
Antiviral Hammerhead Ribozymes Are Effective for Developing Transgenic Suppression of Chikungunya Virus in Aedes aegypti Mosquitoes

Abstract
:1. Background
2. Materials and Method
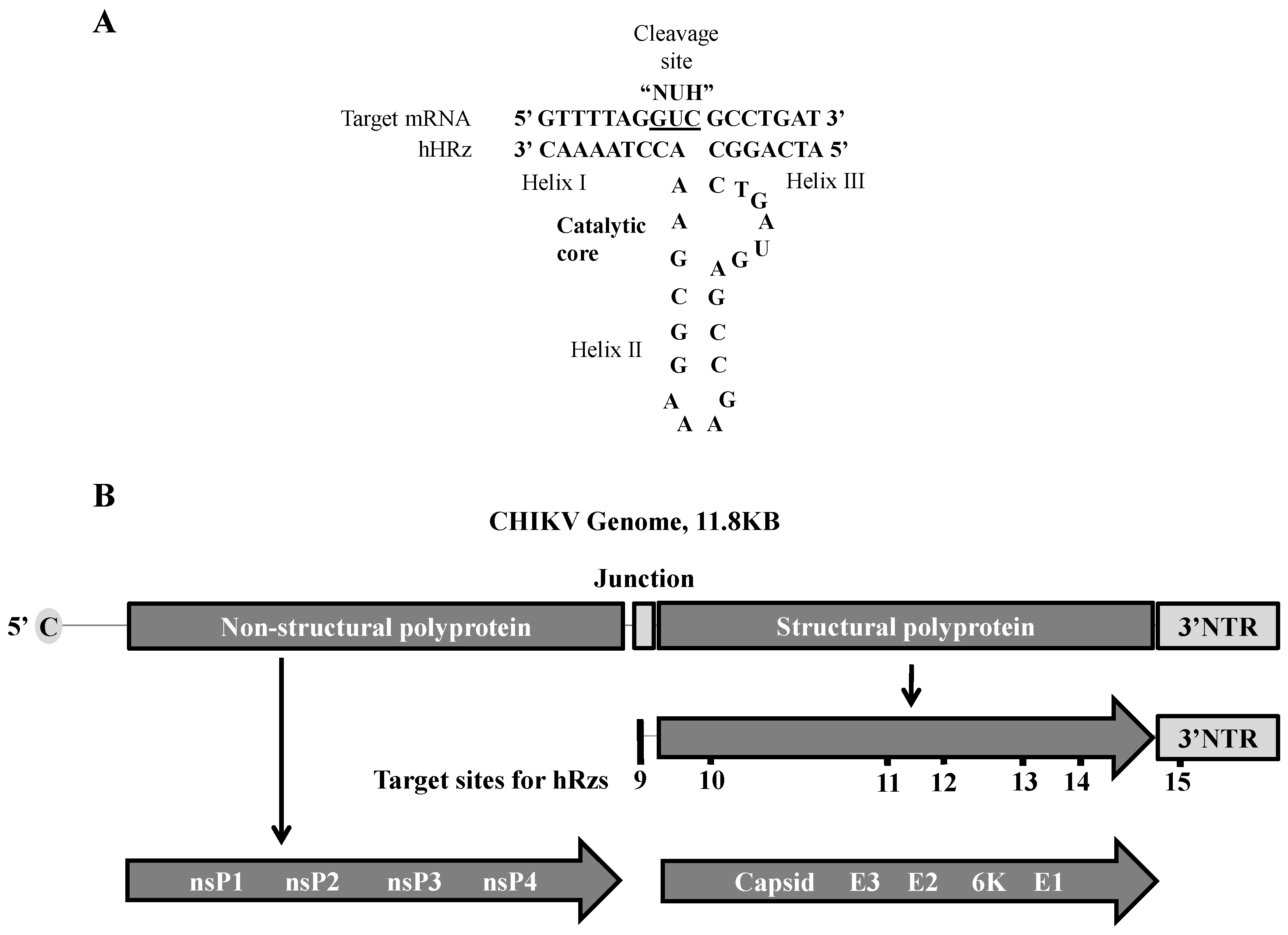
2.1. Selection of hRz Target Sites in the CHIKV Genome
2.2. Plasmid Construction
2.3. Cell Cultures, Transfection, Clonal Generation and Infection
2.4. RT-PCR Analysis
2.5. TCID50 Immunofluorescence Assays
2.6. Caspase 3 Assays
2.7. Quantitative RT-PCR
2.8. Generation of hRz Transgenic Mosquito Lines
2.9. Splinkerette PCR
2.10. Mosquitoes, Infection and Maintenance
2.11. Genotyping of Mosquitoes
2.12. In Situ Indirect Immuno-Fluorescent Assays
2.13. Assays of Virus in Saliva
3. Results
3.1. Construction of Transformation Vectors Carrying Anti-CHIKV hRz
3.2. Single-Cell Sorting for Transformed Selected Cell Populations and Screening for CHIKV Suppression
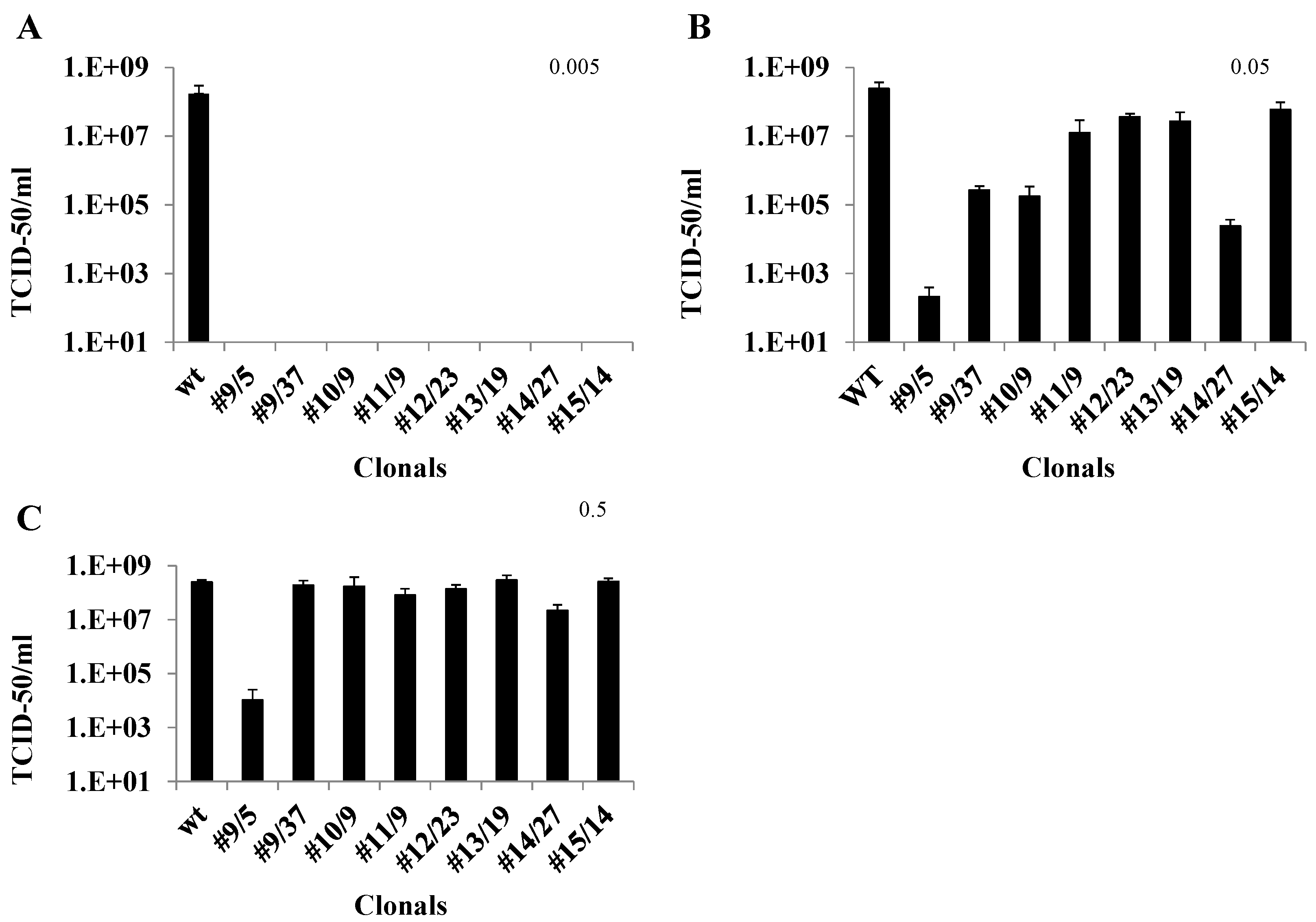
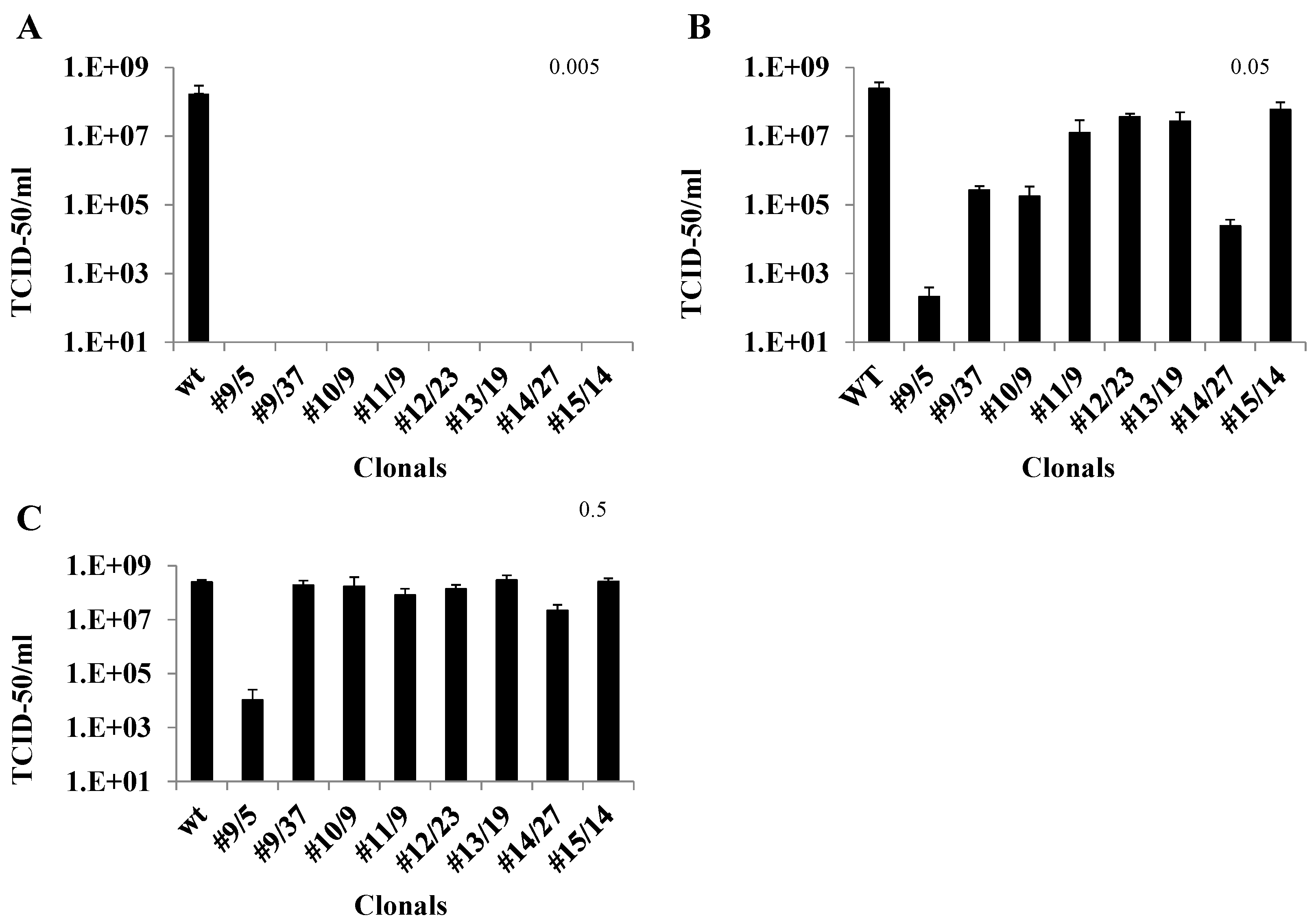
3.3. TCID50-IFA Analysis of CHIKV Suppression in Clones Lacking Visible CPE
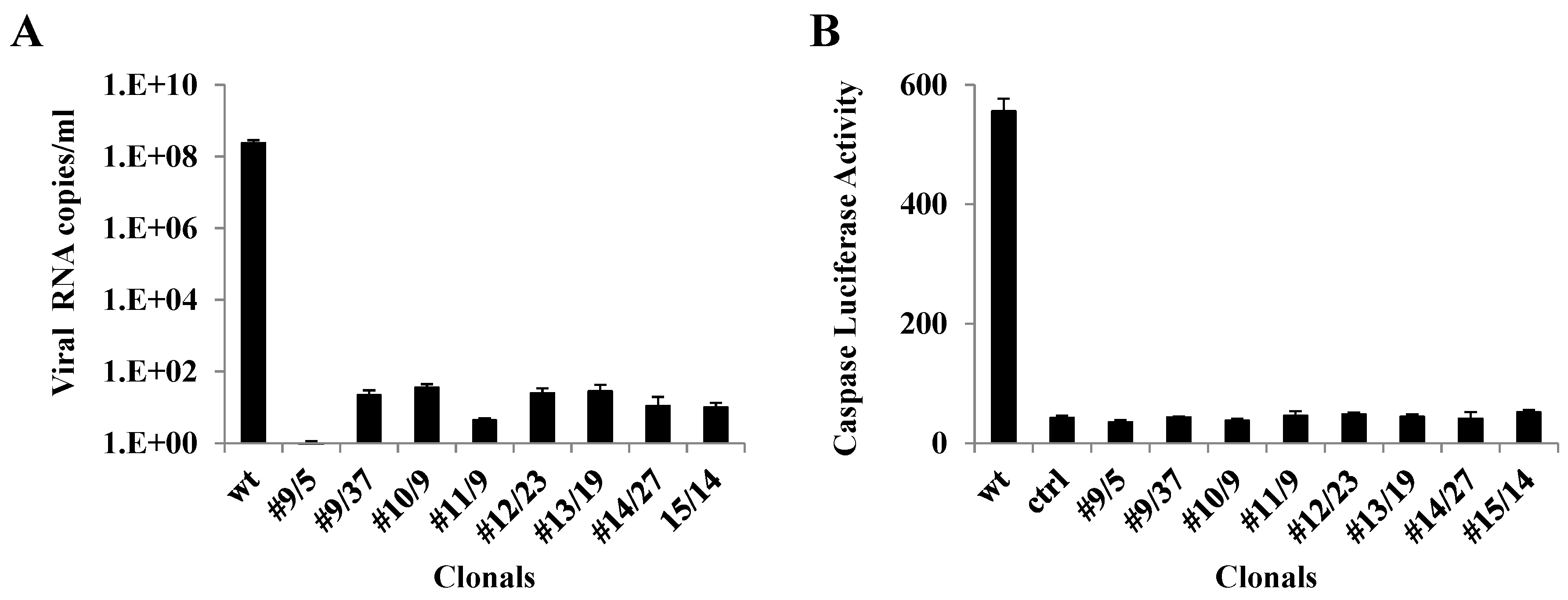
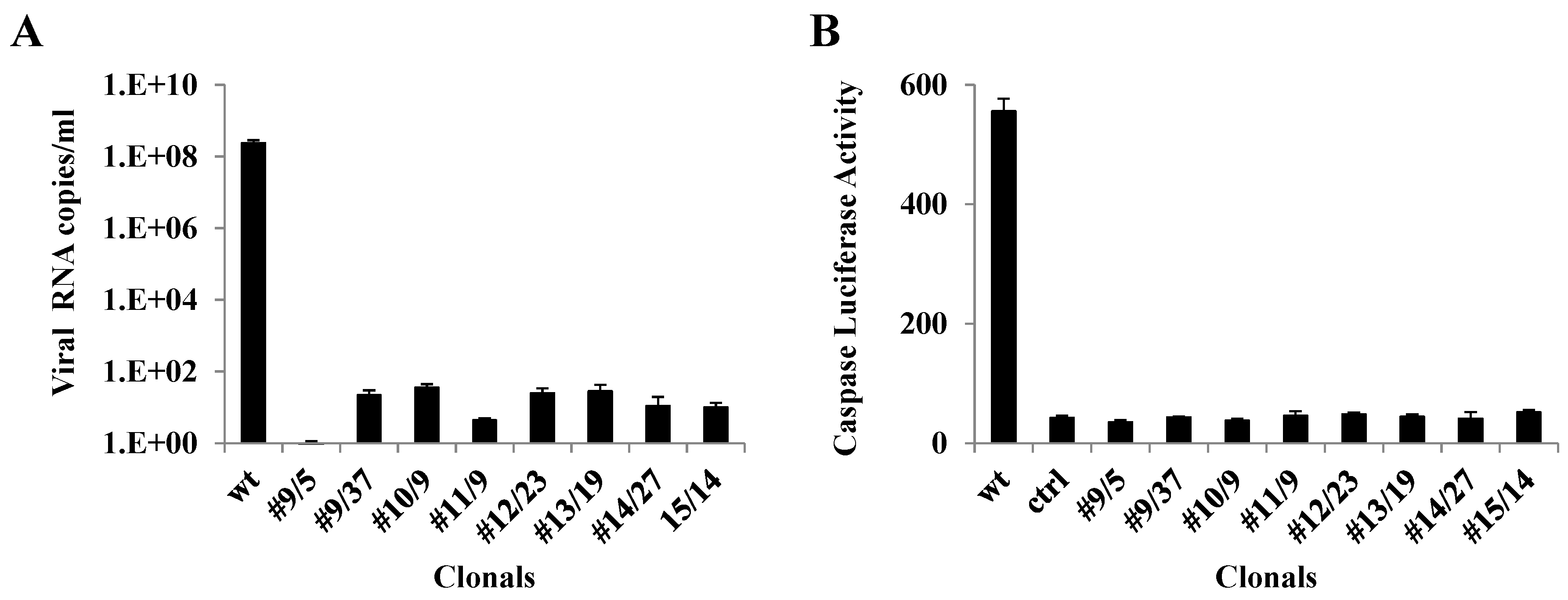
3.4. qRT-PCR Analysis of Ribozyme-Expressing Cell Clones Demonstrates Complete Suppression
3.5. Caspase 3 Assays of Ribozyme-Expressing Cell Clones Confirms Lack of CHIKV Infection
3.6. Construction of PiggyBac Vectors and Establishment of Transgenic Mosquitoes
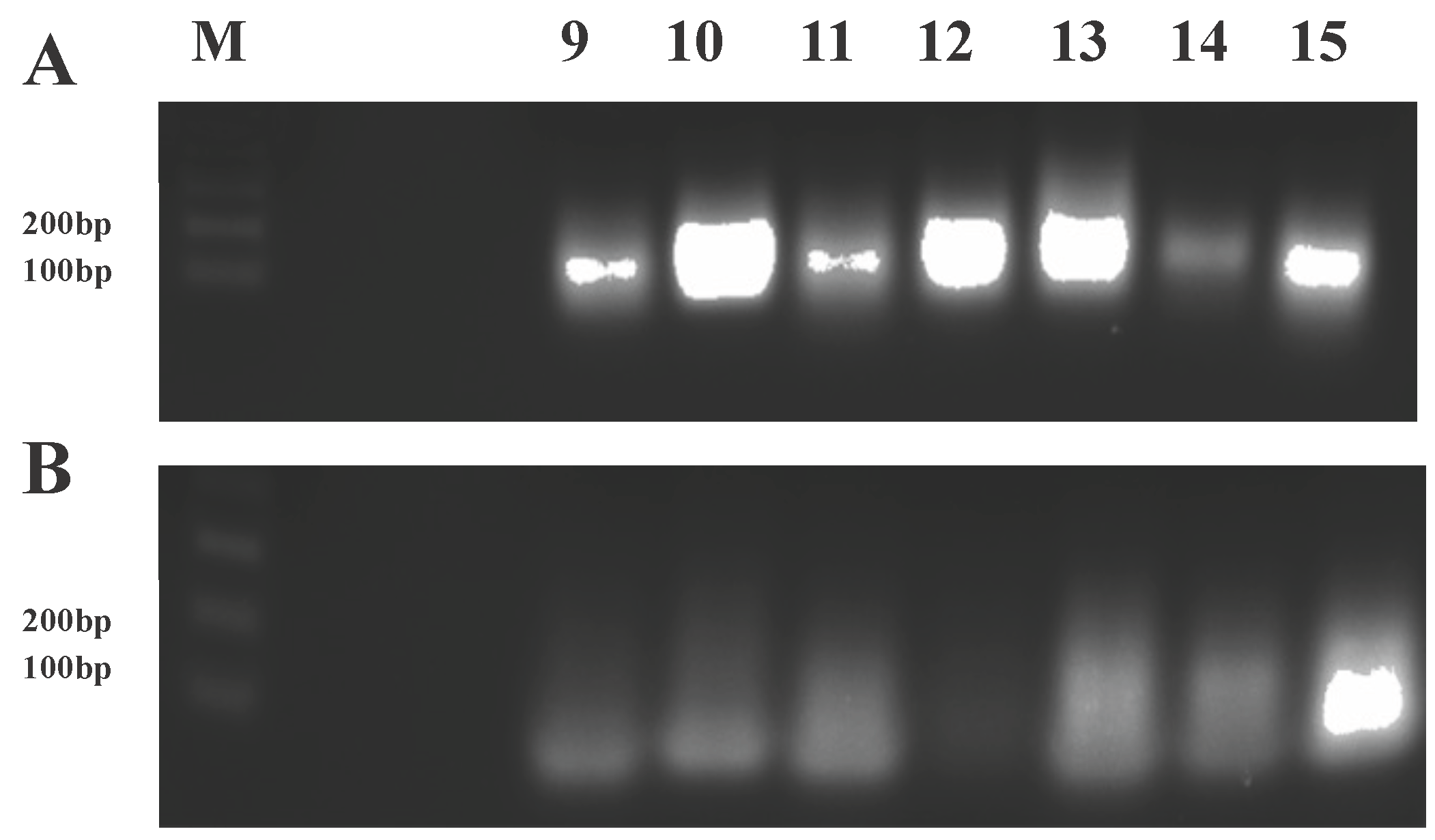
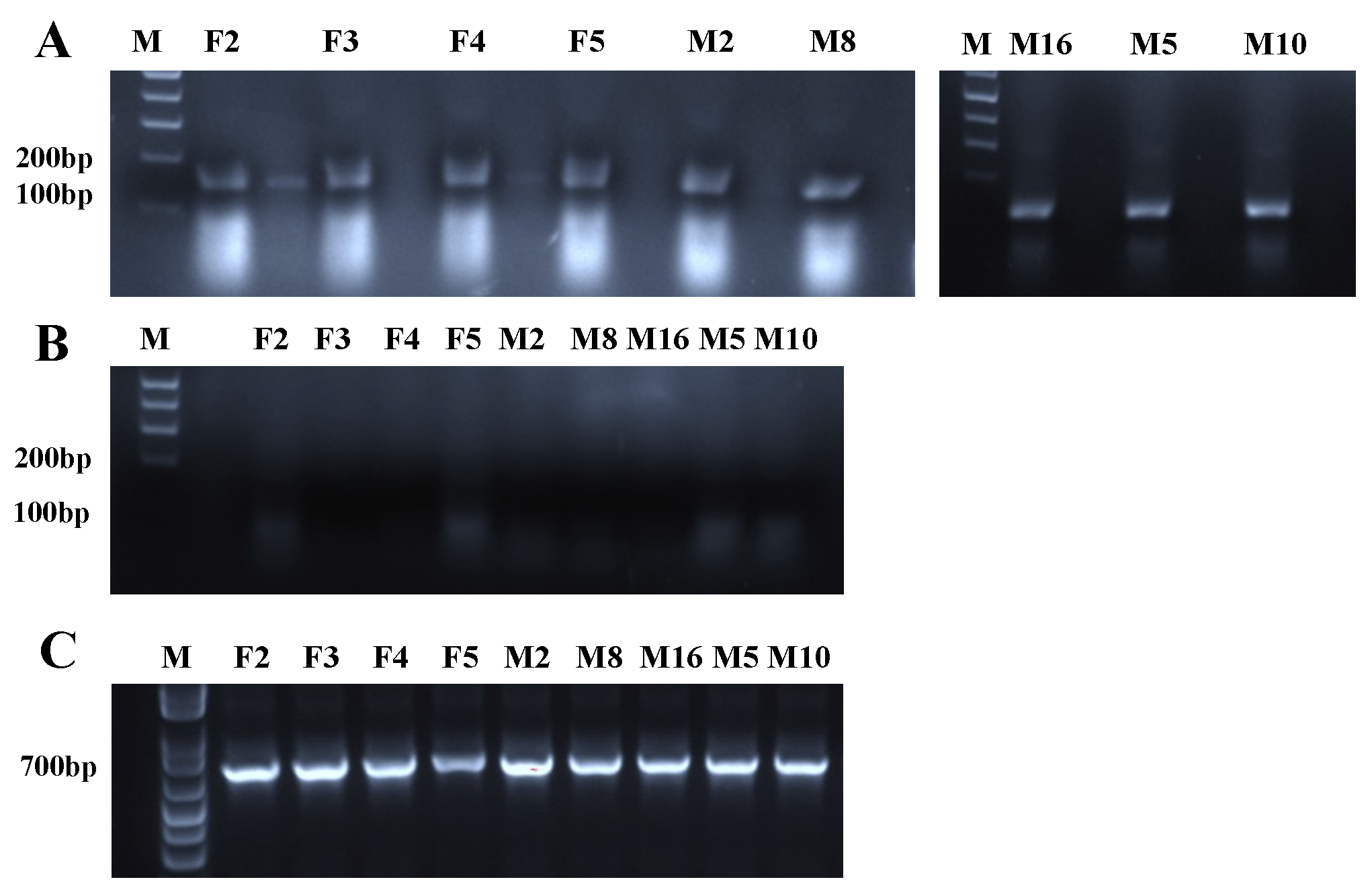
3.7. RT-PCR to Confirm Transgene Expression
