
Mechanisms of Cellular Membrane Reorganization to Support Hepatitis C Virus Replication


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. History and Methods of Studying the HCV Membranous Web
3. Biogenesis of the Membranous Web
3.1. Role of Viral Factors in Membranous Web Biogenesis
3.2. Roles of Host Factors in Membranous Web Biogenesis
3.2.1. PI4KA—PI4P—OSBP and FAPP2: Cholesterol and Glycosphingolipids
3.2.2. Membrane Deforming Proteins
3.2.3. Nuclear Pore Complex Proteins
3.2.4. Autophagy and DMV Formation
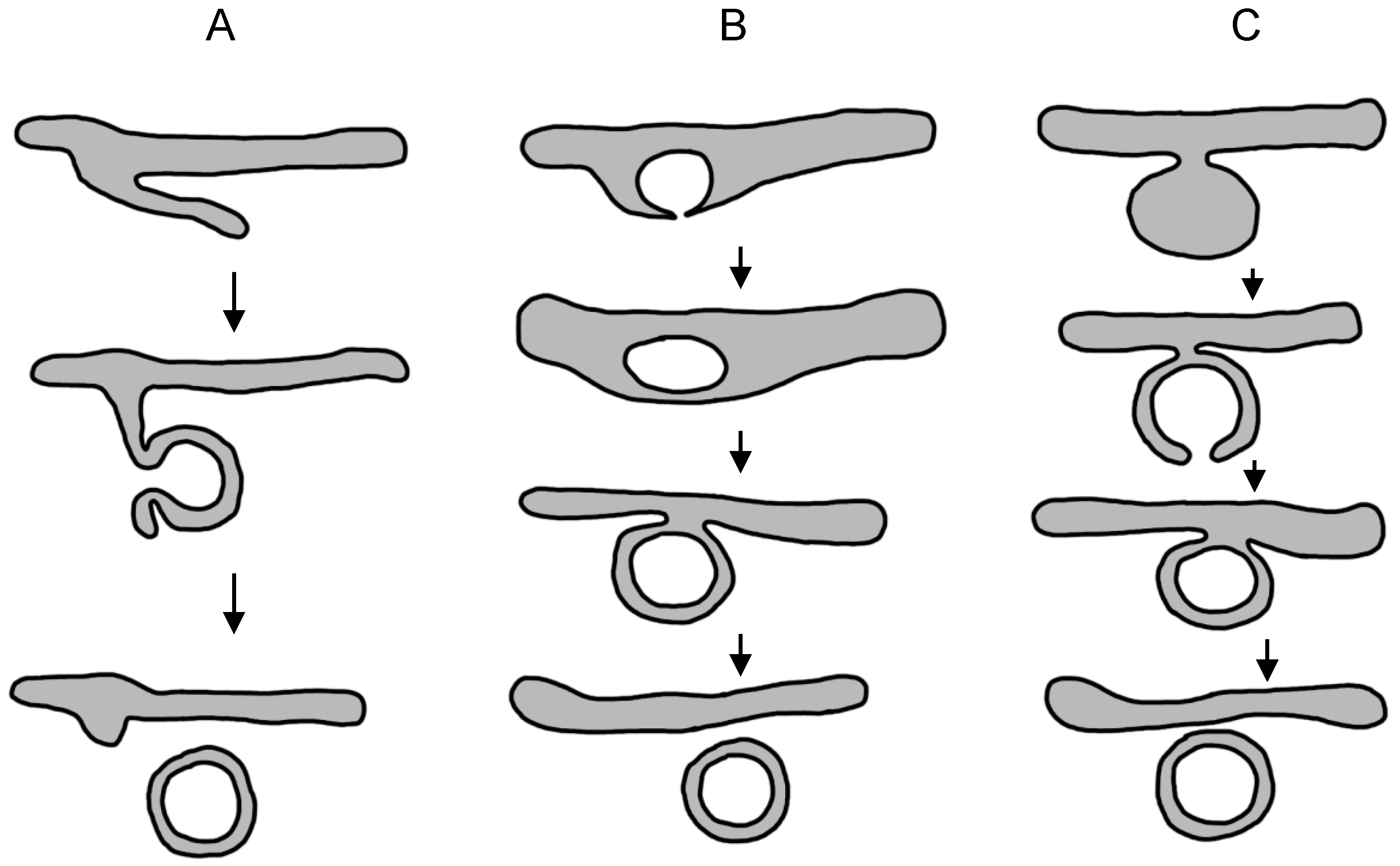
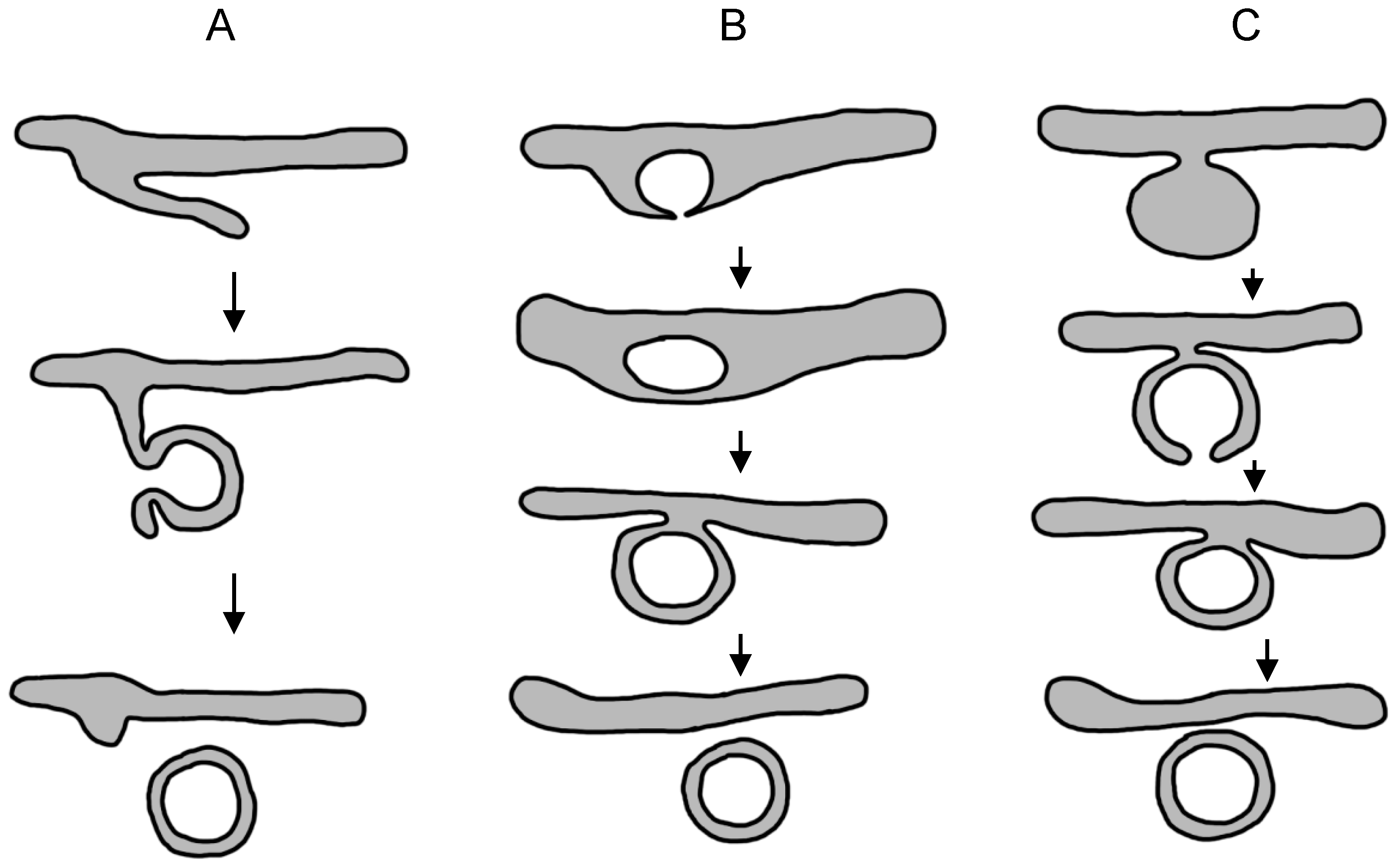
3.2.5. Models of Membranous Web Formation
4. Functions of the Membranous Web
4.1. The Membranous Web and HCV Genome Replication
4.2. Other Potential Functions of the Membranous Web
5. Future Perspectives
Acknowledgments
Conflicts of Interest
Abbreviations
| DMV | double-membrane vesicle |
| DRM | detergent resistant membrane |
| EM | electron microscopy |
| HCV | hepatitis C virus |
| MMV | multi-membrane vesicle |
| MW | membranous web |
| PAMP | pathogen-associated molecular pattern |
| PCH | Pombe Cdc15 homology |
| PI4P | phosphatidylinositol 4-phosphate |
| PRR | pattern-recognition receptor |
| SMV | single-membrane vesicle |
References
- Lindenbach, B.D.; Rice, C.M. Unravelling hepatitis C virus replication from genome to function. Nature 2005, 436, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V. Hepatitis C virus RNA replication. Curr. Top. Microbiol. Immunol. 2013, 369, 167–198. [Google Scholar] [PubMed]
- Bartenschlager, R.; Cosset, F.L.; Lohmann, V. Hepatitis C virus replication cycle. J. Hepatol. 2010, 53, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.T.; Murray, C.L.; Eastman, D.K.; Tassello, J.; Rice, C.M. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J. Virol. 2007, 81, 8374–8383. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, E.; Penin, F.; Kallis, S.; Patel, A.H.; Bartenschlager, R.; Pietschmann, T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007, 3, e103. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.; Tabor, E.; Gerety, R.J. Acute non-A, non-B hepatitis: Specific ultrastructural alterations in endoplasmic reticulum of infected hepatocytes. Lancet 1979, 1, 1249–1250. [Google Scholar] [CrossRef]
