
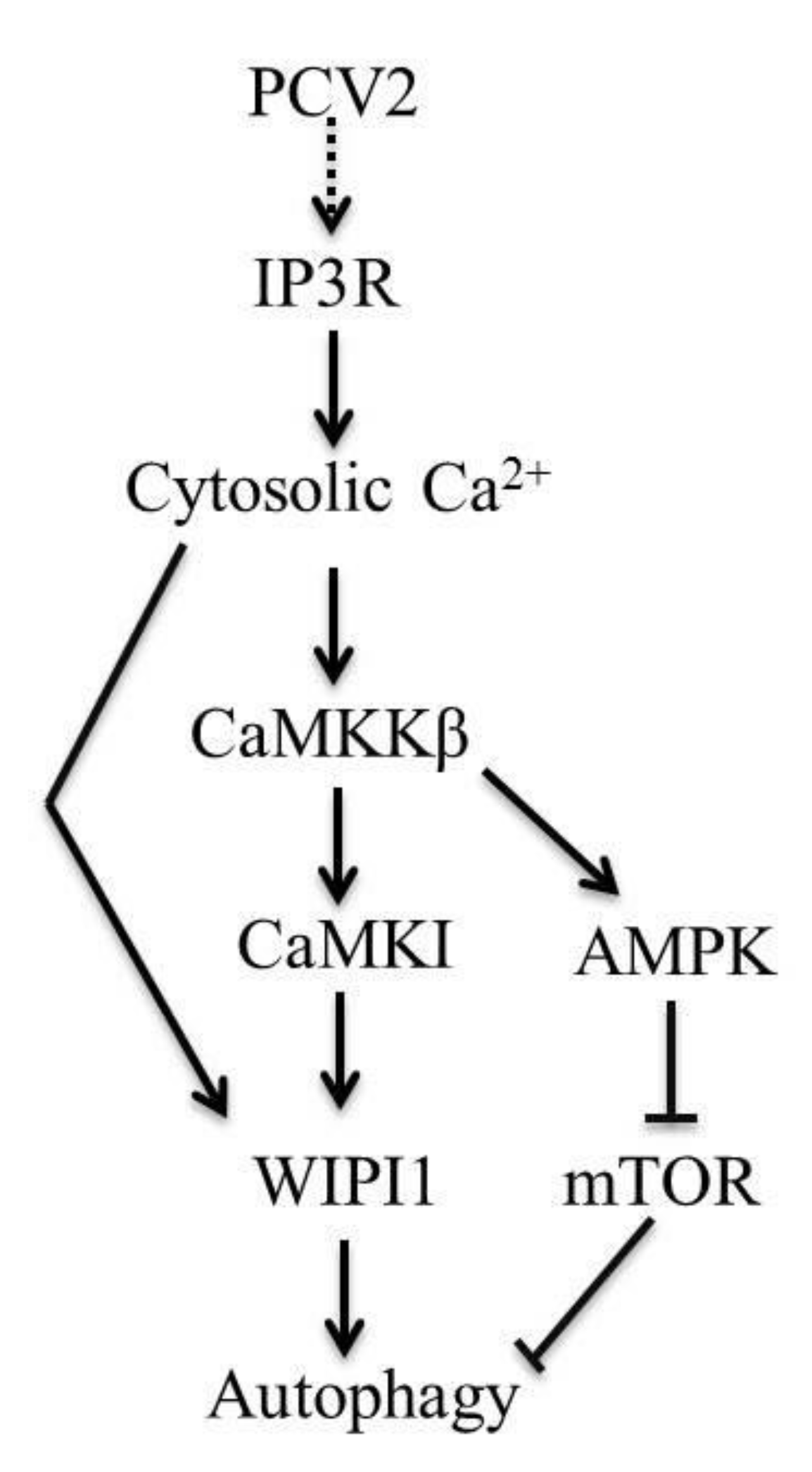
Porcine Circovirus Type 2 Activates CaMMKβ to Initiate Autophagy in PK-15 Cells by Increasing Cytosolic Calcium

