
Stress Beyond Translation: Poxviruses and More


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Poxvirus Infection and Host Translation
2.1. Host Translation Shutoff and Differential Regulation of Host Transcripts
2.2. Heat Shock Responses
2.3. Evade the Surveillance
2.4. Manipulating the Translation Machinery
2.5. Codon Usage
2.6. Reactive Oxygen Species (ROS)
3. Poxvirus Infection, Where Stress Granules and Antiviral Granules Cross Paths
3.1. Stress Granules
3.2. Antiviral Granules (AVGs) and the PKR/eIF2α Axis
3.3. Antiviral Granules and the SAMD9 Pathway
3.4. Antiviral Granules and Innate Sensing Molecules
4. SGs and RNA-stimulated Antiviral Stress Granules as Signaling Hubs
5. Cytoplasmic Bodies Containing Viral DNA: Signaling Beyond Translation
5.1. DNA-Sensing Bodies Containing IFN γ-inducible Protein 16 (IFI16)
5.2. Other DNA-Sensing Bodies
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anderson, P.; Kedersha, N. Stressful initiations. J. Cell Sci. 2002, 115, 3227–3234. [Google Scholar] [PubMed]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Mohr, I. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbio. 2011, 9, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Schnierle, B.S.; Moss, B. Vaccinia virus-mediated inhibition of host protein synthesis involves neither degradation nor underphosphorylation of components of the cap-binding eukaryotic translation initiation factor complex eIF-4F. Virology 1992, 188, 931–933. [Google Scholar] [CrossRef]
- Guerra, S.; Lopez-Fernandez, L.A.; Pascual-Montano, A.; Munoz, M.; Harshman, K.; Esteban, M. Cellular gene expression survey of vaccinia virus infection of human HeLa cells. J. Virol. 2003, 77, 6493–6506. [Google Scholar] [CrossRef] [PubMed]
- Guerra, S.; Aracil, M.; Conde, R.; Bernad, A.; Esteban, M. Wiskott-aldrich syndrome protein is needed for vaccinia virus pathogenesis. J. Virol. 2005, 79, 2133–2140. [Google Scholar] [CrossRef] [PubMed]
- Gershowitz, A.; Boone, R.F.; Moss, B. Multiple roles for ATP in the synthesis and processing of mRNA by vaccinia virus: Specific inhibitory effects of adenosine (beta,gamma-imido) triphosphate. J. Virol. 1978, 27, 399–408. [Google Scholar] [PubMed]
- Shuman, S.; Spencer, E.; Furneaux, H.; Hurwitz, J. The role of ATP in in vitro vaccinia virus RNA synthesis effects of AMP-PNP and ATP gamma S. J. Biol. Chem. 1980, 255, 5396–5403. [Google Scholar] [PubMed]
- Foglesong, P.D.; Bauer, W.R. Effects of ATP and inhibitory factors on the activity of vaccinia virus type i topoisomerase. J. Virol. 1984, 49, 1–8. [Google Scholar] [PubMed]
- Deng, L.; Shuman, S. Vaccinia NPH-I, a dexh-box ATPase, is the energy coupling factor for mRNA transcription termination. Genes Dev. 1998, 12, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Boyle, K.A.; Arps, L.; Traktman, P. Biochemical and genetic analysis of the vaccinia virus d5 protein: Multimerization-dependent ATPase activity is required to support viral DNA replication. J. Virol. 2007, 81, 844–859. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Li, H.C.; Hsu, C.F.; Chang, C.Y.; Lo, S.Y. Increased ATP generation in the host cell is required for efficient vaccinia virus production. J. Biomed. Sci. 2009, 16, 80. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Snijder, B.; Sacher, R.; Burkard, C.; Bleck, C.K.; Stahlberg, H.; Pelkmans, L.; Helenius, A. RNAi screening reveals proteasome- and Cullin3-dependent stages in vaccinia virus infection. Cell Rep. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Sivan, G.; Martin, S.E.; Myers, T.G.; Buehler, E.; Szymczyk, K.H.; Ormanoglu, P.; Moss, B. Human genome-wide RNAi screen reveals a role for nuclear pore proteins in poxvirus morphogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3519–3524. [Google Scholar] [CrossRef] [PubMed]
- Teferi, W.M.; Dodd, K.; Maranchuk, R.; Favis, N.; Evans, D.H. A whole-genome RNA interference screen for human cell factors affecting myxoma virus replication. J. Virol. 2013, 87, 4623–4641. [Google Scholar] [CrossRef] [PubMed]
- Filone, C.M.; Caballero, I.S.; Dower, K.; Mendillo, M.L.; Cowley, G.S.; Santagata, S.; Rozelle, D.K.; Yen, J.; Rubins, K.H.; Hacohen, N.; et al. The master regulator of the cellular stress response (HSF1) is critical for orthopoxvirus infection. PLoS Pathog. 2014, 10, e1003904. [Google Scholar] [CrossRef] [PubMed]