- Shimizu, Y.K.; Feinstone, S.M.; Purcell, R.H.; Alter, H.J.; London, W.T. Non-A, non-B hepatitis: Ultrastructural evidence for two agents in experimentally infected chimpanzees. Science 1979, 205, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Wölk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef] [PubMed]
- El-Hage, N.; Luo, G. Replication of hepatitis C virus RNA occurs in a membrane-bound replication complex containing nonstructural viral proteins and RNA. J. Gen. Virol. 2003, 84, 2761–2769. [Google Scholar] [CrossRef] [PubMed]
- Mottola, G.; Cardinali, G.; Ceccacci, A.; Trozzi, C.; Bartholomew, L.; Torrisi, M.R.; Pedrazzini, E.; Bonatti, S.; Migliaccio, G. Hepatitis C virus nonstructural proteins are localized in a modified endoplasmic reticulum of cells expressing viral subgenomic replicons. Virology 2002, 293, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.T.; Lee, K.J.; Aizaki, H.; Hwang, S.B.; Lai, M.M. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J. Virol. 2003, 77, 4160–4168. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, P.; Blanchard, E.; Roingeard, P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J. Gen. Virol. 2010, 91, 2230–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, D.; Bartenschlager, R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef] [PubMed]
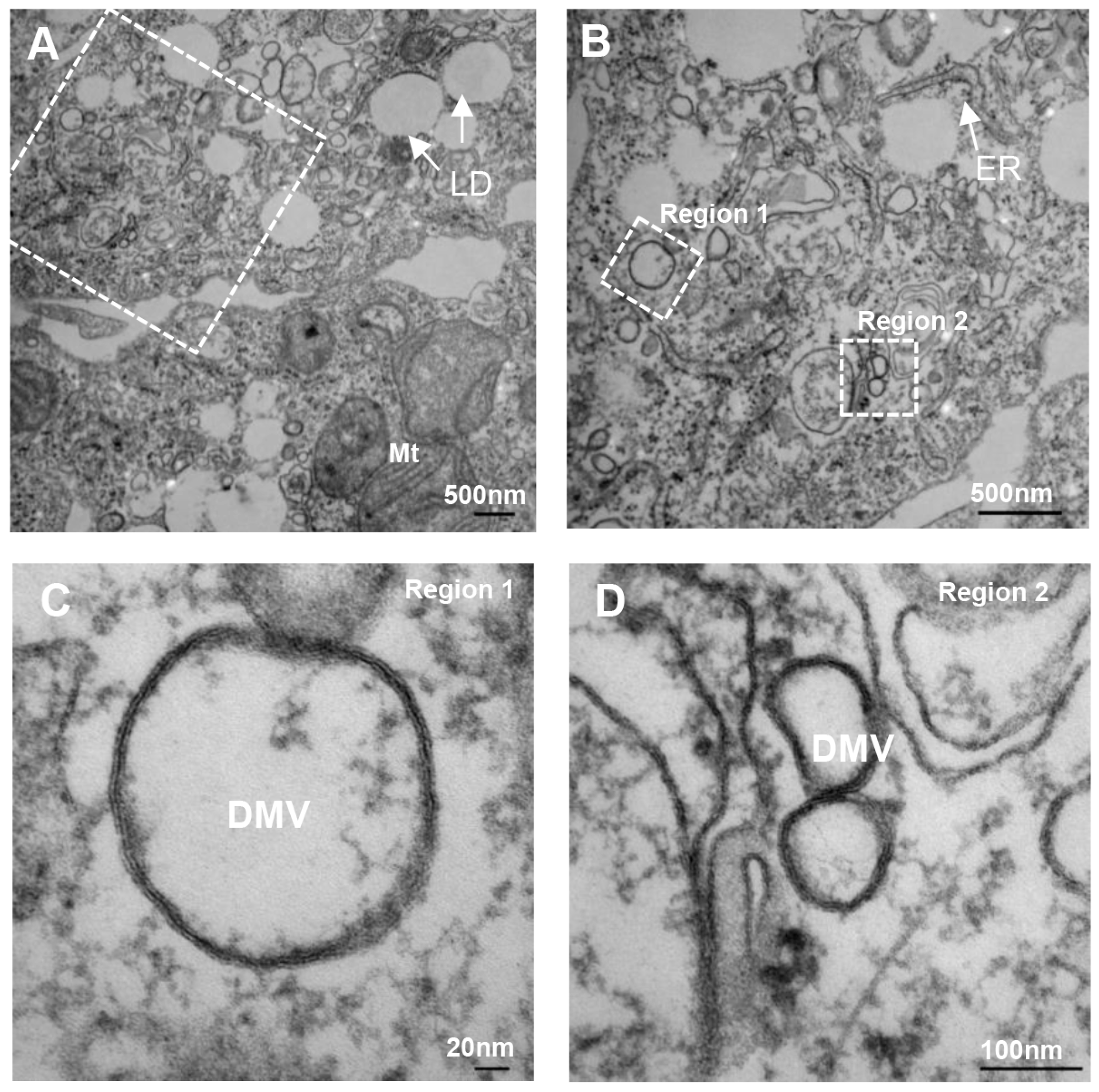
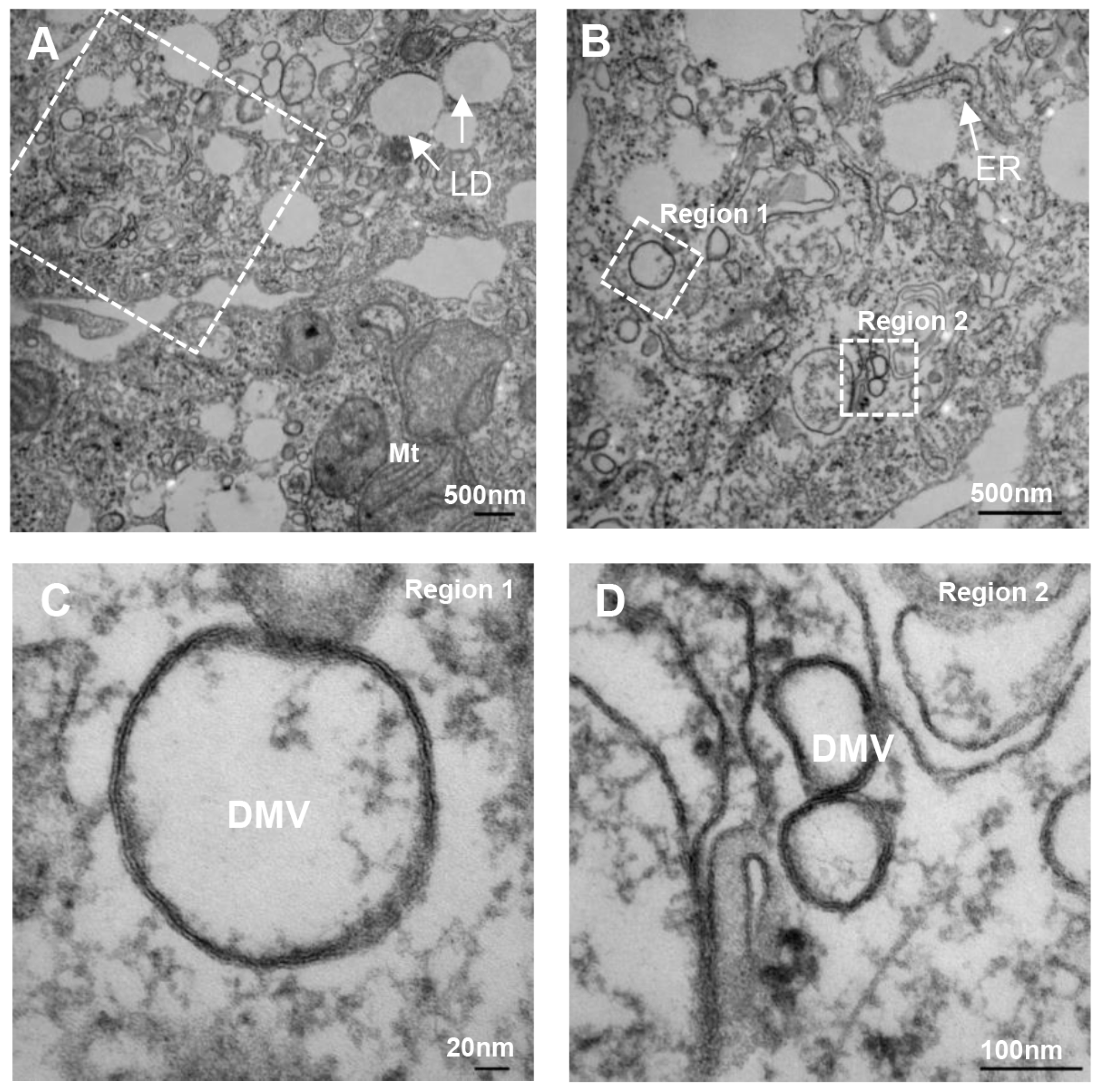
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Evans, M.J.; Gosert, R.; Yuan, Z.; Blum, H.E.; Goff, S.P.; Lindenbach, B.D.; Rice, C.M. Insertion of green fluorescent protein into nonstructural protein 5A allows direct visualization of functional hepatitis C virus replication complexes. J. Virol. 2004, 78, 7400–7409. [Google Scholar] [CrossRef] [PubMed]
- Wölk, B.; Büchele, B.; Moradpour, D.; Rice, C.M. A dynamic view of hepatitis C virus replication complexes. J. Virol. 2008, 82, 10519–10531. [Google Scholar] [CrossRef] [PubMed]
- Eyre, N.S.; Fiches, G.N.; Aloia, A.L.; Helbig, K.J.; McCartney, E.M.; McErlean, C.S.; Li, K.; Aggarwal, A.; Turville, S.G.; Beard, M.R. Dynamic imaging of the hepatitis C virus NS5A protein during a productive infection. J. Virol. 2014, 88, 3636–3652. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Romero-Brey, I.; Radujkovic, D.; Terreux, R.; Zayas, M.; Paul, D.; Harak, C.; Hoppe, S.; Gao, M.; Penin, F.; et al. Daclatasvir-like inhibitors of NS5A block early biogenesis of hepatitis C virus-induced membranous replication factories, independent of RNA replication. Gastroenterology 2014, 147, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Berger, C.; Kallis, S.; Kolovou, A.; Paul, D.; Lohmann, V.; Bartenschlager, R. NS5A domain 1 and polyprotein cleavage kinetics are critical for induction of double-membrane vesicles associated with hepatitis C virus replication. MBio 2015, 6, e00759. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Tardif, K.D.; Siddiqui, A. Cell-free replication of the hepatitis C virus subgenomic replicon. J. Virol. 2002, 76, 12001–12007. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.W.; Marcotrigiano, J.; Blight, K.J.; Majors, J.E.; Rice, C.M. Hepatitis C virus RNA synthesis in a cell-free system isolated from replicon-containing hepatoma cells. J. Virol. 2003, 77, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Lai, V.C.; Dempsey, S.; Lau, J.Y.; Hong, Z.; Zhong, W. In vitro RNA replication directed by replicase complexes isolated from the subgenomic replicon cells of hepatitis C virus. J. Virol. 2003, 77, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Miyanari, Y.; Hijikata, M.; Yamaji, M.; Hosaka, M.; Takahashi, H.; Shimotohno, K. Hepatitis C virus non-structural proteins in the probable membranous compartment function in viral genome replication. J. Biol. Chem. 2003, 278, 50301–50308. [Google Scholar] [CrossRef] [PubMed]
- Hanzal-Bayer, M.F.; Hancock, J.F. Lipid rafts and membrane traffic. FEBS Lett. 2007, 581, 2098–2104. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Quinkert, D.; Bartenschlager, R.; Lohmann, V. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 2005, 79, 13594–13605. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 2013, 87, 10612–10627. [Google Scholar] [CrossRef] [PubMed]
- Salloum, S.; Wang, H.; Ferguson, C.; Parton, R.G.; Tai, A.W. Rab18 binds to hepatitis C virus NS5A and promotes interaction between sites of viral replication and lipid droplets. PLoS Pathog. 2013, 9, e1003513. [Google Scholar] [CrossRef] [PubMed]