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Virus, Cell Lines, and Plasmids
2.2. Recombinant Plasmids, Small Interfering RNAs and Transfection
2.3. Virus Infection, Treatments with Chemicals and Transfection
2.4. Western Blotting
2.5. Indirect Immunofluorescence, and Quantification of Virus Titer and Viral DNA
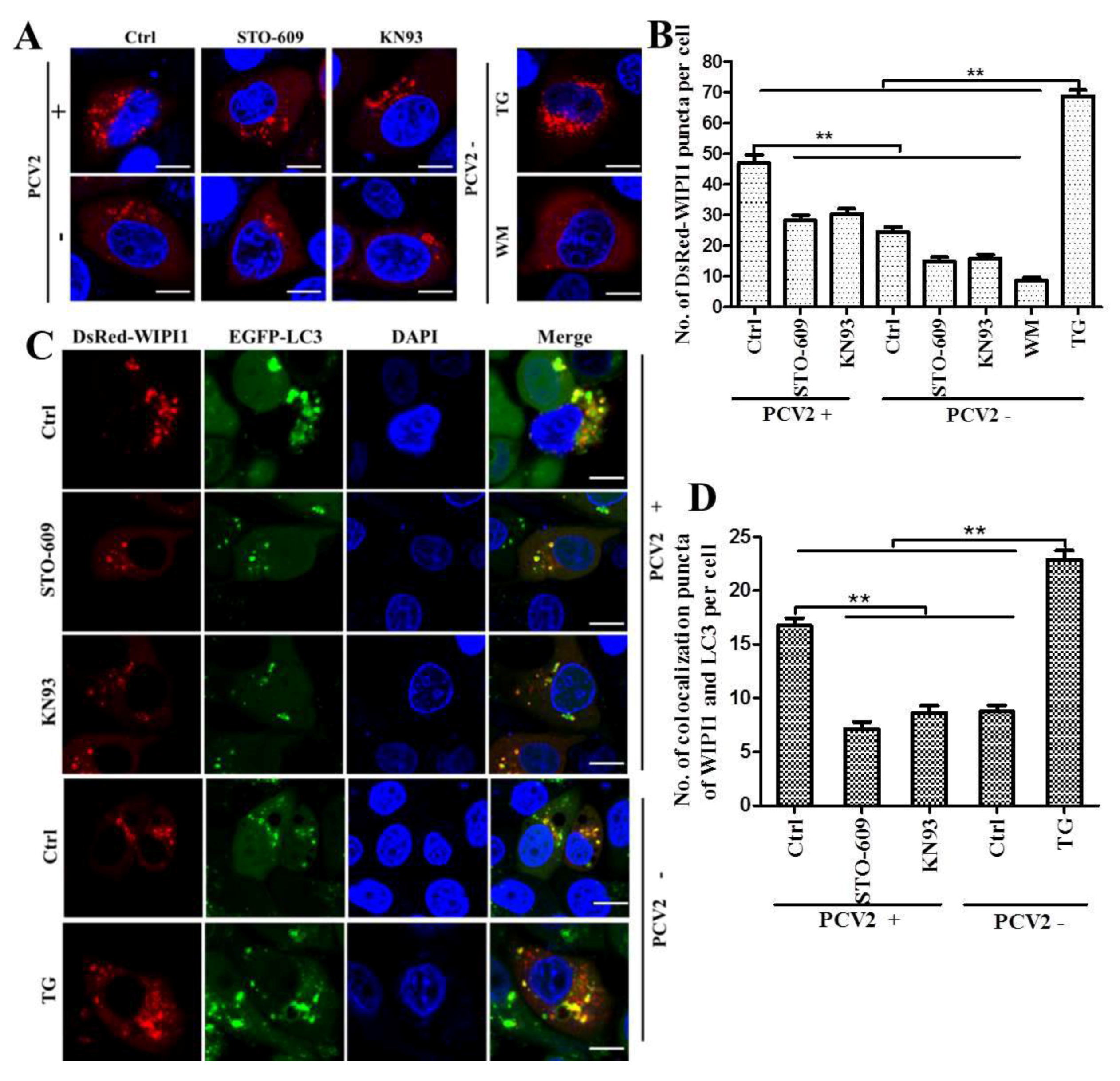
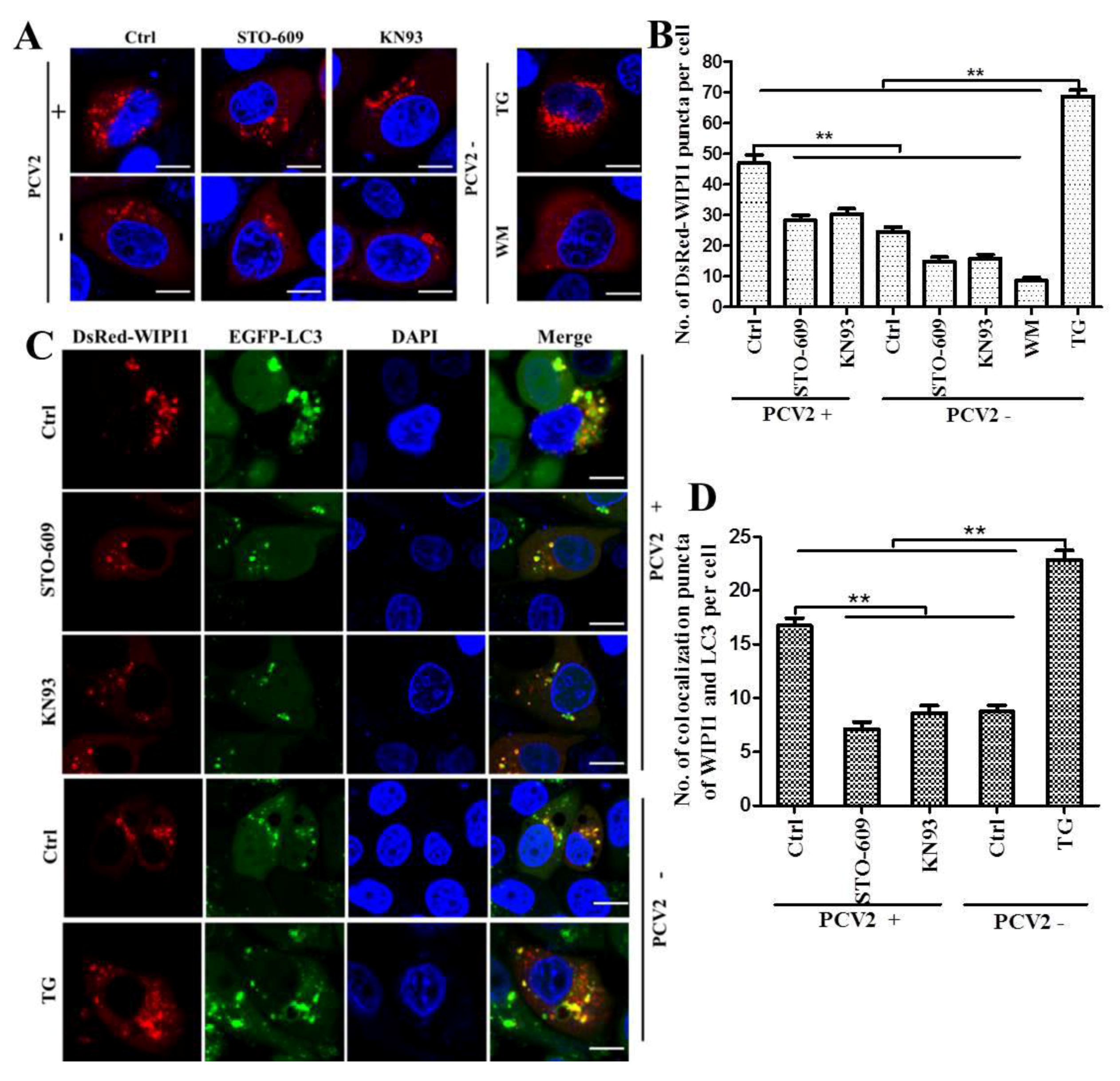
2.6. Confocal Microscopic Analysis of