- Guettouche, T.; Boellmann, F.; Lane, W.S.; Voellmy, R. Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem. 2005, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- de Magalhaes, J.C.; Andrade, A.A.; Silva, P.N.; Sousa, L.P.; Ropert, C.; Ferreira, P.C.; Kroon, E.G.; Gazzinelli, R.T.; Bonjardim, C.A. A mitogenic signal triggered at an early stage of vaccinia virus infection: Implication of mek/erk and protein kinase A in virus multiplication. J. Biol. Chem. 2001, 276, 38353–38360. [Google Scholar] [CrossRef] [PubMed]
- Andrade, A.A.; Silva, P.N.; Pereira, A.C.; De Sousa, L.P.; Ferreira, P.C.; Gazzinelli, R.T.; Kroon, E.G.; Ropert, C.; Bonjardim, C.A. The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication. Biochem. J. 2004, 381, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Jindal, S.; Young, R.A. Vaccinia virus infection induces a stress response that leads to association of Hsp70 with viral proteins. J. Virol. 1992, 66, 5357–5362. [Google Scholar] [PubMed]
- Sedger, L.; Ruby, J. Heat shock response to vaccinia virus infection. J. Virol. 1994, 68, 4685–4689. [Google Scholar] [PubMed]
- Sedger, L.; Ramshaw, I.; Condie, A.; Medveczky, J.; Braithwaite, A.; Ruby, J. Vaccinia virus replication is independent of cellular Hsp72 expression which is induced during virus infection. Virology 1996, 225, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.J.; Chung, C.S.; Chang, W. Molecular chaperone Hsp90 is important for vaccinia virus growth in cells. J. Virol. 2002, 76, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Stohr, N.; Lederer, M.; Reinke, C.; Meyer, S.; Hatzfeld, M.; Singer, R.H.; Huttelmaier, S. Zbp1 regulates mRNA stability during cellular stress. J. Cell Biol. 2006, 175, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Baguet, A.; Degot, S.; Cougot, N.; Bertrand, E.; Chenard, M.P.; Wendling, C.; Kessler, P.; Le Hir, H.; Rio, M.C.; Tomasetto, C. The exon-junction-complex-component metastatic lymph node 51 functions in stress-granule assembly. J. Cell Sci. 2007, 120, 2774–2784. [Google Scholar] [CrossRef] [PubMed]
- Yost, H.J.; Petersen, R.B.; Lindquist, S. RNA metabolism: Strategies for regulation in the heat shock response. Trends Genet. TIG 1990, 6, 223–227. [Google Scholar] [CrossRef]
- Rubtsova, M.P.; Sizova, D.V.; Dmitriev, S.E.; Ivanov, D.S.; Prassolov, V.S.; Shatsky, I.N. Distinctive properties of the 5'-untranslated region of human Hsp70 mRNA. J. Biol. Chem. 2003, 278, 22350–22356. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Duncan, R.F. Translational regulation of Hsp90 mRNA. Aug-proximal 5'-untranslated region elements essential for preferential heat shock translation. J. Biol. Chem. 2004, 279, 49919–49930. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.F. Rapamycin conditionally inhibits Hsp90 but not Hsp70 mRNA translation in drosophila: Implications for the mechanisms of Hsp mRNA translation. Cell Stress Chaperones 2008, 13, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, K.; Mohamed, M.R.; Zhang, L.; Villa, N.Y.; Werden, S.J.; Liu, J.; McFadden, G. Poxvirus proteomics and virus-host protein interactions. Microbiol. Mol. Biol. Rev. MMBR 2009, 73, 730–749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Villa, N.Y.; Rahman, M.M.; Smallwood, S.; Shattuck, D.; Neff, C.; Dufford, M.; Lanchbury, J.S.; Labaer, J.; McFadden, G. Analysis of vaccinia virus-host protein-protein interactions: Validations of yeast two-hybrid screenings. J. Proteome Res. 2009, 8, 4311–4318. [Google Scholar] [CrossRef] [PubMed]
- Ensinger, M.J.; Martin, S.A.; Paoletti, E.; Moss, B. Modification of the 5′-terminus of mRNA by soluble guanylyl and methyl transferases from vaccinia virus. Proc. Natl. Acad. Sci. USA 1975, 72, 2525–2529. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; Moss, B. Modification of RNA by mRNA guanylyltransferase and mRNA (guanine-7-) methyltransferase from vaccinia virions. J. Biol. Chem. 1975, 250, 9330–9335. [Google Scholar] [PubMed]
- Boone, R.F.; Moss, B. Methylated 5'-terminal sequences of vaccinia virus mRNA species made in vivo at early and late times after infection. Virology 1977, 79, 67–80. [Google Scholar] [CrossRef]
- Venkatesan, S.; Gershowitz, A.; Moss, B. Modification of the 5' end of mRNA. Association of RNA triphosphatase with the RNA guanylyltransferase-RNA (guanine-7-)methyltransferase complex from vaccinia virus. J. Biol. Chem. 1980, 255, 903–908. [Google Scholar] [PubMed]
- Atherton, K.T.; Darby, G. Patterns of transcription of messengers containing poly A in vaccinia virus-infected cells. J. Gen. Virol. 1974, 22, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.E.; Hillner, P.E.; Vale, R.D.; Sachs, A.B. Circularization of mRNA by eukaryotic translation initiation factors. Mol. Cell 1998, 2, 135–140. [Google Scholar] [CrossRef]
