Afonso, C.L.; Amarasinghe, G.K.; Banyai, K.; Bao, Y.; Basler, C.F.; Bavari, S.; Bejerman, N.; Blasdell, K.R.; Briand, F.X.; Briese, T.; et al. Taxonomy of the order Mononegavirales: Update 2016. Arch. Virol. 2016, 161, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Badrane, H.; Bahloul, C.; Perrin, P.; Tordo, N. Evidence of two Lyssavirus phylogroups with distinct pathogenicity and immunogenicity. J. Virol. 2001, 75, 3268–3276. [Google Scholar] [CrossRef] [PubMed]
- Delmas, O.; Holmes, E.C.; Talbi, C.; Larrous, F.; Dacheux, L.; Bouchier, C.; Bourhy, H. Genomic diversity and evolution of the lyssaviruses. PLoS ONE 2008, 3, e2057. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, I.V.; Novella, I.S.; Dietzgen, R.G.; Padhi, A.; Rupprecht, C.E. The rhabdoviruses: Biodiversity, phylogenetics, and evolution. Infect. Genet. Evol. 2009, 9, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, I.V.; Wu, X.; Tordo, N.; Rupprecht, C.E. Complete genomes of Aravan, Khujand, Irkut and West Caucasian bat viruses, with special attention to the polymerase gene and non-coding regions. Virus Res. 2008, 136, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Malerczyk, C.; Freuling, C.; Gniel, D.; Giesen, A.; Selhorst, T.; Muller, T. Cross-neutralization of antibodies induced by vaccination with Purified Chick Embryo Cell Vaccine (PCECV) against different Lyssavirus species. Hum. Vaccin. Immunother. 2014, 10, 2799–2804. [Google Scholar] [CrossRef] [PubMed]
- Fooks, A.R.; Banyard, A.C.; Horton, D.L.; Johnson, N.; McElhinney, L.M.; Jackson, A.C. Current status of rabies and prospects for elimination. Lancet 2014, 384, 1389–1399. [Google Scholar] [CrossRef]
- Fooks, A.R.; McElhinney, L.M.; Pounder, D.J.; Finnegan, C.J.; Mansfield, K.; Johnson, N.; Brookes, S.M.; Parsons, G.; White, K.; McIntyre, P.G.; et al. Case report: Isolation of a European bat lyssavirus type 2a from a fatal human case of rabies encephalitis. J. Med. Virol. 2003, 71, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Familusi, J.B.; Osunkoya, B.O.; Moore, D.L.; Kemp, G.E.; Fabiyi, A. A fatal human infection with Mokola virus. Am. J. Trop. Med. Hyg. 1972, 21, 959–963. [Google Scholar] [PubMed]
- Francis, J.R.; Nourse, C.; Vaska, V.L.; Calvert, S.; Northill, J.A.; McCall, B.; Mattke, A.C. Australian Bat Lyssavirus in a child: The first reported case. Pediatrics 2014, 133, e1063–e1067. [Google Scholar] [CrossRef] [PubMed]
- Paweska, J.T.; Blumberg, L.H.; Liebenberg, C.; Hewlett, R.H.; Grobbelaar, A.A.; Leman, P.A.; Croft, J.E.; Nel, L.H.; Nutt, L.; Swanepoel, R. Fatal human infection with rabies-related Duvenhage virus, South Africa. Emerg. Infect. Dis. 2006, 12, 1965–1967. [Google Scholar] [CrossRef] [PubMed]
- Dietzschold, B.; Li, J.; Faber, M.; Schnell, M. Concepts in the pathogenesis of rabies. Future Virol. 2008, 3, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.H.; Dietzschold, B.; Schneider, L.G. Rabies virus glycoprotein. II. Biological and serological characterization. Infect. Immun. 1977, 16, 754–759. [Google Scholar] [PubMed]
- Evans, J.S.; Horton, D.L.; Easton, A.J.; Fooks, A.R.; Banyard, A.C. Rabies virus vaccines: Is there a need for a pan-lyssavirus vaccine? Vaccine 2012, 30, 7447–7454. [Google Scholar] [CrossRef] [PubMed]
- Hampson, K.; Coudeville, L.; Lembo, T.; Sambo, M.; Kieffer, A.; Attlan, M.; Barrat, J.; Blanton, J.D.; Briggs, D.J.; Cleaveland, S.; et al. Estimating the global burden of endemic canine rabies. PLoS Negl. Trop. Dis. 2015, 9, e0003709. [Google Scholar]
- Knobel, D.L.; Cleaveland, S.; Coleman, P.G.; Fevre, E.M.; Meltzer, M.I.; Miranda, M.E.; Shaw, A.; Zinsstag, J.; Meslin, F.X. Re-evaluating the burden of rabies in Africa and Asia. Bull. World Health Organ. 2005, 83, 360–368. [Google Scholar] [PubMed]