WIPI1 Puncta and WIPI1-LC3 Colocalization
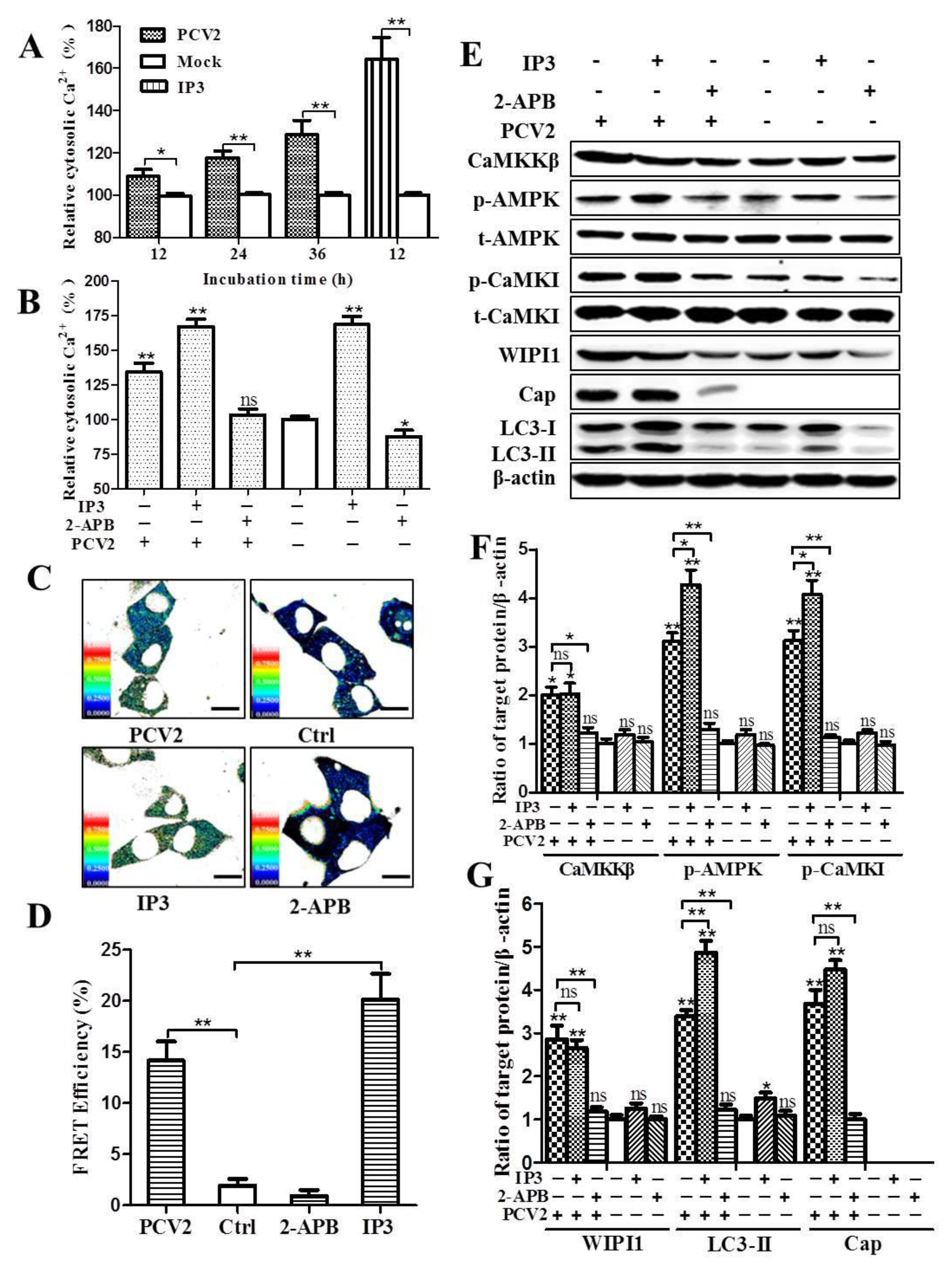
2.7. Analysis of Cytosolic Ca2+ by Flow Cytometry and FRET