- Pause, A.; Belsham, G.J.; Gingras, A.C.; Donze, O.; Lin, T.A.; Lawrence, J.C., Jr.; Sonenberg, N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5'-cap function. Nature 1994, 371, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Reineke, L.C.; Lloyd, R.E. Animal virus schemes for translation dominance. Curr. Opin. Virol. 2011, 1, 363–372. [Google Scholar] [CrossRef] [PubMed]
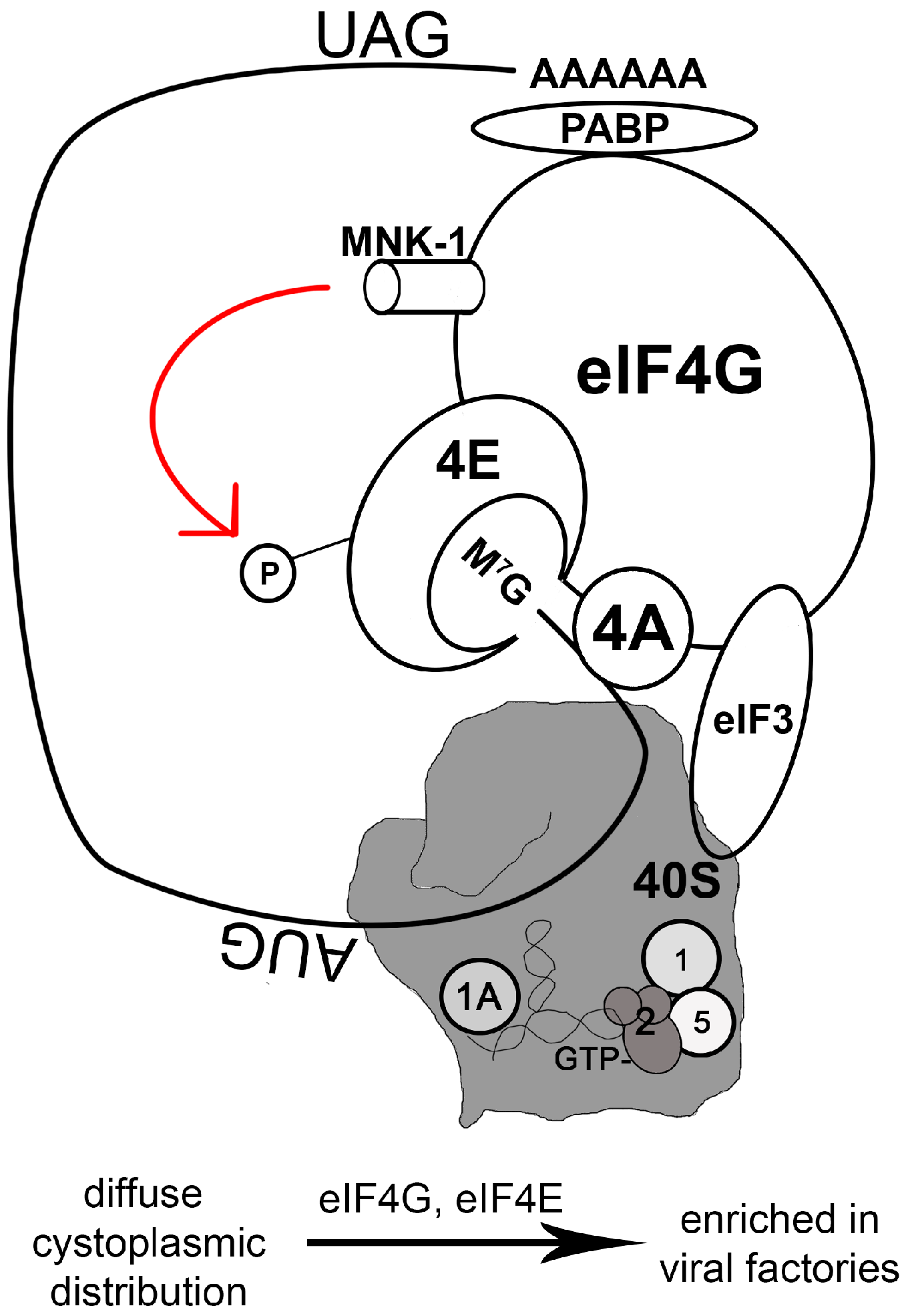
- Zaborowska, I.; Kellner, K.; Henry, M.; Meleady, P.; Walsh, D. Recruitment of host translation initiation factor eif4g by the vaccinia virus ssDNA-binding protein I3. Virology 2012, 425, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Katsafanas, G.C.; Moss, B. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2007, 2, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Arias, C.; Perez, C.; Halladin, D.; Escandon, M.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R.; Mohr, I. Eukaryotic translation initiation factor 4f architectural alterations accompany translation initiation factor redistribution in poxvirus-infected cells. Mol. Cell. Biol. 2008, 28, 2648–2658. [Google Scholar] [CrossRef] [PubMed]
- Castello, A.; Quintas, A.; Sanchez, E.G.; Sabina, P.; Nogal, M.; Carrasco, L.; Revilla, Y. Regulation of host translational machinery by african swine fever virus. PLoS Pathog. 2009, 5, e1000562. [Google Scholar] [CrossRef] [PubMed]
- Rozelle, D.K.; Filone, C.M.; Kedersha, N.; Connor, J.H. Activation of stress response pathways promotes formation of antiviral granules and restricts virus replication. Mol. Cell. Biol. 2014, 34, 2003–2016. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; McFadden, G. SAMD9 is an innate antiviral host factor with stress response properties that can be antagonized by poxviruses. J. Virol. 2015, 89, 1925–1931. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.; Walsh, D.; Harbell, J.; Wilson, A.C.; Mohr, I. Activation of host translational control pathways by a viral developmental switch. PLoS Pathog. 2009, 5, e1000334. [Google Scholar] [CrossRef] [PubMed]
- Kudchodkar, S.B.; Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mtor kinase. J. Virol. 2004, 78, 11030–11039. [Google Scholar] [CrossRef] [PubMed]
- McMahon, R.; Zaborowska, I.; Walsh, D. Noncytotoxic inhibition of viral infection through eif4f-independent suppression of translation by 4EGI-1. J. Virol. 2011, 85, 853–864. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, C.; Klupsch, K.; Choi, S.; Bagus, B.; Soria, C.; Shen, J.; McCormick, F.; Stokoe, D. Adenoviral proteins mimic nutrient/growth signals to activate the mtor pathway for viral replication. EMBO J. 2005, 24, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Moorman, N.J.; Shenk, T. Rapamycin-resistant mtorc1 kinase activity is required for herpesvirus replication. J. Virol. 2010, 84, 5260–5269. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D. Manipulation of the host translation initiation complex eif4f by DNA viruses. Biochem. Soc. Trans. 2010, 38, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Gierman, T.M.; Frederickson, R.M.; Sonenberg, N.; Pickup, D.J. The eukaryotic translation initiation factor 4e is not modified during the course of vaccinia virus replication. Virology 1992, 188, 934–937. [Google Scholar] [CrossRef]
- Rochester, S.C.; Traktman, P. Characterization of the single-stranded DNA binding protein encoded by the vaccinia virus I3 gene. J. Virol. 1998, 72, 2917–2926. [Google Scholar] [PubMed]
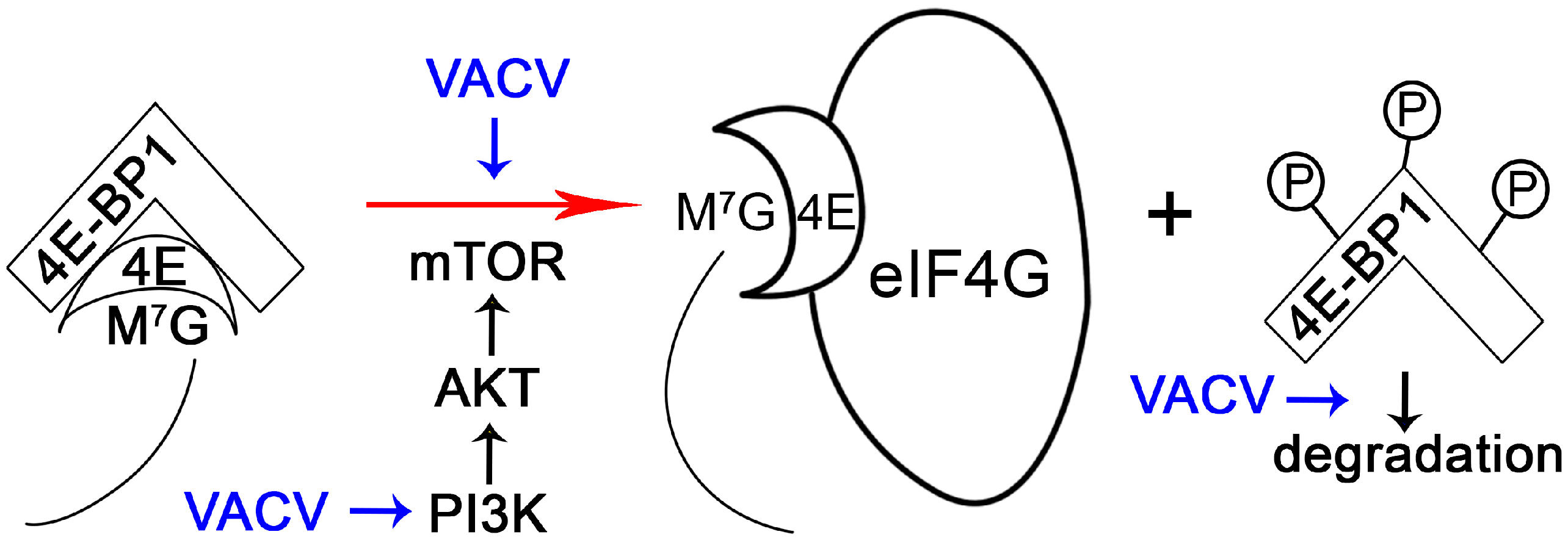
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4e-bp1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Barrett, J.W.; Stanford, M.; Werden, S.J.; Johnston, J.B.; Gao, X.; Sun, M.; Cheng, J.Q.; McFadden, G. Infection of human cancer cells with myxoma virus requires akt activation via interaction with a viral ankyrin-repeat host range factor. Proc. Natl. Acad. Sci. USA 2006, 103, 4640–4645. [Google Scholar] [CrossRef] [PubMed]
- Stanford, M.M.; Barrett, J.W.; Nazarian, S.H.; Werden, S.; McFadden, G. Oncolytic virotherapy synergism with signaling inhibitors: Rapamycin increases myxoma virus tropism for human tumor cells. J. Virol. 2007, 81, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Zaborowska, I.; Walsh, D. PI3k signaling regulates rapamycin-insensitive translation initiation complex formation in vaccinia virus-infected cells. J. Virol. 2009, 83, 3988–3992. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mtor, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. CB 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, R.; Hunter, T. Mnk1, a new map kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 1997, 16, 1921–1933. [Google Scholar] [CrossRef] [PubMed]