- Cliquet, F.; Aubert, M.; Sagne, L. Development of a fluorescent antibody virus neutralisation test (FAVN test) for the quantitation of rabies-neutralising antibody. J. Immunol. Methods 1998, 212, 79–87. [Google Scholar] [CrossRef]
- Smith, J.S.; Yager, P.A.; Baer, G.M. A rapid reproducible test for determining rabies neutralizing antibody. Bull. World Health Organ. 1973, 48, 535–541. [Google Scholar] [PubMed]
- Moore, S.M.; Hanlon, C.A. Rabies-specific antibodies: Measuring surrogates of protection against a fatal disease. PLoS Negl. Trop. Dis. 2010, 4, e595. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.; McNabb, S.; Goddard, T.; Horton, D.L.; Lembo, T.; Nel, L.H.; Weiss, R.A.; Cleaveland, S.; Fooks, A.R. A robust lentiviral pseudotype neutralisation assay for in-field serosurveillance of rabies and lyssaviruses in Africa. Vaccine 2009, 27, 7178–7186. [Google Scholar] [CrossRef] [PubMed]
- Hanika, A.; Larisch, B.; Steinmann, E.; Schwegmann-Wessels, C.; Herrler, G.; Zimmer, G. Use of influenza C virus glycoprotein HEF for generation of vesicular stomatitis virus pseudotypes. J. Gen. Virol. 2005, 86, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Genz, B.; Nolden, T.; Negatsch, A.; Teifke, J.P.; Conzelmann, K.K.; Finke, S. Chimeric rabies viruses for trans-species comparison of lyssavirus glycoprotein ectodomain functions in virus replication and pathogenesis. Berl. Munchener Tierarztliche Wochenschr. 2012, 125, 219–227. [Google Scholar]
- Conzelmann, K.K.; Cox, J.H.; Schneider, L.G.; Thiel, H.J. Molecular cloning and complete nucleotide sequence of the attenuated rabies virus SAD B19. Virology 1990, 175, 485–499. [Google Scholar] [CrossRef]
- Marston, D.A.; McElhinney, L.M.; Johnson, N.; Muller, T.; Conzelmann, K.K.; Tordo, N.; Fooks, A.R. Comparative analysis of the full genome sequence of European bat lyssavirus type 1 and type 2 with other lyssaviruses and evidence for a conserved transcription termination and polyadenylation motif in the G-L 3′ non-translated region. J. Gen. Virol. 2007, 88, 1302–1314. [Google Scholar] [CrossRef] [PubMed]
- Mebatsion, T.; Schnell, M.J.; Conzelmann, K.K. Mokola virus glycoprotein and chimeric proteins can replace rabies virus glycoprotein in the rescue of infectious defective rabies virus particles. J. Virol. 1995, 69, 1444–1451. [Google Scholar] [PubMed]
- Kalhoro, N.H.; Veits, J.; Rautenschlein, S.; Zimmer, G. A recombinant vesicular stomatitis virus replicon vaccine protects chickens from highly pathogenic avian influenza virus (H7N1). Vaccine 2009, 27, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Berger Rentsch, M.; Zimmer, G. A vesicular stomatitis virus replicon-based bioassay for the rapid and sensitive determination of multi-species type I interferon. PLoS ONE 2011, 6, e25858. [Google Scholar] [CrossRef] [PubMed]
- Krämer, B.; Bruckner, L.; Daas, A.; Milne, C. Collaborative study for validation of a serological potency assay for rabies vaccine (inactivated) for veterinary use. Pharmeuropa Bio Sci. Notes 2010, 2010, 37–55. [Google Scholar]