3.8. Identification of Each Transgene Integration Site via Splinkerette PCR
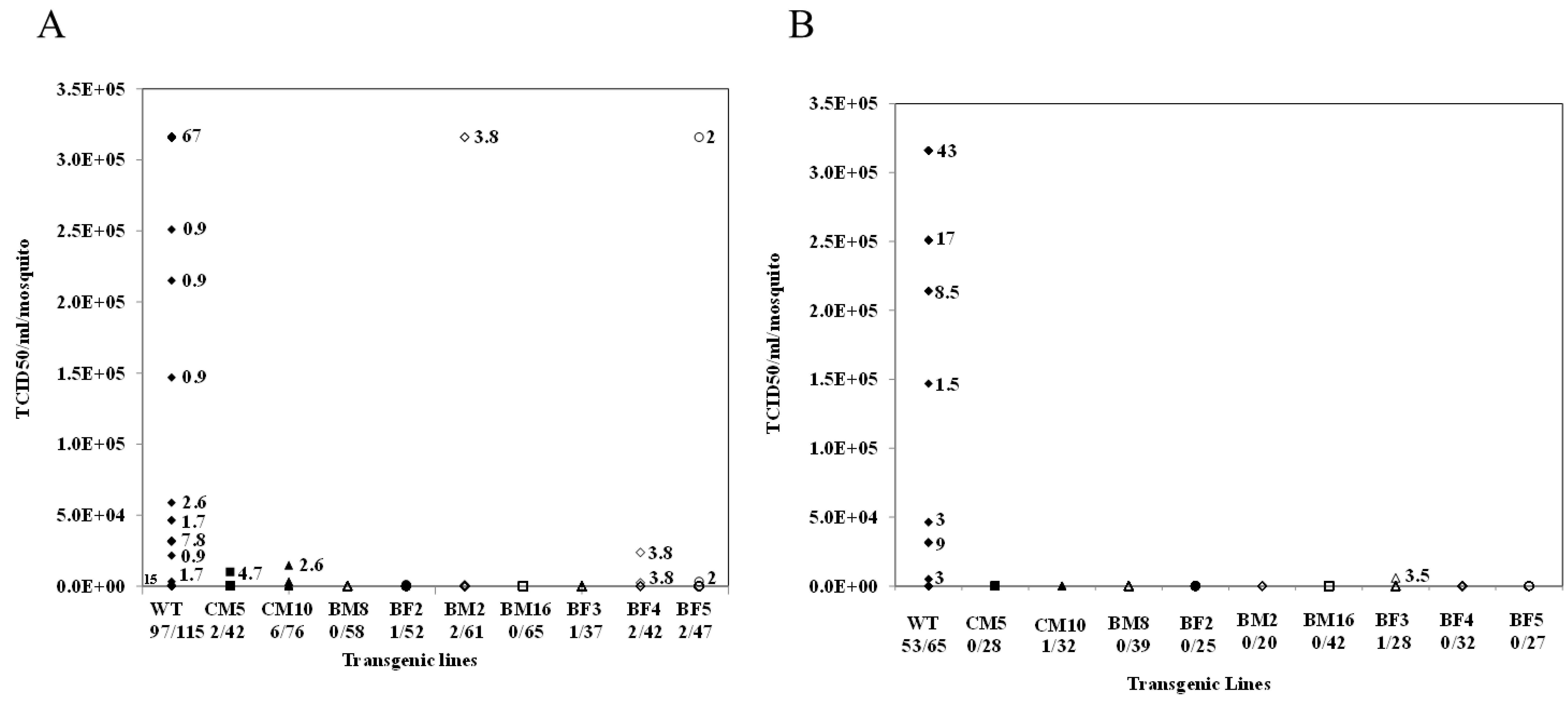
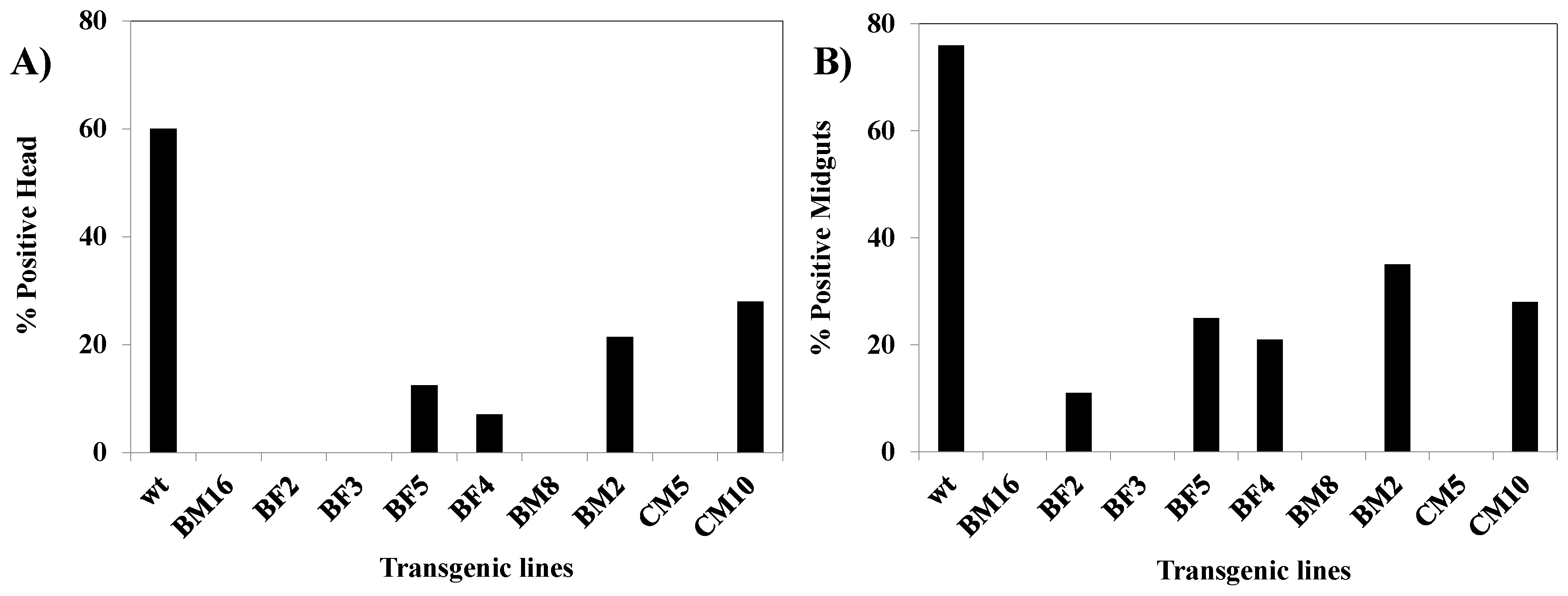
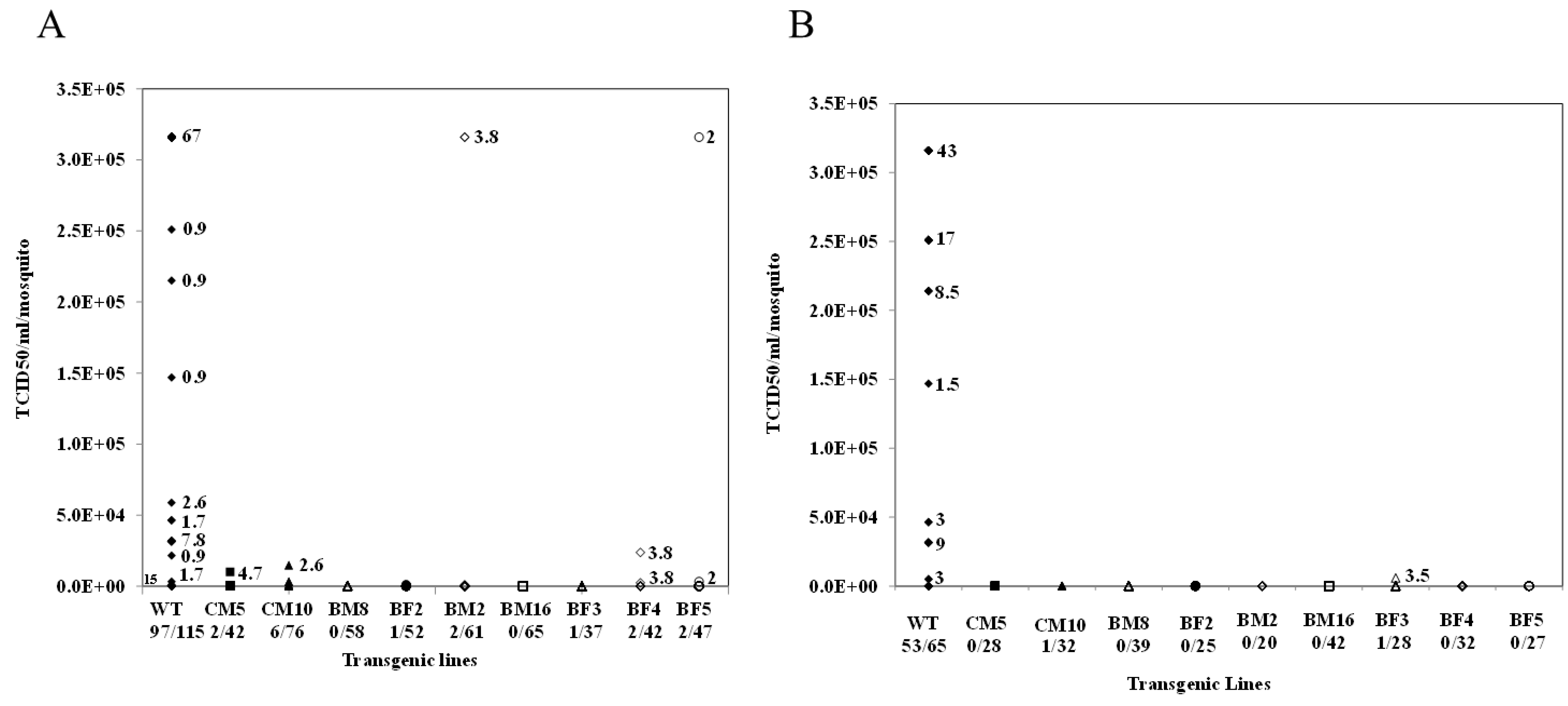
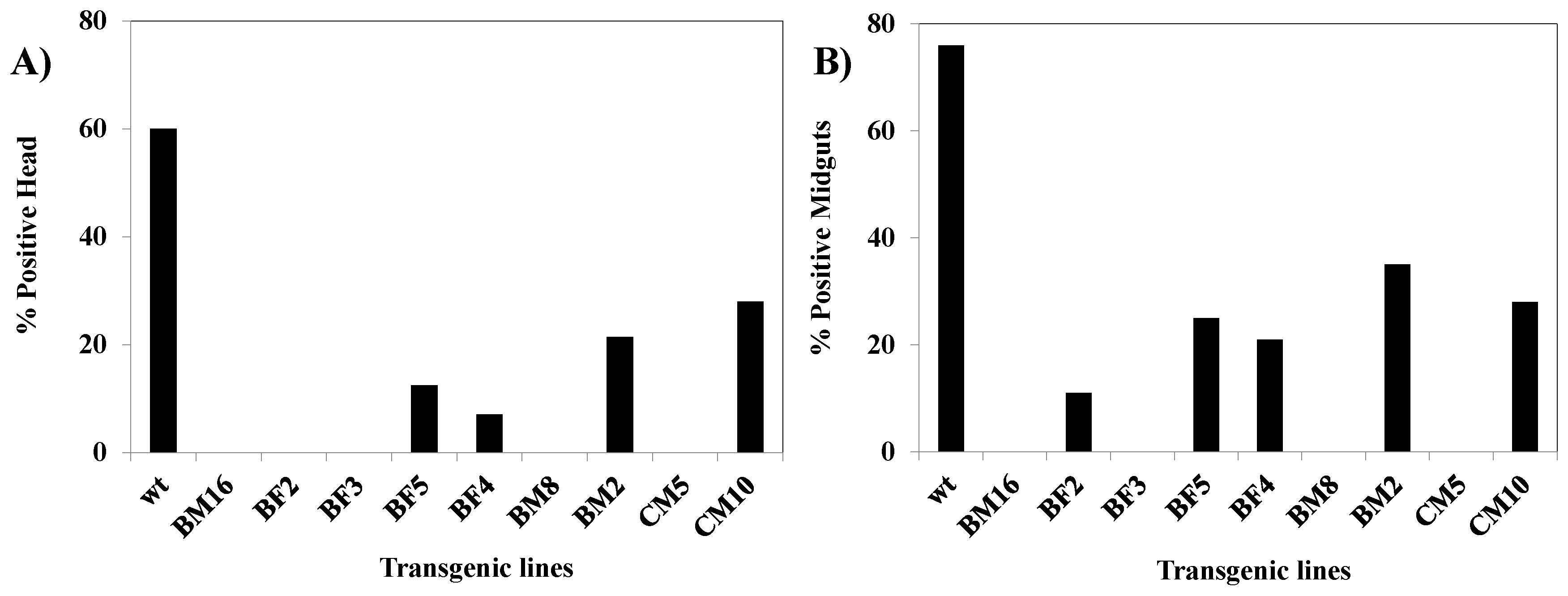
3.9. Analysis of CHIKV Infection Suppression in hRz Transgenic Mosquitoes
3.10. Direct PCR Analysis Confirms Haploid Genotype of G6 mosquitoes
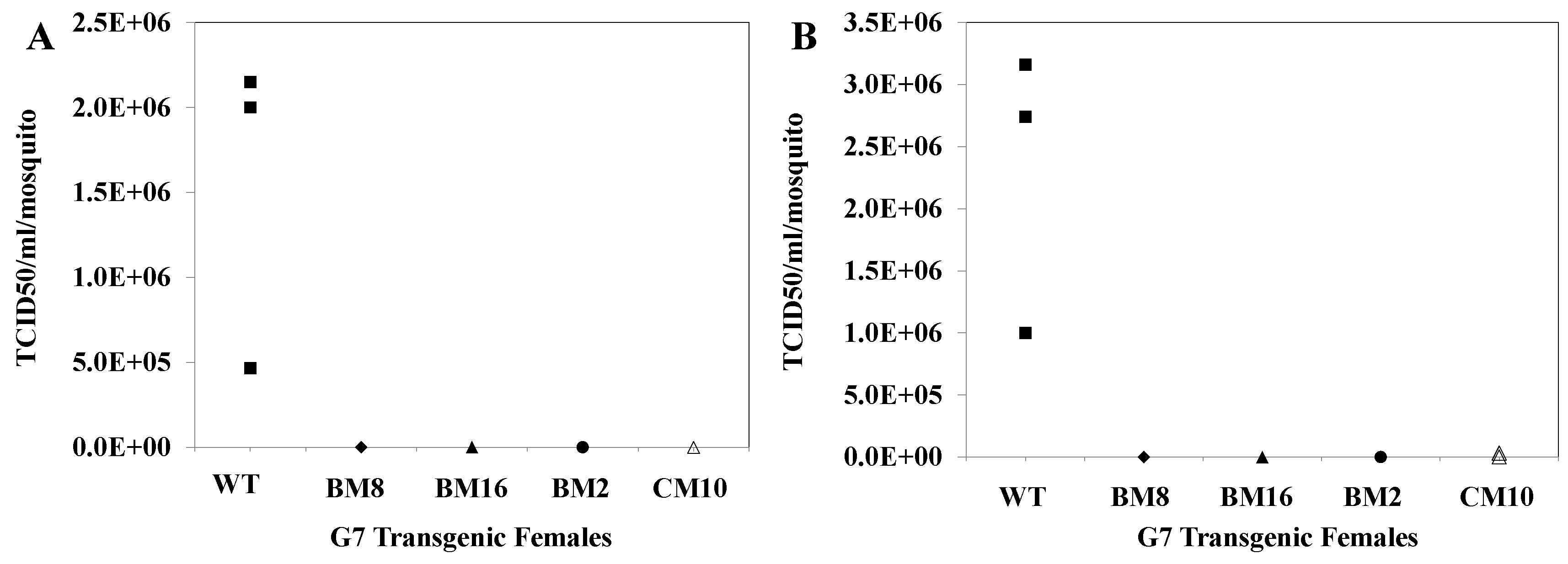
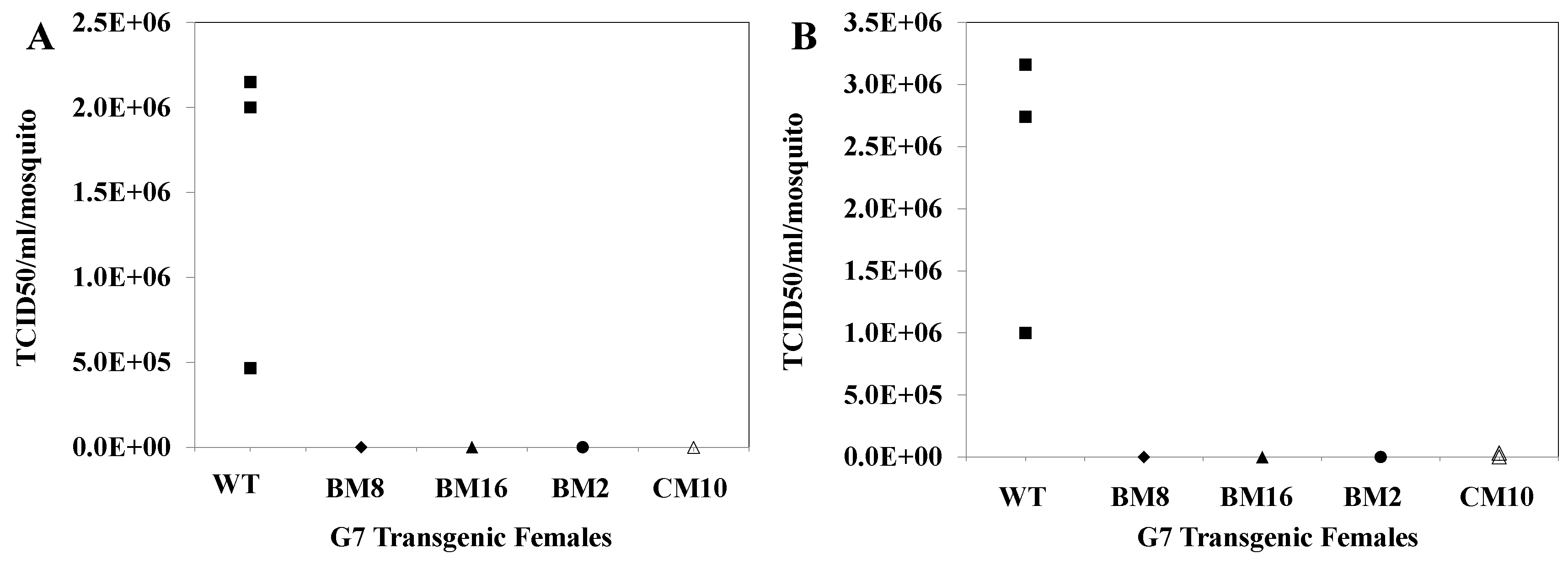
3.11. CHIKV Titers in Saliva Collected from Challenged Transgenic Female Mosquitoes
4. Discussion
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
References
- Pialoux, G.; Gauzere, B.A.; Jaureguiberry, S.; Strobel, M. Chikungunya, an epidemic arbovirosis. Lancet Infect. Dis. 2007, 7, 319–327. [Google Scholar] [CrossRef]
- Farnon, E.C.; Sejvar, J.J.; Staples, J.E. Severe disease manifestations associated with acute chikungunya virus infection. Crit. Care Med. 2008, 36, 2682–2683. [Google Scholar] [CrossRef] [PubMed]
- Schuffenecker, I.; Iteman, I.; Michault, A.; Murri, S.; Frangeul, L.; Vaney, M.C.; Lavenir, R.; Pardigon, N.; Reynes, J.M.; Pettinelli, F.; et al. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLOS Med. 2006, 3, e263. [Google Scholar] [CrossRef] [PubMed]
- Mavalankar, D.; Shastri, P.; Raman, P. Chikungunya epidemic in India: A major public-health disaster. Lancet Infect. Dis. 2007, 7, 306–307. [Google Scholar] [CrossRef]