- Waskiewicz, A.J.; Flynn, A.; Proud, C.G.; Cooper, J.A. Mitogen-activated protein kinases activate the serine/threonine kinases mnk1 and mnk2. EMBO J. 1997, 16, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kedersha, N. Visibly stressed: The role of eif2, TIA-1, and stress granules in protein translation. Cell Stress Chaperones 2002, 7, 213–221. [Google Scholar] [CrossRef]
- Dar, A.C.; Dever, T.E.; Sicheri, F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell 2005, 122, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Dey, M.; Cao, C.; Dar, A.C.; Tamura, T.; Ozato, K.; Sicheri, F.; Dever, T.E. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell 2005, 122, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, M.D.; Malik, H.S. Rules of engagement: Molecular insights from host-virus arms races. Ann. Rev. Genet. 2012, 46, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R. Signal integration via PKR. Science’s STKE: Signal transduction knowledge environment 2001, 2001, re2. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Kawagishi-Kobayashi, M.; Cao, C.; Lu, J.; Ozato, K.; Dever, T.E. Pseudosubstrate inhibition of protein kinase PKR by swine pox virus c8l gene product. Virology 2000, 276, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Elde, N.C.; Child, S.J.; Geballe, A.P.; Malik, H.S. Protein kinase r reveals an evolutionary model for defeating viral mimicry. Nature 2009, 457, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Rothenburg, S.; Seo, E.J.; Gibbs, J.S.; Dever, T.E.; Dittmar, K. Rapid evolution of protein kinase PKR alters sensitivity to viral inhibitors. Nat. Struct. Mol. Biol. 2009, 16, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Ramelot, T.A.; Cort, J.R.; Yee, A.A.; Liu, F.; Goshe, M.B.; Edwards, A.M.; Smith, R.D.; Arrowsmith, C.H.; Dever, T.E.; Kennedy, M.A. Myxoma virus immunomodulatory protein m156r is a structural mimic of eukaryotic translation initiation factor eIF2alpha. J. Mol. Biol. 2002, 322, 943–954. [Google Scholar] [CrossRef]
- Peng, C.; Haller, S.L.; Rahman, M.M.; McFadden, G.; Rothenburg, S. Myxoma virus m156 is a specific inhibitor of rabbit PKR but contains a loss-of-function mutation in australian virus isolates. Proc. Natl. Acad. Sci. USA 2016, 113, 3855–3860. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.W.; Watson, J.C.; Jacobs, B.L. The e3l gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1992, 89, 4825–4829. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Liu, J.; Chan, W.M.; Rothenburg, S.; McFadden, G. Myxoma virus protein m029 is a dual function immunomodulator that inhibits PKR and also conscripts rha/dhx9 to promote expanded host tropism and viral replication. PLoS Pathog. 2013, 9, e1003465. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.M.; Santa Maria, F.G.; Lahiri, T.; Friedman, E.; Marie, I.J.; Levy, D.E. PKR transduces mda5-dependent signals for type i ifn induction. PLoS Pathog. 2016, 12, e1005489. [Google Scholar] [CrossRef] [PubMed]
- Vijaysri, S.; Talasela, L.; Mercer, A.A.; McInnes, C.J.; Jacobs, B.L.; Langland, J.O. The orf virus e3l homologue is able to complement deletion of the vaccinia virus e3l gene in vitro but not in vivo. Virology 2003, 314, 305–314. [Google Scholar] [CrossRef]
- Brandt, T.; Heck, M.C.; Vijaysri, S.; Jentarra, G.M.; Cameron, J.M.; Jacobs, B.L. The n-terminal domain of the vaccinia virus E3l-protein is required for neurovirulence, but not induction of a protective immune response. Virology 2005, 333, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Parrish, S.; Moss, B. Characterization of a vaccinia virus mutant with a deletion of the D10r gene encoding a putative negative regulator of gene expression. J. Virol. 2006, 80, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Parrish, S.; Moss, B. Characterization of a second vaccinia virus mRNA-decapping enzyme conserved in poxviruses. J. Virol. 2007, 81, 12973–12978. [Google Scholar] [CrossRef] [PubMed]