- Krämer, B.; Schildger, H.; Behrensdorf-Nicol, H.A.; Hanschmann, K.M.; Duchow, K. The rapid fluorescent focus inhibition test is a suitable method for batch potency testing of inactivated rabies vaccines. Biologicals 2009, 37, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Wulff, N.H.; Tzatzaris, M.; Young, P.J. Monte Carlo simulation of the Spearman-Kaerber TCID50. J. Clin. Bioinform. 2012, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Wu, Y.J.; Gerber, M.; Berger-Rentsch, M.; Heimrich, B.; Schwemmle, M.; Zimmer, G. Fusion-active glycoprotein G mediates the cytotoxicity of vesicular stomatitis virus M mutants lacking host shut-off activity. J. Gen. Virol. 2010, 91, 2782–2793. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Vos, A.; Freuling, C.; Tordo, N.; Fooks, A.R.; Muller, T. Human rabies due to lyssavirus infection of bat origin. Vet. Microbiol. 2010, 142, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.B.; Lu, Z.L.; Wei, X.K.; Zhong, Y.Z.; Zhong, T.Z.; Pan, Y.; Luo, Y.; Liao, S.H.; Minamoto, N.; Luo, T.R. A recombinant rabies virus expressing a phosphoprotein-eGFP fusion is rescued and applied to the rapid virus neutralization antibody assay. J. Virol. Methods 2015, 219, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Zheng, X.; Liang, H.; Feng, N.; Zhao, Y.; Gao, Y.; Wang, H.; Yang, S.; Xia, X. Generation of recombinant rabies Virus CVS-11 expressing eGFP applied to the rapid virus neutralization test. Viruses 2014, 6, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Khawplod, P.; Inoue, K.; Shoji, Y.; Wilde, H.; Ubol, S.; Nishizono, A.; Kurane, I.; Morimoto, K. A novel rapid fluorescent focus inhibition test for rabies virus using a recombinant rabies virus visualizing a green fluorescent protein. J. Virol. Methods 2005, 125, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Conzelmann, K.K. G gene-deficient single-round rabies viruses for neuronal circuit analysis. Virus Res. 2016, 216, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Tani, H.; Morikawa, S.; Matsuura, Y. Development and Applications of VSV Vectors Based on Cell Tropism. Front. Microbiol. 2011, 2, 272. [Google Scholar] [CrossRef] [PubMed]
- Kontsekova, E.; Macikova, I.; Novak, M.; Dedek, L.; Vrzal, V.; Kontsek, P. Conformation-dependent accessibility of the linear epitopes located on the rabies virus glycoprotein. Viral Immunol. 1992, 5, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.R.; Minamoto, N.; Hishida, M.; Yamamoto, K.; Fujise, T.; Hiraga, S.; Ito, N.; Sugiyama, M.; Kinjo, T. Antigenic and functional analyses of glycoprotein of rabies virus using monoclonal antibodies. Microbiol. Immunol. 1998, 42, 187–193. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neutralization Titer (ND50/mL) for VSV*∆G(Luc) Pseudotyped with the G protein of | ||||||
|---|---|---|---|---|---|---|
| Antiserum/Antibody | VSV (Indiana) | RABV (SAD B18) | RABV (CVS-11) | EBLV-1 | EBLV-2 | MOKV (Eth-16) |
| α-VSV | 1333 | <4 | <4 | <4 | <4 | <4 |
| α-RABV(RAI) a | <4 | 191 | 224 | 19 | 38 | <4 |
| mAb 16DB4 b | <4 | 13,881 | 8913 | 3452 | 5673 | <4 |
| α-CVS-11 G c | <4 | 1608 | 5392 | 7 | 19 | <4 |
| α-MOKV G c | <4 | <4 | <4 | <4 | <4 | 1831 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moeschler, S.; Locher, S.; Conzelmann, K.-K.; Krämer, B.; Zimmer, G. Quantification of Lyssavirus-Neutralizing Antibodies Using Vesicular Stomatitis Virus Pseudotype Particles. Viruses 2016, 8, 254. https://doi.org/10.3390/v8090254
Moeschler S, Locher S, Conzelmann K-K, Krämer B, Zimmer G. Quantification of Lyssavirus-Neutralizing Antibodies Using Vesicular Stomatitis Virus Pseudotype Particles. Viruses. 2016; 8(9):254. https://doi.org/10.3390/v8090254
Chicago/Turabian StyleMoeschler, Sarah, Samira Locher, Karl-Klaus Conzelmann, Beate Krämer, and Gert Zimmer. 2016. "Quantification of Lyssavirus-Neutralizing Antibodies Using Vesicular Stomatitis Virus Pseudotype Particles" Viruses 8, no. 9: 254. https://doi.org/10.3390/v8090254
APA StyleMoeschler, S., Locher, S., Conzelmann, K.-K., Krämer, B., & Zimmer, G. (2016). Quantification of Lyssavirus-Neutralizing Antibodies Using Vesicular Stomatitis Virus Pseudotype Particles. Viruses, 8(9), 254. https://doi.org/10.3390/v8090254