- Hochedez, P.; Hausfater, P.; Jaureguiberry, S.; Gay, F.; Datry, A.; Danis, M.; Bricaire, F.; Bossi, P. Cases of chikungunya fever imported from the islands of the South West Indian Ocean to Paris, France. Eur. Surveill. 2007, 12, 679. [Google Scholar]
- Morrison, T.E. Reemergence of Chikungunya Virus. J. Virol. 2014, 88, 11644–11647. [Google Scholar] [CrossRef] [PubMed]
- Nasci, R.S. Movement of Chikungunya Virus into the Western Hemisphere. Emerging Infect. Dis. 2014, 20, 1394–1395. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Volkova, E.; Adams, A.P.; Forrester, N.; Xiao, S.; Frolov, I.I.; Weaver, S.C. Chimeric alphavirus vaccine candidates for chikungunya. Vaccine 2008, 26, 5030–5039. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Sudeep, A.B.; Arankalle, V.A. Evaluation of recombinant E2 protein based and whole virus inactivated candidate vaccine against chikungunya virus. Vaccine 2012, 30, 6142–6149. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Suhribier, A.; Penn-Nicholson, A.; Woaratanadharm, J.; Gardner, J.; Luo, M.; Le, T.T.; Anraku, I.; Sakalian, M.; Einfeld, D.; et al. A complex adenovirus vaccine against chikungunya virus provides complete protection against viraemia and arthritis. Vaccine 2011, 29, 2803–2809. [Google Scholar] [CrossRef] [PubMed]
- Metz, S.W.; Gardner, J.; Geertsema, C.; Le, T.T.; Goh, L.; Vlak, J.M.; Suhrbier, A.; Pijlman, G.P. Effective chikungunya virus like particle vaccine produced in insect cells. PLoS Negl. Trop. Dis. 2013, 7, e2124. [Google Scholar] [CrossRef] [PubMed]