- Parrish, S.; Resch, W.; Moss, B. Vaccinia virus D10 protein has mRNA decapping activity, providing a mechanism for control of host and viral gene expression. Proc. Natl. Acad. Sci. USA 2007, 104, 2139–2144. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.W.; Wyatt, L.S.; Orandle, M.S.; Minai, M.; Moss, B. The D10 decapping enzyme of vaccinia virus contributes to decay of cellular and viral mRNAs and to virulence in mice. J. Virol. 2014, 88, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.W.; Katsafanas, G.C.; Liu, R.; Wyatt, L.S.; Moss, B. Poxvirus decapping enzymes enhance virulence by preventing the accumulation of dsRNA and the induction of innate antiviral responses. Cell Host Microbe 2015, 17, 320–331. [Google Scholar] [PubMed]
- Burgess, H.M.; Mohr, I. Cellular 5′-3′ mRNA exonuclease xrn1 controls double-stranded RNA accumulation and anti-viral responses. Cell Host Microbe 2015, 17, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Strnadova, P.; Ren, H.; Valentine, R.; Mazzon, M.; Sweeney, T.R.; Brierley, I.; Smith, G.L. Inhibition of translation initiation by protein 169: A vaccinia virus strategy to suppress innate and adaptive immunity and alter virus virulence. PLoS Pathog. 2015, 11, e1005151. [Google Scholar] [CrossRef] [PubMed]
- Pavon-Eternod, M.; David, A.; Dittmar, K.; Berglund, P.; Pan, T.; Bennink, J.R.; Yewdell, J.W. Vaccinia and influenza a viruses select rather than adjust tRNAs to optimize translation. Nucl. Acids Res. 2013, 41, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Martinez, O.; Miranda, E.; Ramirez, M.; Santos, S.; Rivera, C.; Vazquez, L.; Sanchez, T.; Tremblay, R.L.; Rios-Olivares, E.; Otero, M. Immunomodulator-based enhancement of anti smallpox immune responses. PLoS ONE 2015, 10, e0123113. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Netzer, N.; Strader, M.B.; Das, S.R.; Chen, C.Y.; Gibbs, J.; Pierre, P.; Bennink, J.R.; Yewdell, J.W. RNA binding targets aminoacyl-tRNA synthetases to translating ribosomes. J. Biol. Chem. 2011, 286, 20688–20700. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, K.A.; Camarda, R.; Lagunoff, M. Vaccinia virus requires glutamine but not glucose for efficient replication. J. Virol. 2014, 88, 4366–4374. [Google Scholar] [CrossRef] [PubMed]
- Greseth, M.D.; Traktman, P. De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for vaccinia virus infection. PLoS Pathog. 2014, 10, e1004021. [Google Scholar] [CrossRef] [PubMed]
- Teoh, M.L.; Turner, P.V.; Evans, D.H. Tumorigenic poxviruses up-regulate intracellular superoxide to inhibit apoptosis and promote cell proliferation. J. Virol. 2005, 79, 5799–5811. [Google Scholar] [CrossRef] [PubMed]
- D’Autreaux, B.; Toledano, M.B. Ros as signalling molecules: Mechanisms that generate specificity in ros homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Almazan, F.; Tscharke, D.C.; Smith, G.L. The vaccinia virus superoxide dismutase-like protein (a45r) is a virion component that is nonessential for virus replication. J. Virol. 2001, 75, 7018–7029. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.X.; Teoh, M.L.; Moon, M.; McFadden, G.; Evans, D.H. Leporipoxvirus cu-zn superoxide dismutase homologs inhibit cellular superoxide dismutase, but are not essential for virus replication or virulence. Virology 2002, 296, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Teoh, M.L.; Walasek, P.J.; Evans, D.H. Leporipoxvirus cu,zn-superoxide dismutase (sod) homologs are catalytically inert decoy proteins that bind copper chaperone for sod. J. Biol. Chem. 2003, 278, 33175–33184. [Google Scholar] [CrossRef] [PubMed]
- Paiva, C.N.; Bozza, M.T. Are reactive oxygen species always detrimental to pathogens? Antioxid. Redox Signal. 2014, 20, 1000–1037. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, M.I. Influence of air pollutants on allergic sensitization: The paradox of increased allergies and decreased resistance to infection. Toxicol. Pathol. 2012, 40, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Fortin, C.; Huang, X.; Yang, Y. Nk cell response to vaccinia virus is regulated by myeloid-derived suppressor cells. J. Immunol. 2012, 189, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kim, D.G.; Kim, B.G.; Yang, W.S.; Hong, J.; Kang, T.; Oh, Y.S.; Kim, K.R.; Han, B.W.; Hwang, B.J.; et al. Promiscuous methionyl-tRNA synthetase mediates adaptive mistranslation to protect cells against oxidative stress. J. Cell Sci. 2014, 127, 4234–4245. [Google Scholar] [CrossRef] [PubMed]