- Amako, Y.; Sarkeshik, A.; Hotta, H.; Yates, J., 3rd; Siddiqui, A. Role of oxysterol binding protein in hepatitis C virus infection. J. Virol. 2009, 83, 9237–9246. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.; Dixit, U.; Manvar, D.; Chaturvedi, N.; Pandey, V.N. Affinity capture and identification of host cell factors associated with hepatitis C virus (+) strand subgenomic RNA. Mol. Cell. Proteom. 2013, 12, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Sarker, S.; Siddiqui, A. Two-step affinity purification of the hepatitis C virus ribonucleoprotein complex. RNA 2004, 10, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Gouttenoire, J.; Roingeard, P.; Penin, F.; Moradpour, D. Amphipathic a-helix AH2 is a major determinant for the oligomerization of hepatitis C virus nonstructural protein 4B. J. Virol. 2010, 84, 12529–12537. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Romero-Brey, I.; Gouttenoire, J.; Stoitsova, S.; Krijnse-Locker, J.; Moradpour, D.; Bartenschlager, R. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J. Virol. 2011, 85, 6963–6976. [Google Scholar] [CrossRef] [PubMed]
- Gouttenoire, J.; Montserret, R.; Paul, D.; Castillo, R.; Meister, S.; Bartenschlager, R.; Penin, F.; Moradpour, D. Aminoterminal amphipathic a-helix AH1 of hepatitis C virus nonstructural protein 4B possesses a dual role in RNA replication and virus production. PLoS Pathog. 2014, 10, e1004501. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.L.; Cooper, J.D.; Heaton, N.S.; Yoon, R.; Oakland, T.E.; Jordan, T.X.; Mateu, G.; Grakoui, A.; Randall, G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 7577–7582. [Google Scholar] [CrossRef] [PubMed]
- Borawski, J.; Troke, P.; Puyang, X.; Gibaja, V.; Zhao, S.; Mickanin, C.; Leighton-Davies, J.; Wilson, C.J.; Myer, V.; Cornellataracido, I.; et al. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J. Virol. 2009, 83, 10058–10074. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.W.; Benita, Y.; Peng, L.F.; Kim, S.S.; Sakamoto, N.; Xavier, R.J.; Chung, R.T. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 2009, 5, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Vaillancourt, F.H.; Pilote, L.; Cartier, M.; Lippens, J.; Liuzzi, M.; Bethell, R.C.; Cordingley, M.G.; Kukolj, G. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology 2009, 387, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.S.; et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011, 9, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.L.; Kelly, S.M.; Jordan, T.X.; Tartell, M.A.; Randall, G. Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J. Virol. 2011, 85, 8870–8883. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.W.; Salloum, S. The role of the phosphatidylinositol 4-kinase PI4KA in hepatitis C virus-induced host membrane rearrangement. PLoS ONE 2011, 6, e26300. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Reghellin, V.; Donnici, L.; Fenu, S.; Alvarez, R.; Baruffa, C.; Peri, F.; Pagani, M.; Abrignani, S.; Neddermann, P.; et al. Metabolism of phosphatidylinositol 4-kinase III alpha-dependent PI4P is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLoS Pathog. 2012, 8, e1002576. [Google Scholar] [CrossRef] [PubMed]
- Furse, S.; Brooks, N.J.; Seddon, A.M.; Woscholski, R.; Templer, R.H.; Tate, E.W.; Gaffney, P.R.J.; Ces, O. Lipid membrane curvature induced by distearoyl phosphatidylinositol 4-phosphate. Soft Matter 2012, 8, 3090–3093. [Google Scholar] [CrossRef]
- Santiago-Tirado, F.H.; Bretscher, A. Membrane-trafficking sorting hubs: Cooperation between PI4P and small GTPases at the trans-golgi network. Trends Cell Biol. 2011, 21, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Katikaneni, D.S.; Han, Q.; Sanchez-Felipe, L.; Hanada, K.; Ambrose, R.L.; Mackenzie, J.M.; Konan, K.V. Modulation of hepatitis C virus genome replication by glycosphingolipids and four-phosphate adaptor protein 2. J. Virol. 2014, 88, 12276–12295. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Perry, J.W.; Lauring, A.S.; Neddermann, P.; de Francesco, R.; Tai, A.W. Oxysterol-binding protein is a phosphatidylinositol 4-kinase effector required for HCV replication membrane integrity and cholesterol trafficking. Gastroenterology 2014, 146, 1373–1385. [Google Scholar] [CrossRef] [PubMed]
- Lev, S. Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Amako, Y.; Syed, G.H.; Siddiqui, A. Protein kinase D negatively regulates hepatitis C virus secretion through phosphorylation of oxysterol-binding protein and ceramide transfer protein. J. Biol. Chem. 2011, 286, 11265–11274. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.C.; Su, W.C.; Huang, J.Y.; Chen, Y.C.; Jeng, K.S.; Wang, H.D.; Lai, M.M. PSTPIP2, proline-serine-threonine phosphatase interacting protein 2, a host membrane-deforming protein, is critical for membranous web formation in hepatitis C virus replication. J. Virol. 