- Livengood, J.A.; Partidos, C.D.; Plante, K.; Seymour, R.; Gorchakoc, R.; Varga, L.; Paykel, J.; Weger, J.; Haller, A.; Stinchcomb, D.T.; et al. Preclinical evaluation of live attenuated chikungunya vaccine. Procedia Vaccinol. 2012, 6, 141–149. [Google Scholar] [CrossRef]
- Sánchez-Vargas, I.; Scott, J.C.; Poole-Smith, B.K.; Franz, A.W.; Barbosa-Solomieu, V.; Wilusz, J.; Olson, K.E.; Blair, C.D. Dengue Virus Type 2 Infections of Aedes aegypti Are Modulated by the Mosquito’s RNA Interference Pathway. PLoS Pathog. 2009, 5, e1000299. [Google Scholar]
- Gu, J.; Liu, M.; Deng, Y.; Peng, H.; Chen, X. Development of an Efficient Recombinant Mosquito Densovirus-Mediated RNA Interference System and Its Preliminary Application in Mosquito Control. PLoS ONE 2011, 6, e21329. [Google Scholar] [CrossRef] [PubMed]
- Franz, A.W.E.; Sanchez-Vargas, I.; Raban, R.R.; Black, W.C.; James, A.A.; Olson, K.E. Fitness Impact and Stability of a Transgene Conferring Resistance to Dengue-2 Virus following Introgression into a Genetically Diverse Aedes aegypti Strain. PLoS Negl. Trop. Dis. 2014, 8, e2833. [Google Scholar] [CrossRef] [PubMed]
- Blair, C.D. Mosquito RNAi is the major innate immune pathway controlling arbovirus infection and transmission. Future Microbiol. 2011, 6, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Brackney, D.E.; Beane, J.E.; Ebel, G.D. RNAi Targeting of West Nile Virus in Mosquito Midguts Promotes Virus Diversification. PLoS Pathog. 2009, 5, e1000502. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.F.; Lin, J.J.; Shi, Y.; Jin, Y.X.; Fu, Z.Q.; Jin, Y.M.; Zhou, Y.C.; Cai, Y.M. Dose-dependent inhibition of gynecophoral canal protein gene expression in vitro in the schistosome (Schistosoma japonicum) by RNA interference. Acta Biochim. Biophys. Sin. (Shanghai) 2005, 37, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vargas, I.; Travanty, E.A.; Keene, K.M.; Franz, A.W.; Beaty, B.J.; Blair, C.D.; Olson, K.E. RNA interference, arthropod-borne viruses, and mosquitoes. Virus Res. 2004, 102, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Adelman, Z.N.; Anderson, M.A.; Wiley, M.R.; Murreddu, M.G.; Samuel, G.H.; Morazzani, E.M.; Myles, K.M. Cooler temperature destabilizes RNA interference and increase susceptibility of disease vector mosquitoes to viral infection. PLoS Negl. Trop. Dis. 2013, 7, e2239. [Google Scholar] [CrossRef] [PubMed]
- Saksmerprome, V.; Roychowdhury-Saha, M.; Jayasena, S.; Khvorova, A.; Bruke, D.H. Artificial tertiary motifs stabilize trans-cleaving hammerhead ribozymes under conditions of submillimolar divalent ions and high temperatures. RNA 2004, 10, 1916–1924. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, M.; Passman, M.; Kew, M.; Arbuthnot, P. Hammerhead ribozyme-mediated inhibition of hepatitis B virus X gene expression in cultured cells. J. Hepatol. 2000, 33, 142–151. [Google Scholar] [CrossRef]
- Lieber, A.; He, C.Y.; Polyak, S.J.; Gretch, D.R.; Barr, D.; Kay, M.A. Elimination of hepatitis C virus RNA in infected human hepatocytes by adenovirus-mediated expression of ribozymes. J. Virol. 1996, 70, 8782–8791. [Google Scholar] [PubMed]