- Netzer, N.; Goodenbour, J.M.; David, A.; Dittmar, K.A.; Jones, R.B.; Schneider, J.R.; Boone, D.; Eves, E.M.; Rosner, M.R.; Gibbs, J.S.; et al. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature 2009, 462, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Pan, T. Methionine mistranslation bypasses the restraint of the genetic code to generate mutant proteins with distinct activities. PLoS Genet. 2015, 11, e1005745. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kedersha, N. Stress granules: The tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-binding proteins TIA-1 and tiar link the phosphorylation of eif-2 alpha to the assembly of mammalian stress granules. J. Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.; Calabrese, J.M.; Sharp, P.A. Quantitative analysis of argonaute protein reveals microRNA-dependent localization to stress granules. Proc. Natl. Acad. Sci. USA 2006, 103, 18125–18130. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Peng, Y.; Murray, E.L.; Otsuka, Y.; Kedersha, N.; Schoenberg, D.R. Polysome-bound endonuclease pmr1 is targeted to stress granules via stress-specific binding to TIA-1. Mol. Cell. Biol. 2006, 26, 8803–8813. [Google Scholar] [CrossRef] [PubMed]
- Tourriere, H.; Chebli, K.; Zekri, L.; Courselaud, B.; Blanchard, J.M.; Bertrand, E.; Tazi, J. The rasgap-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 2003, 160, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.; Xu, Y.; Wang, B.; David, M.D.; Schubert, P.; Kennedy, D.; Schrader, J.W. Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol. Cell. Biol. 2007, 27, 2324–2342. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Back, S.H.; Kim, V.; Ryu, I.; Jang, S.K. Sequestration of traf2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol. Cell. Biol. 2005, 25, 2450–2462. [Google Scholar] [CrossRef] [PubMed]
- Kroschwald, S.; Maharana, S.; Mateju, D.; Malinovska, L.; Nuske, E.; Poser, I.; Richter, D.; Alberti, S. Promiscuous interactions and protein disaggregases determine the material state of stress-inducible rnp granules. eLife 2015, 4, e06807. [Google Scholar] [CrossRef] [PubMed]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef] [PubMed]
- Panas, M.D.; Varjak, M.; Lulla, A.; Eng, K.E.; Merits, A.; Karlsson Hedestam, G.B.; McInerney, G.M. Sequestration of G3BP coupled with efficient translation inhibits stress granules in semliki forest virus infection. Mol. Biol. Cell 2012, 23, 4701–4712. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Okamoto, T.; Fukuhara, T.; Kambara, H.; Morita, E.; Mori, Y.; Kamitani, W.; Matsuura, Y. Japanese encephalitis virus core protein inhibits stress granule formation through an interaction with caprin-1 and facilitates viral propagation. J. Virol. 2013, 87, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Fros, J.J.; Domeradzka, N.E.; Baggen, J.; Geertsema, C.; Flipse, J.; Vlak, J.M.; Pijlman, G.P. Chikungunya virus nsp3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J. Virol. 2012, 86, 10873–10879. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Lloyd, R.E. Poliovirus unlinks TIA1 aggregation and mRNA stress granule formation. J. Virol. 2011, 85, 12442–12454. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, J.; Hansen, S.J.; Park, N.; Jamka, K.; Sarnow, P.; Gustin, K.E. Stable formation of compositionally unique stress granules in virus-infected cells. J. Virol. 2010, 84, 3654–3665. [Google Scholar] [CrossRef] [PubMed]
- Mok, B.W.; Song, W.; Wang, P.; Tai, H.; Chen, Y.; Zheng, M.; Wen, X.; Lau, S.Y.; Wu, W.L.; Matsumoto, K.; et al. The ns1 protein of influenza a virus interacts with cellular processing bodies and stress granules through RNA-associated protein 55 (rap55) during virus infection. J. Virol. 2012, 86, 12695–12707. [Google Scholar] [CrossRef] [PubMed]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical role of an antiviral stress granule containing rig-i and PKR in viral detection and innate immunity. PLoS ONE 2012, 7, e43031. [Google Scholar] [CrossRef]
- Simpson-Holley, M.; Kedersha, N.; Dower, K.; Rubins, K.H.; Anderson, P.; Hensley, L.E.; Connor, J.H. Formation of antiviral cytoplasmic granules during orthopoxvirus infection. J. Virol. 2011, 85, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wennier, S.; Zhang, L.; McFadden, G. M062 is a host range factor essential for myxoma virus pathogenesis and functions as an antagonist of host SAMD9 in human cells. J. Virol. 2011, 85, 3270–3282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Langland, J.O.; Jacobs, B.L.; Samuel, C.E. Protein kinase PKR-dependent activation of mitogen-activated protein kinases occurs through mitochondrial adapter ips-1 and is antagonized by vaccinia virus e3l. J. Virol. 2009, 83, 5718–5725. [Google Scholar] [CrossRef] [PubMed]