2.8. IP3 Measurement by ELISA
2.9. Cell Viability Measurement
2.10. Statistical Analysis
3. Results
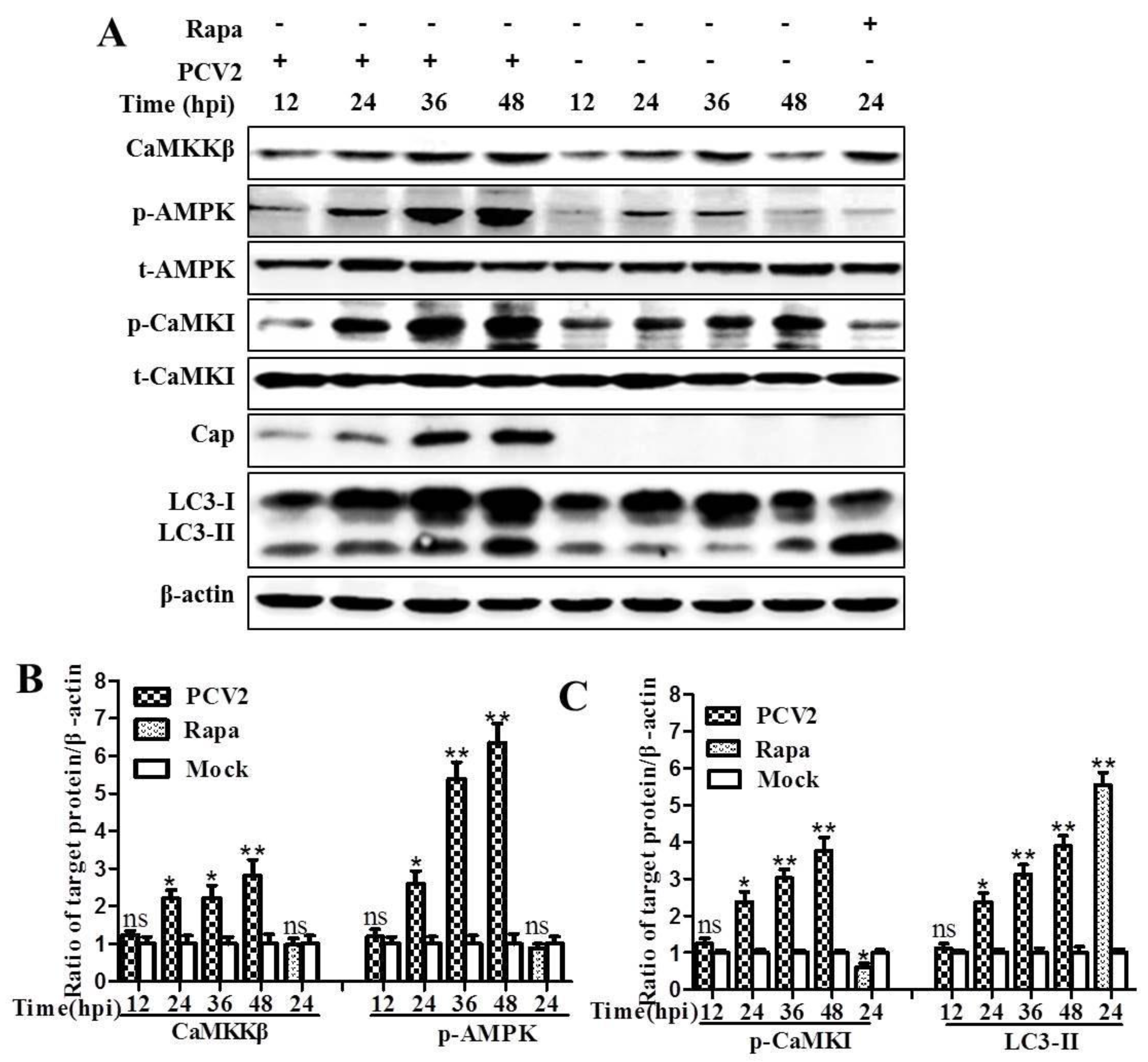