- Jackson, W.H., Jr.; Moscoso, H.; Nechtman, J.F.; Galileo, D.S.; Garver, F.A.; Lanclos, K.D. Inhibition of HIV-1 replication by an anti-tat hammerhead ribozyme. Biochem. Biophys. Res. Commun. 1998, 245, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Nawtaisong, P.; Keith, J.; Fraser, T.; Balaraman, V.; Kolokoltsov, A.; Davey, R.A.; Higgs, S.; Mohammed, A.; Rongsriyam, Y.; Komalamisra, N.; et al. Effective suppression of Dengue fever virus in mosquito cell cultures using retroviral transduction of hammerhead ribozymes targeting the viral genome. Virol. J. 2009, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Harrell, R.A.; Handler, A.M.; Beam, T.; Hennessy, K.; Fraser, M.J., Jr. PiggyBac internal sequences are necessary for efficient transformation of target genomes. Insect Mol. Biol. 2005, 14, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Staley, M.; Dorman, K.S.; Bartholomay, L.C.; Salas, I.; Farfan-Ale, J.A.; Loroño-Pino, M.A.; Garcia-Rejon, J.E.; Ibarra-Juarez, L.; Blitvich, B.J. Universal Primers for the amplification and sequence analysis of actin-1 from diverse mosquito species. J. Am. Mosq. Control Assoc. 2010, 26, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Karber, G. 50% end-point calculation. Arch. Exp. Pathol. Pharmak. 1931, 162, 480–483. [Google Scholar]