- Lemos de Matos, A.; Liu, J.; McFadden, G.; Esteves, P.J. Evolution and divergence of the mammalian SAMD9/SAMD9l gene family. BMC Evol. Biol. 2013, 13, 121. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Chao, J.; Xiang, Y. Identification from diverse mammalian poxviruses of host-range regulatory genes functioning equivalently to vaccinia virus C7l. Virology 2008, 372, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rothenburg, S.; McFadden, G. The poxvirus C7l host range factor superfamily. Curr. Opin. Virol. 2012, 2, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Perkus, M.E.; Goebel, S.J.; Davis, S.W.; Johnson, G.P.; Limbach, K.; Norton, E.K.; Paoletti, E. Vaccinia virus host range genes. Virology 1990, 179, 276–286. [Google Scholar] [CrossRef]
- Meng, X.; Krumm, B.; Li, Y.; Deng, J.; Xiang, Y. Structural basis for antagonizing a host restriction factor by C7 family of poxvirus host-range proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 14858–14863. [Google Scholar] [CrossRef] [PubMed]
- Sivan, G.; Ormanoglu, P.; Buehler, E.C.; Martin, S.E.; Moss, B. Identification of restriction factors by human genome-wide RNA interference screening of viral host range mutants exemplified by discovery of SAMD9 and wdr6 as inhibitors of the vaccinia virus k1l-C7l- mutant. mBio 2015, 6, e01122. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.S.; Jogi, M.; Yoo, J.S.; Onomoto, K.; Koike, S.; Iwasaki, T.; Yoneyama, M.; Kato, H.; Fujita, T. Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J. Virol. 2013, 87, 9511–9522. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Schoggins, J.; Rose, L.; Cao, J.; Ploss, A.; Rice, C.M.; Xiang, Y. C7l family of poxvirus host range genes inhibits antiviral activities induced by type i interferons and interferon regulatory factor 1. J. Virol. 2012, 86, 4538–4547. [Google Scholar] [CrossRef] [PubMed]
- Reineke, L.C.; Lloyd, R.E. The stress granule protein G3BP1 recruits protein kinase r to promote multiple innate immune antiviral responses. J. Virol. 2015, 89, 2575–2589. [Google Scholar] [CrossRef] [PubMed]
- Langereis, M.A.; Feng, Q.; van Kuppeveld, F.J. Mda5 localizes to stress granules, but this localization is not required for the induction of type i interferon. J. Virol. 2013, 87, 6314–6325. [Google Scholar] [CrossRef] [PubMed]
- Iordanov, M.S.; Wong, J.; Bell, J.C.; Magun, B.E. Activation of nf-kappab by double-stranded RNA (dsRNA) in the absence of protein kinase r and RNAse l demonstrates the existence of two separate dsRNA-triggered antiviral programs. Mol. Cell. Biol. 2001, 21, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Khaperskyy, D.A.; Hatchette, T.F.; McCormick, C. Influenza a virus inhibits cytoplasmic stress granule formation. FASEB J. 2012, 26, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Hato, S.V.; Ricour, C.; Schulte, B.M.; Lanke, K.H.; de Bruijni, M.; Zoll, J.; Melchers, W.J.; Michiels, T.; van Kuppeveld, F.J. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell. Microbiol. 2007, 9, 2921–2930. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Kawabata, R.; Honda, T.; Tomonaga, K.; Sakaguchi, T.; Irie, T. Ifn-beta-inducing, unusual viral RNA species produced by paramyxovirus infection accumulated into distinct cytoplasmic structures in an RNA-type-dependent manner. Front. Microbiol. 2015, 6, 804. [Google Scholar] [CrossRef] [PubMed]
- Peters, N.E.; Ferguson, B.J.; Mazzon, M.; Fahy, A.S.; Krysztofinska, E.; Arribas-Bosacoma, R.; Pearl, L.H.; Ren, H.; Smith, G.L. A mechanism for the inhibition of DNA-pk-mediated DNA sensing by a virus. PLoS Pathog. 2013, 9, e1003649. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.; Stein, S.; Falck-Pedersen, E. Adenovirus detection by the cgas/sting/tbk1 DNA sensing cascade. J. Virol. 2014, 88, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. Ifi16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, M.R.; Bak, R.O.; Andersen, A.; Berg, R.K.; Jensen, S.B.; Tengchuan, J.; Laustsen, A.; Hansen, K.; Ostergaard, L.; Fitzgerald, K.A.; et al. Ifi16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. USA 2013, 110, E4571–4580. [Google Scholar] [CrossRef] [PubMed]