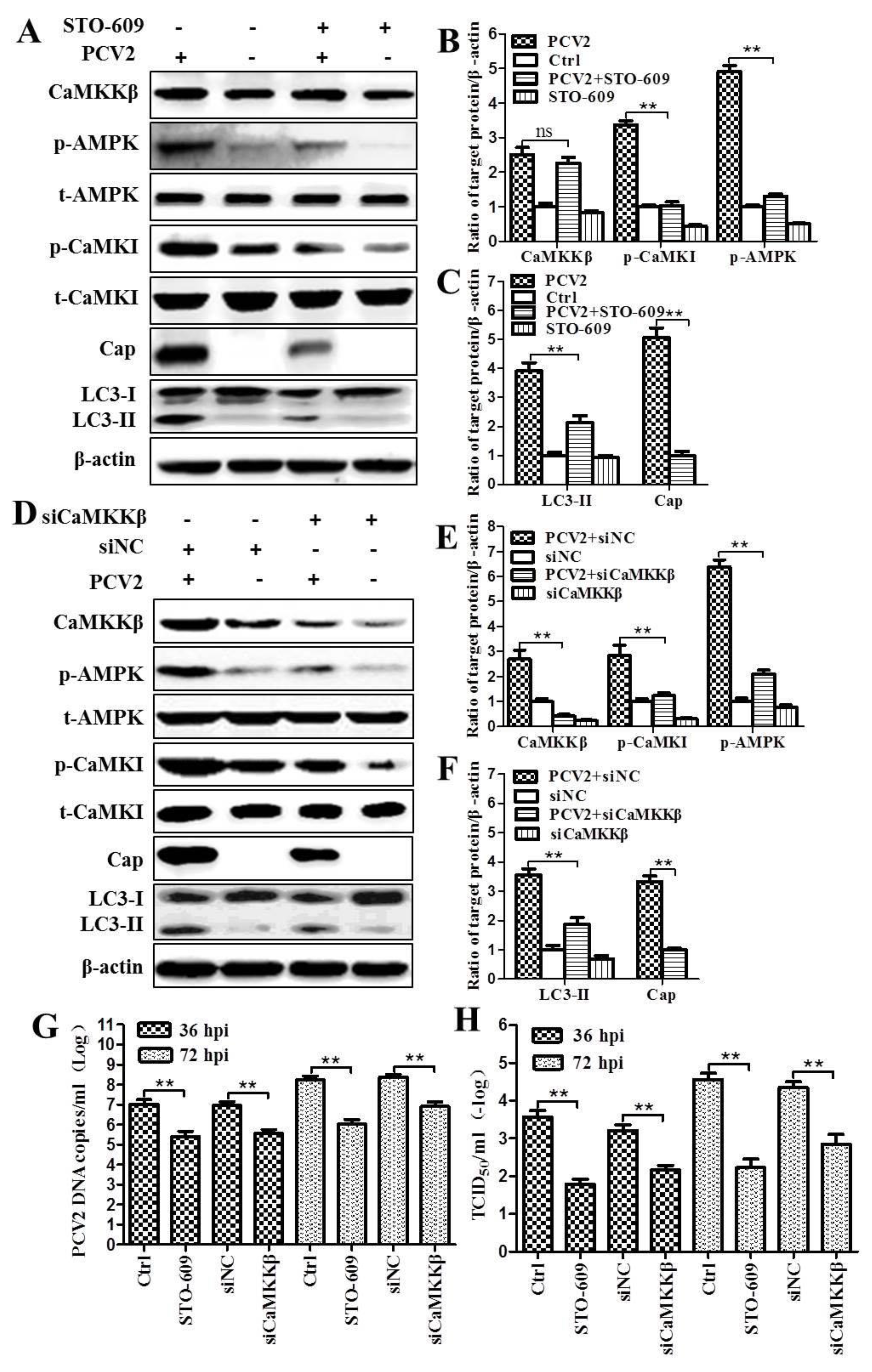
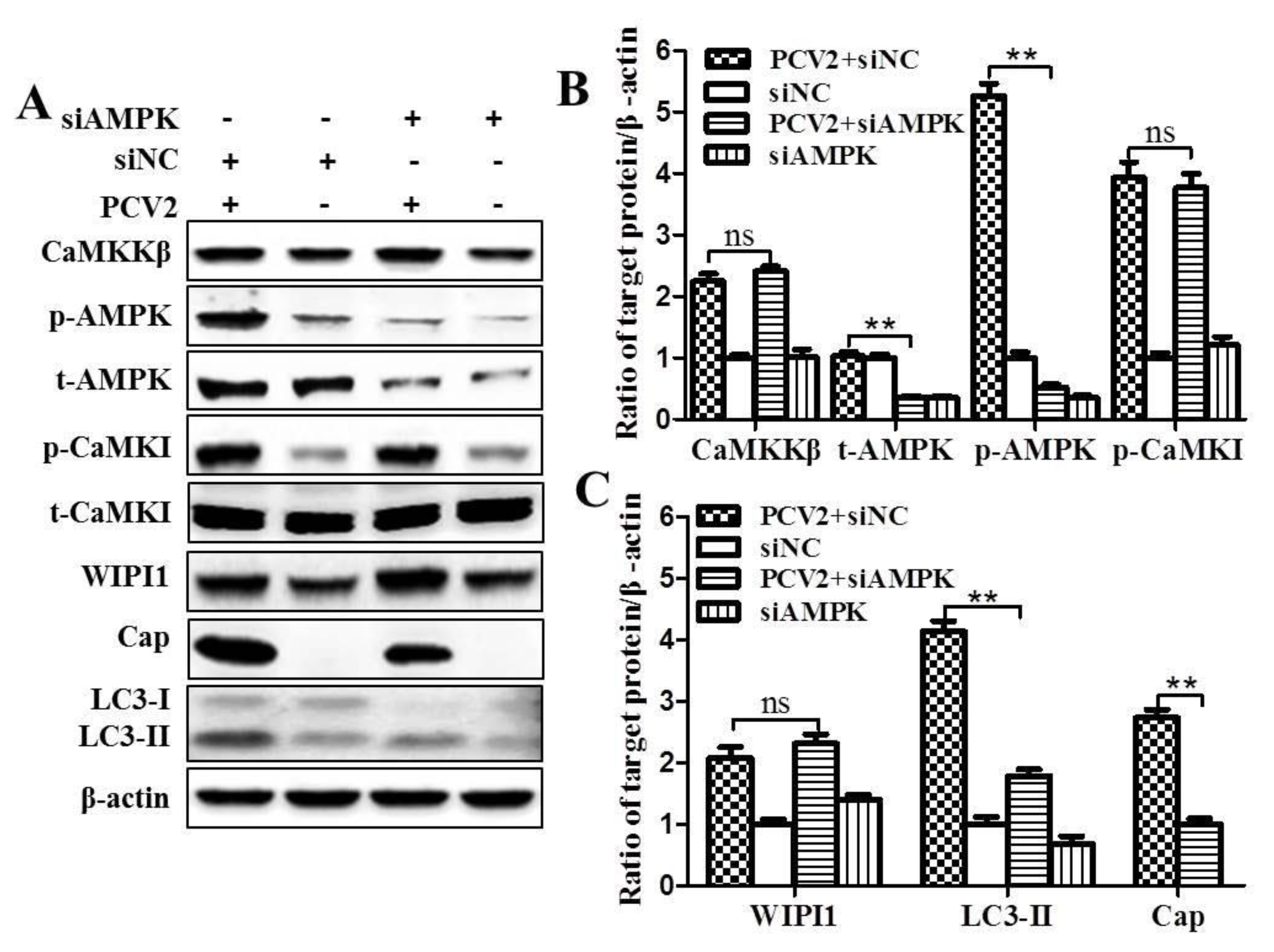
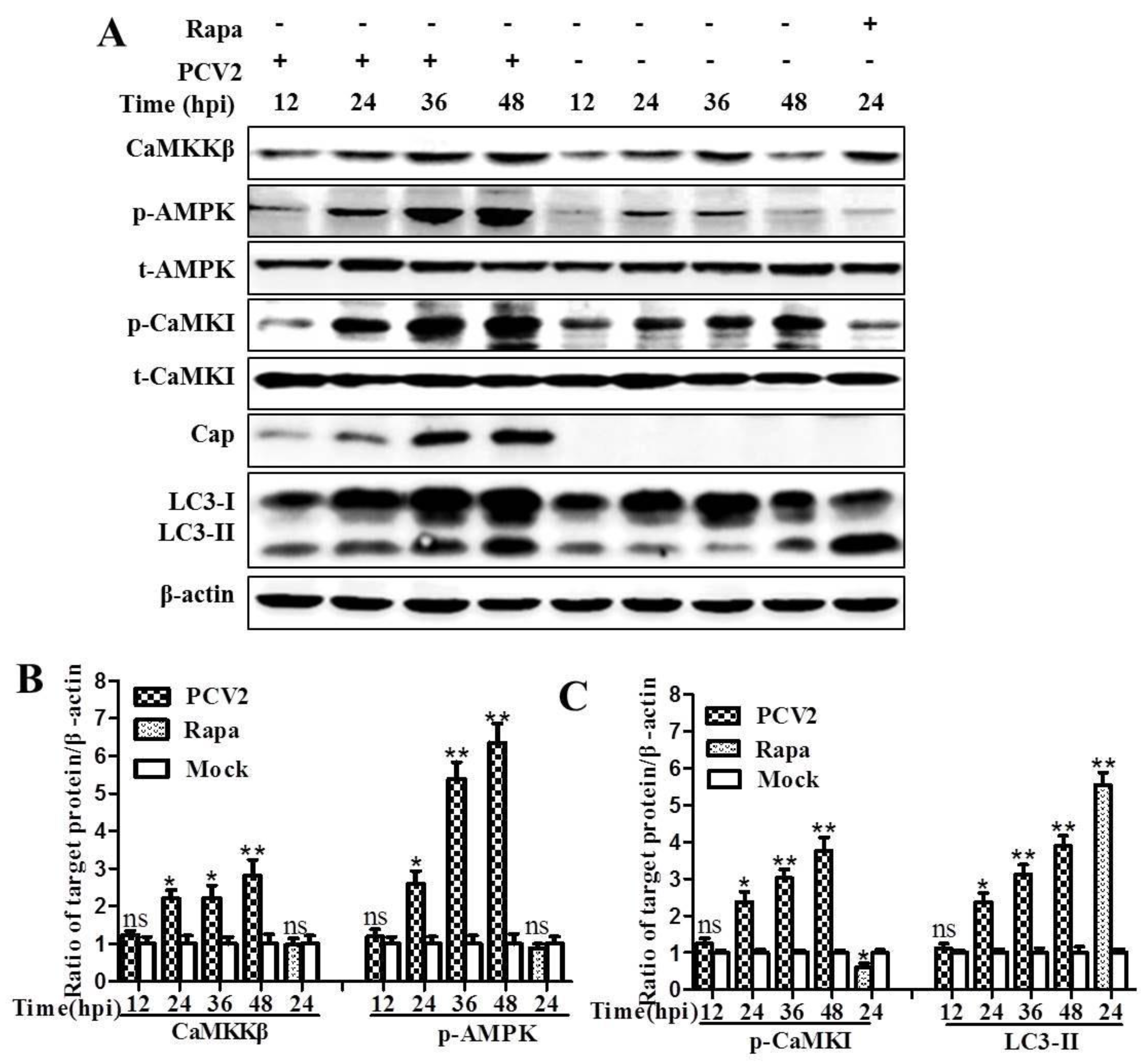
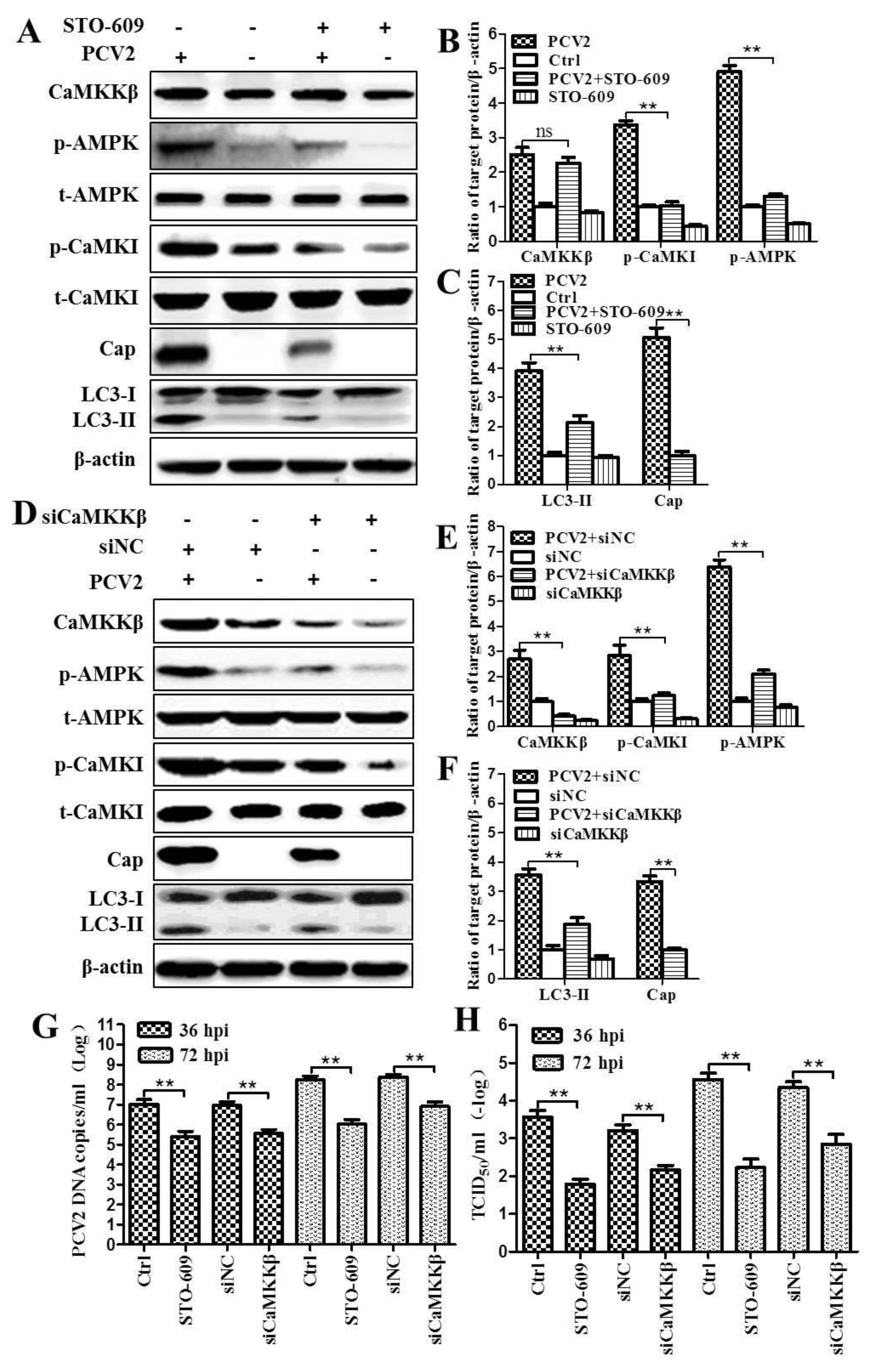