- Ho, P.S.; Ng, M.M.; Chu, J.J. Establishment of one-step SYBR green-based real time-PCR assay for rapid detection and quantification of chikungunya virus infection. Virol. J. 2010, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Handler, A.M.; Mccombs, S.D.; Fraser, M.J.; Saul, S.H. The lepidopteran transposon vector, piggyBac mediates germLine transformation in the Mediterranean fruit fly. PNAS 1998, 95, 7520–7525. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.J.; Luo, L. Splinkerette PCR for Mapping Transposable Elements in Drosophila. PLoS ONE 2010, 5, e10168. [Google Scholar] [CrossRef] [PubMed]
- VectorBase Bioinformatic resource for invertebrate vectors of human pathogen. Available online: http://aaegypti.vectorbase.org (accessed on 2 June 2016).
- Weaver, S.C.; Osorio, J.E.; LiVengood, J.A.; Chen, R.; Stinchcomb, D.T. Chikungunya virus and prospects for a vaccine. Expert Rev. Vaccines 2012, 11, 1087–1101. [Google Scholar] [CrossRef] [PubMed]
- Pesko, K.; Westbrook, C.J.; Mores, C.N.; Lounibos, L.P.; Reiskind, M.H. Effects of Infectious Virus Dose and Blood meal Delivery Method on Susceptibility of Aedes aegypti and Aedes albopictus to Chikungunya Virus. J. Med. Entomol. 2009, 46, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Section VIII-F: Arboviruses and Related Zoonotic Viruses. Available online: https://www.cdc.gov/biosafety/publications/bmbl5/bmbl5_sect_viii_f.pdf (accessed on 2 June 2016).
- Appendix B. Classification of human etiologic agents on the basis of hazard. Available online: https://www.unh.edu/research/sites/www.unh.edu.research/files/docs/EHS/Biosafety/NIH_Guidelines_Risk_Groups.pdf (accessed on 2 June 2016).