2012, 86, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Nanda, S.K.; Herion, D.; Liang, T.J. The SH3 binding motif of HCV NS5A protein interacts with Bin1 and is important for apoptosis and infectivity. Gastroenterology 2006, 130, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Bonamassa, B.; Ciccarese, F.; Antonio, V.D.; Contarini, A.; Palu, G.; Alvisi, G. Hepatitis C virus and host cell nuclear transport machinery: A clandestine affair. Front. Microbiol 2015, 6, 619. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Sakamoto, S.; Tsutsumi, T.; Rikimaru, A.; Tanaka, K.; Shimoike, T.; Moriishi, K.; Iwasaki, T.; Mizumoto, K.; Matsuura, Y.; et al. Molecular determinants for subcellular localization of hepatitis C virus core protein. J. Virol. 2005, 79, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Neufeldt, C.J.; Pang, D.; Wilson, K.; Loewen-Dobler, D.; Joyce, M.A.; Wozniak, R.W.; Tyrrell, D.L. Functional characterization of nuclear localization and export signals in hepatitis C virus proteins and their role in the membranous web. PLoS ONE 2014, 9, e114629. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, M.A.; Imache, M.R.; Higgs, M.R.; Carmouse, S.; Pawlotsky, J.M.; Lerat, H. Regulation of hepatitis C virus replication by nuclear translocation of nonstructural 5A protein and transcriptional activation of host genes. J. Virol. 2013, 87, 5523–5539. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, A.; Maillard, P.; Minisini, R.; Vidalain, P.O.; Roohvand, F.; Pecheur, E.I.; Pirisi, M.; Budkowska, A. Identification of a functional, CRM-1-dependent nuclear export signal in hepatitis C virus core protein. PLoS ONE 2011, 6, e25854. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.E. Nuclear proteins hijacked by mammalian cytoplasmic plus strand RNA viruses. Virology 2015, 479–480, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, C.J.; Joyce, M.A.; Levin, A.; Steenbergen, R.H.; Pang, D.; Shields, J.; Tyrrell, D.L.; Wozniak, R.W. Hepatitis C virus-induced cytoplasmic organelles use the nuclear transport machinery to establish an environment conducive to virus replication. PLoS Pathog. 2013, 9, e1003744. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Tooze, S.A.; Reggiori, F. The puzzling origin of the autophagosomal membrane. F1000 Biol. Rep. 2011, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; James Ou, J.H. Hepatitis C virus and autophagy. Biol. Chem. 2015, 396, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Ait-Goughoulte, M.; Kanda, T.; Meyer, K.; Ryerse, J.S.; Ray, R.B.; Ray, R. Hepatitis C virus genotype 1a growth and induction of autophagy. J. Virol. 2008, 82, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Su, W.C.; Chao, T.C.; Huang, Y.L.; Weng, S.C.; Jeng, K.S.; Lai, M.M. Rab5 and class III phosphoinositide 3-kinase vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 2011, 85, 10561–10571. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Bhanja Chowdhury, J.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus upregulates beclin1 for induction of autophagy and activates mTOR signaling. J. Virol. 2012, 86, 8705–8712. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Guevin, C.; Manna, D.; Belanger, C.; Konan, K.V.; Mak, P.; Labonte, P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010, 405, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sir, D.; Kuo, C.F.; Tian, Y.; Liu, H.M.; Huang, E.J.; Jung, J.U.; Machida, K.; Ou, J.H. Replication of hepatitis C virus RNA on autophagosomal membranes. J. Biol. Chem. 2012, 287, 18036–18043. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.Y.; Chen, S.S. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investig. 2011, 121, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Fukasawa, M.; Ueno, T.; Kominami, E.; Wakita, T.; Hanada, K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009, 5, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.W.; van der Meer, Y.; Roos, N.; Snijder, E.J. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J. Virol. 1999, 73, 2016–2026. [Google Scholar] [PubMed]
- Ferraris, P.; Beaumont, E.; Uzbekov, R.; Brand, D.; Gaillard, J.; Blanchard, E.; Roingeard, P. Sequential biogenesis of host cell membrane rearrangements induced by hepatitis C virus infection. Cell. Mol. Life Sci. 2013, 70, 1297–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limpens, R.W.; van der Schaar, H.M.; Kumar, D.; Koster, A.J.; Snijder, E.J.; van Kuppeveld, F.J.; Barcena, M. The transformation of enterovirus replication structures: A three-dimensional study of single- and double-membrane compartments. MBio 2011, 2, e00166-11. [Google Scholar] [CrossRef] [PubMed]