- Doitsh, G.; Galloway, N.L.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Munoz-Arias, I.; et al. Cell death by pyroptosis drives cd4 t-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Monroe, K.M.; Yang, Z.; Johnson, J.R.; Geng, X.; Doitsh, G.; Krogan, N.J.; Greene, W.C. Ifi16 DNA sensor is required for death of lymphoid cd4 t cells abortively infected with HIV. Science 2014, 343, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Dutta, S.; Veettil, M.V.; Dutta, D.; Iqbal, J.; Kumar, B.; Roy, A.; Chikoti, L.; Singh, V.V.; Chandran, B. Herpesvirus genome recognition induced acetylation of nuclear ifi16 is essential for its cytoplasmic translocation, inflammasome and ifn-beta responses. PLoS Pathog. 2015, 11, e1005019. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Arias, I.; Doitsh, G.; Yang, Z.; Sowinski, S.; Ruelas, D.; Greene, W.C. Blood-derived cd4 t cells naturally resist pyroptosis during abortive HIV-1 infection. Cell Host Microbe 2015, 18, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; Broekema, N.M.; Diner, B.A.; Hancks, D.C.; Elde, N.C.; Cristea, I.M.; Knipe, D.M. Cgas-mediated stabilization of ifi16 promotes innate signaling during herpes simplex virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, E1773–E1781. [Google Scholar] [CrossRef] [PubMed]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Soby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Veeranki, S.; Choubey, D. Interferon-inducible p200-family protein ifi16, an innate immune sensor for cytosolic and nuclear double-stranded DNA: Regulation of subcellular localization. Mol. Immunol. 2012, 49, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. Sting regulates intracellular DNA-mediated, type i interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.; Kim, S.; Bumeister, R.; Loo, Y.M.; Kwon, S.W.; Johnson, C.L.; Balakireva, M.G.; Romeo, Y.; Kopelovich, L.; Gale, M., Jr.; et al. Ralb gtpase-mediated activation of the ikappab family kinase tbk1 couples innate immune signaling to tumor cell survival. Cell 2006, 127, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Fons, R.D.; Bogert, B.A.; Hegde, R.S. Substrate-specific function of the translocon-associated protein complex during translocation across the er membrane. J. Cell Biol. 2003, 160, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. Sting is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. ATG9A controls dsDNA-driven dynamic translocation of sting and the innate immune response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar] [CrossRef] [PubMed]
- Berg, R.K.; Rahbek, S.H.; Kofod-Olsen, E.; Holm, C.K.; Melchjorsen, J.; Jensen, D.G.; Hansen, A.L.; Jorgensen, L.B.; Ostergaard, L.; Tolstrup, M.; et al. T cells detect intracellular DNA but fail to induce type i ifn responses: Implications for restriction of HIV replication. PLoS ONE 2014, 9, e84513. [Google Scholar] [CrossRef] [PubMed]
- Stetson, D.B.; Medzhitov, R. Recognition of cytosolic DNA activates an irf3-dependent innate immune response. Immunity 2006, 24, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-pk is a DNA sensor for irf-3-dependent innate immunity. eLife 2012, 1, e00047. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.J. The helicase ddx41 senses intracellular DNA mediated by the adaptor sting in dendritic cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Kobayashi, J.; Saitoh, T.; Maruyama, K.; Ishii, K.J.; Barber, G.N.; Komatsu, K.; Akira, S.; Kawai, T. DNA damage sensor mre11 recognizes cytosolic double-stranded DNA and induces type i interferon by regulating sting trafficking. Proc. Natl. Acad. Sci. USA 2013, 110, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic gmp-amp synthase is a cytosolic DNA sensor that activates the type i interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Baptiste, N.; Prives, C. P53 in the cytoplasm: A question of overkill? Cell 2004, 116, 487–489. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. Aim2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with asc. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.L.; Idris, A.; Dunn, J.A.; Kelly, G.M.; Burnton, C.M.; Hodgson, S.; Hardy, L.L.; Garceau, V.; Sweet, M.J.; Ross, I.L.; et al. Hin-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009, 323, 1057–1060. [Google Scholar] [CrossRef] [PubMed]
- Kis-Toth, K.; Szanto, A.; Thai, T.H.; Tsokos, G.C. Cytosolic DNA-activated human dendritic cells are potent activators of the adaptive immune response. J. Immunol. 2011, 187, 1222–1234. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liem, J.; Liu, J. Stress Beyond Translation: Poxviruses and More. Viruses 2016, 8, 169. https://doi.org/10.3390/v8060169
Liem J, Liu J. Stress Beyond Translation: Poxviruses and More. Viruses. 2016; 8(6):169. https://doi.org/10.3390/v8060169
Chicago/Turabian StyleLiem, Jason, and Jia Liu. 2016. "Stress Beyond Translation: Poxviruses and More" Viruses 8, no. 6: 169. https://doi.org/10.3390/v8060169
APA StyleLiem, J., & Liu, J. (2016). Stress Beyond Translation: Poxviruses and More. Viruses, 8(6), 169. https://doi.org/10.3390/v8060169