- Kuberski, T.T.; Rosen, L. A simple technique for the detection of Dengue Antigen in Mosquitoes by Immunofluorescence. Am. J. Trop. Med. Hyg. 1977, 26, 533–537. [Google Scholar] [PubMed]
- Franz, A.W.E.; Sanchez-Vargas, I.; Adelman, Z.N.; Blair, C.D.; Beaty, B.J.; James, A.A.; Olson, K.E. Engineering RNA interference-based resistance to dengue virus type 2 in genetically modified Aedes aegypti. PNAS 2006, 103, 4198–4203. [Google Scholar] [CrossRef] [PubMed]
- Amarzguioui, M.; Prydz, H. Hammerhead ribozymes design and application. Cell Mol. Life Sci. 1998, 54, 1175–1202. [Google Scholar] [CrossRef] [PubMed]
- Koseki, S.; Tanabe, T.; Tani, K.; Asano, S.; Shioda, T.; Nagai, Y.; Shimada, T.; Ohkawa, J.; Taira, K. Factors governing the activity in vivo of ribozymes transcribed by RNA polymerase III. J. Virol. 1999, 73, 1868–1877. [Google Scholar] [PubMed]
- Akashi, H.; Matsumoto, S.; Taira, K. Gene discovery by ribozyme and siRNA libraries. Nat. Rev. Mol. Cell Bio. 2005, 6, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, J.; Allsopp, T.E. Programmed cell death in virus infection of the nervous system. Curr. Top. Microb. Immunol. 2001, 253, 95–119. [Google Scholar]
- Levine, B.; Huang, Q.I.; Isaacs, J.T.; Reed, J.C.; Griffin, D.E.; Hardwick, M. Conversion of lytic to persistent alphavirus infection by the bcl-2 cellular oncogene. Lett. Nat. 1993, 361, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Krejbich-Trotot, P.; Denizot, M.; Hoarau, J.J.; Jaffar-Bandjee, M.C.; Das, T.; Gasque, P. Chikungunya virus mobilizes the apoptotic machinery to invade host cell defenses. FASEB J. 2011, 25, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Sourisseau, M.; Schilte, C.; Casartelli, N.; Trouillet, C.; Guivel-Benhassine, F.; Rudnicka, D.; Foulon, N.S.; Roux, K.L.; Prevost, M.C.; Fsihi, H.; et al. Characterization of reemerging chikungunya virus. PLoS Pathog. 2007, 3, e89. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.G.; Siripanyaphinyo, U.; Tumkosit, U.; Noranate, N.; A-nueqoonpipat, A.; Tao, R.; Kurosu, T.; Ikuta, K.; Takeda, N.; Anantapreecha, S. Chikungunya virus induces a more moderate cytopathic effect in mosquito cells than in mammalian cells. Intervirology 2013, 56, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.R.; Keith, J.H.; Fraser, T.S.; Dawson, J.L.; Kucharski, C.A.; Home, K.M.; Higgs, S.; Fraser, M.J., Jr. Effective suppression of Dengue virus using a novel group-I intron that induces apoptotic cell death upon infection through conditional expression of the Bax C-terminal domain. Virol. J. 2014, 13, 111. [Google Scholar] [CrossRef] [PubMed]
- Adelman, Z.N.; Sanchez-Vargas, I.; Travanty, E.A.; Carlson, J.O.; Beaty, B.J.; Blair, C.D.; Olson, K.E. RNA Silencing of Dengue Virus Type 2 Replication in Transformed C6/36 Mosquito Cells Transcribing an Inverted-Repeat RNA derived from the Virus genome. J. Virol. 2002, 76, 12925–12933. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Micorb. Rev. 1994, 58, 491–562. [Google Scholar]
- Gavin, D.K.; Gupta, K.C. Efficient Hammerhead Ribozymes Targeted to the Polycistronic Sendai Virus P/C mRNA. Structure and function relationship. J. Biol. Chem. 1997, 272, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Kennerdell, J.R.; Carthew, R.W. Use of dsRNA-mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell 1998, 95, 1017–1026. [Google Scholar] [CrossRef]
- Rossi, J.J. Controlled, targeted, intracellular expression of ribozymes: Progress and problems. Trends Biotechnol. 1995, 13, 301–306. [Google Scholar] [CrossRef]
- Sethuraman, N.; Fraser, M.J.; Eggleston, P.; O’Brochta, D. Post-integration stability of piggyBac in Aedes aegypti. Insect Biochem. Mol. Biol. 2007, 37, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Dubrulle, M.; Mousson, L.; Moutailler, S.; Vazeille, M.; Failloux, A.B. Chikungunya virus and Aedes mosquitoes: Saliva is infectious as soon as two days after oral infection. PLoS ONE 2009, 4, e5895. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.K.; Majumdar, S.S.; Alam, P.; Gulati, N.; Brahmachari, V. Epigenetic regulation of cytomegalovirus major immediate-early promoter activity in transgenic mice. Gene 2009, 428, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Bissinger, P.; Bullock, D.W.; Damak, S.; Wallace, R.; Whitelaw, C.B.; Yull, F. Chromosomal position effects and the modulation of transgene expression. Reprod. Fertil. Dev. 1994, 6, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Marrelli, M.T.; Li, C.; Rasgon, J.L.; Jacobs-Lorena, M. Trasgenic malaria-resistant mosquitoes have a fitness advantage when feeding on plasmodium-infected blood. Proc. Natl. Acad. Sci. USA 2007, 104, 5580–5583. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hRz | Template Sequence | Target Sequence (5’–3’) |
|---|---|---|
| hRz#9 | cgaacccgggcactacaaaaaccaacaaCTATTTAGCTGATGAGGCCGAAAGGCCGAAACCGCCGTACgggcccgggcccaaaaa | gtacggcggtcctaaatag |
| hRz#10 | cgaacccgggcactacaaaaaccaacaaAAGCGTCGCTGATGAGGCCGAAAGGCCGAAACTTCATGTGggggcccgggcccaaaaa | cacatgaagtccgacgctt |
| hRz#11 | cgaacccgggcactacaaaaaccaacaaTTACGCGGCTGATGAGGCCGAAAGGCCGAAACCAGAGGGgggcccgggcccaaaaa | ccctctggtcccgcgtaa |
| hRz#12 | cgaacccgggcactacaaaaaccaacaaAGCATGATCTGATGAGGCCGAAAGGCCGAAACTTGGTTTTggggcccgggcccaaaaa | aaaaccaagtcatcatgct |
| hRz#13 | cgaacccgggcactacaaaaaccaacaaAATGGGTACTGATGAGGCCGAAAGGCCGAAACGCCGGTGAgggcccgggcccaaaaa | tcaccggcgtctacccatt |
| hRz#14 | cgaacccgggcactacaaaaaccaacaaAGGCTGAACTGATGAGGCCGAAAGGCCGAAACATTGGCCCgggcccgggcccaaaaa | gggccaatgtcttcagcct |
| hRz#15 | cgaacccgggcactacaaaaaccaacaaTCTTAGGGCTGATGAGGCCGAAAGGCCGAAACACATATACgggcccgggcccaaaaa | gtatatgtgtcccctaaga |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, P.; Furey, C.; Balaraman, V.; Fraser, M.J. Antiviral Hammerhead Ribozymes Are Effective for Developing Transgenic Suppression of Chikungunya Virus in Aedes aegypti Mosquitoes. Viruses 2016, 8, 163. https://doi.org/10.3390/v8060163
Mishra P, Furey C, Balaraman V, Fraser MJ. Antiviral Hammerhead Ribozymes Are Effective for Developing Transgenic Suppression of Chikungunya Virus in Aedes aegypti Mosquitoes. Viruses. 2016; 8(6):163. https://doi.org/10.3390/v8060163
Chicago/Turabian StyleMishra, Priya, Colleen Furey, Velmurugan Balaraman, and Malcolm J. Fraser. 2016. "Antiviral Hammerhead Ribozymes Are Effective for Developing Transgenic Suppression of Chikungunya Virus in Aedes aegypti Mosquitoes" Viruses 8, no. 6: 163. https://doi.org/10.3390/v8060163
APA StyleMishra, P., Furey, C., Balaraman, V., & Fraser, M. J. (2016). Antiviral Hammerhead Ribozymes Are Effective for Developing Transgenic Suppression of Chikungunya Virus in Aedes aegypti Mosquitoes. Viruses, 8(6), 163. https://doi.org/10.3390/v8060163