- Shulla, A.; Randall, G. Spatiotemporal analysis of hepatitis C virus infection. PLoS Pathog. 2015, 11, e1004758. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Aizaki, H.; Lee, K.J.; Sung, V.M.; Ishiko, H.; Lai, M.M. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 2004, 324, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Imamura, H.; Suzuki, R.; Aizaki, H.; Watanabe, T.; Wakita, T.; Suzuki, T. Visualization and measurement of ATP levels in living cells replicating hepatitis C virus genome RNA. PLoS Pathog. 2012, 8, e1002561. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Pasamontes, L.; Bolten, R.; Boyko, V.; Bienz, K. Reversible dissociation of the poliovirus replication complex: Functions and interactions of its components in viral RNA synthesis. J. Virol. 1996, 70, 8675–8683. [Google Scholar] [PubMed]
- Hagemeijer, M.C.; Verheije, M.H.; Ulasli, M.; Shaltiel, I.A.; de Vries, L.A.; Reggiori, F.; Rottier, P.J.; de Haan, C.A. Dynamics of coronavirus replication-transcription complexes. J. Virol. 2010, 84, 2134–2149. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, N.S.; Vazquez, C.; Horner, S.M. Hepatitis C virus. Strategies to evade antiviral responses. Future Virol. 2014, 9, 1061–1075. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, C.J.; Joyce, M.A.; Van Buuren, N.; Levin, A.; Kirkegaard, K.; Gale, M., Jr.; Tyrrell, D.L.; Wozniak, R.W. The hepatitis C virus-induced membranous web and associated nuclear transport machinery limit access of pattern recognition receptors to viral replication sites. PLoS Pathog. 2016, 12, e1005428. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Tai, A.W. Mechanisms of Cellular Membrane Reorganization to Support Hepatitis C Virus Replication. Viruses 2016, 8, 142. https://doi.org/10.3390/v8050142
Wang H, Tai AW. Mechanisms of Cellular Membrane Reorganization to Support Hepatitis C Virus Replication. Viruses. 2016; 8(5):142. https://doi.org/10.3390/v8050142
Chicago/Turabian StyleWang, Hongliang, and Andrew W. Tai. 2016. "Mechanisms of Cellular Membrane Reorganization to Support Hepatitis C Virus Replication" Viruses 8, no. 5: 142. https://doi.org/10.3390/v8050142
APA StyleWang, H., & Tai, A. W. (2016). Mechanisms of Cellular Membrane Reorganization to Support Hepatitis C Virus Replication. Viruses, 8(5), 142. https://doi.org/10.3390/v8050142