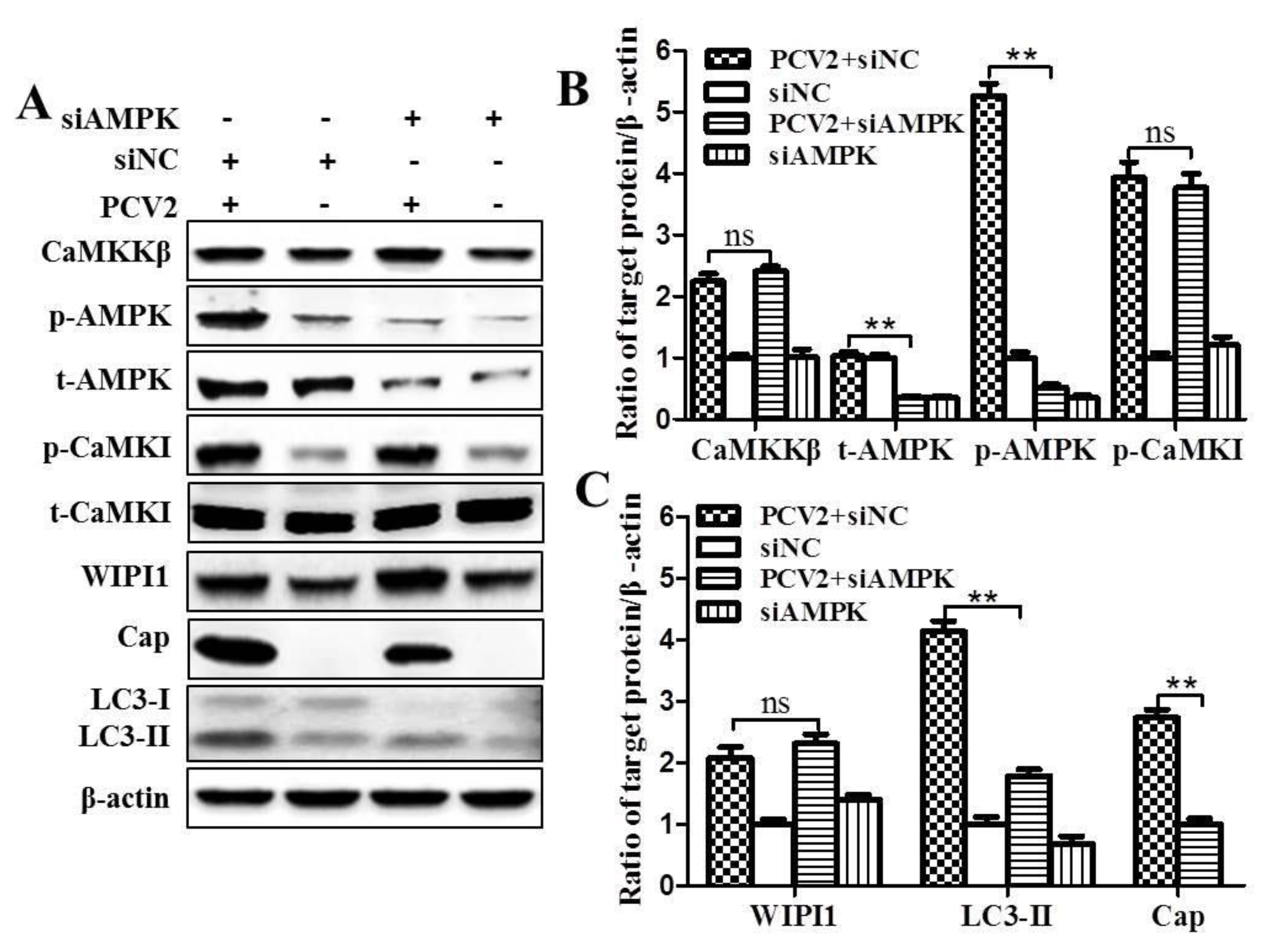
3.1. CaMKKβ Is the Upstream Activator of AMPK in PCV2-Induced Autophagy
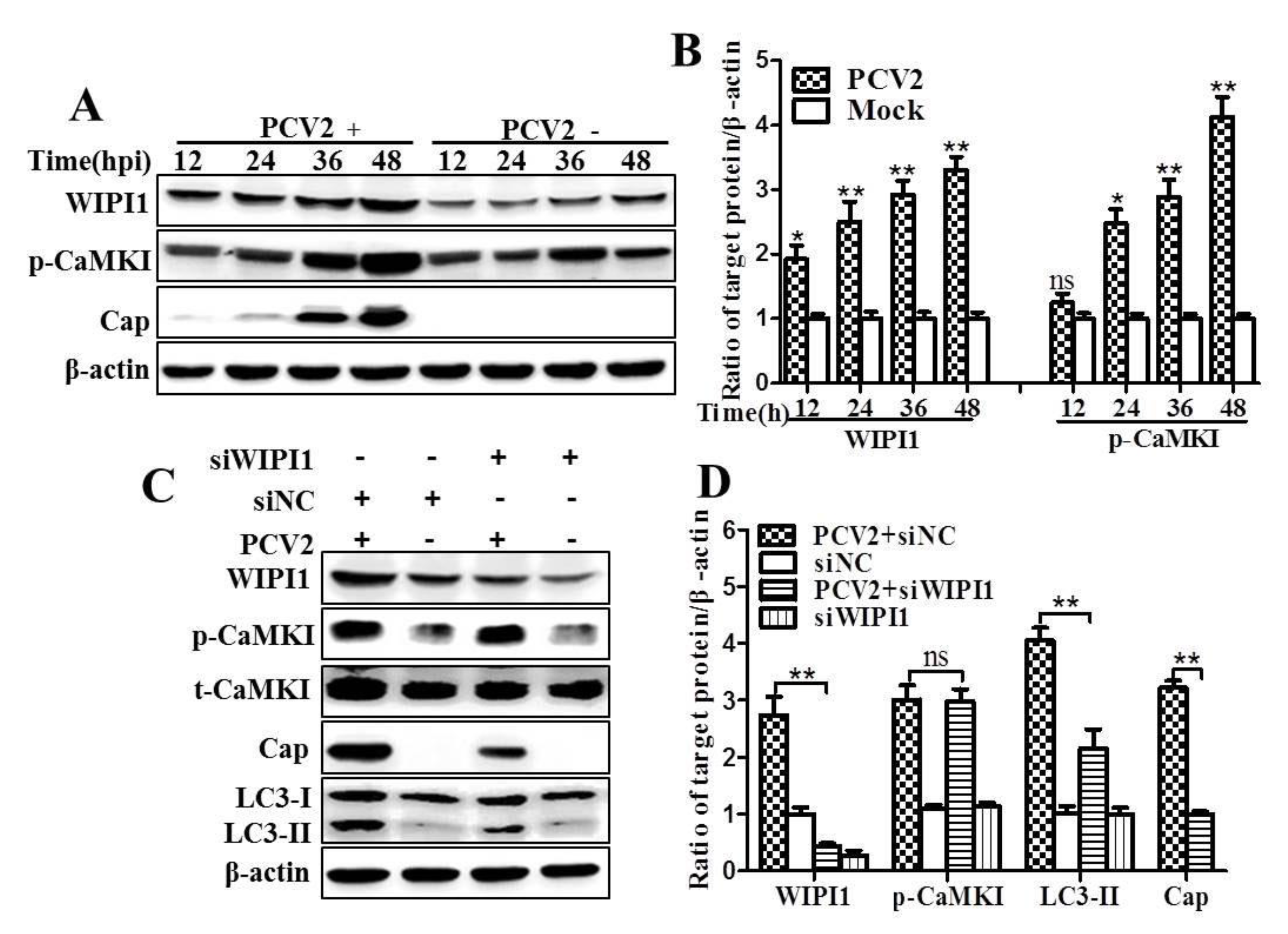
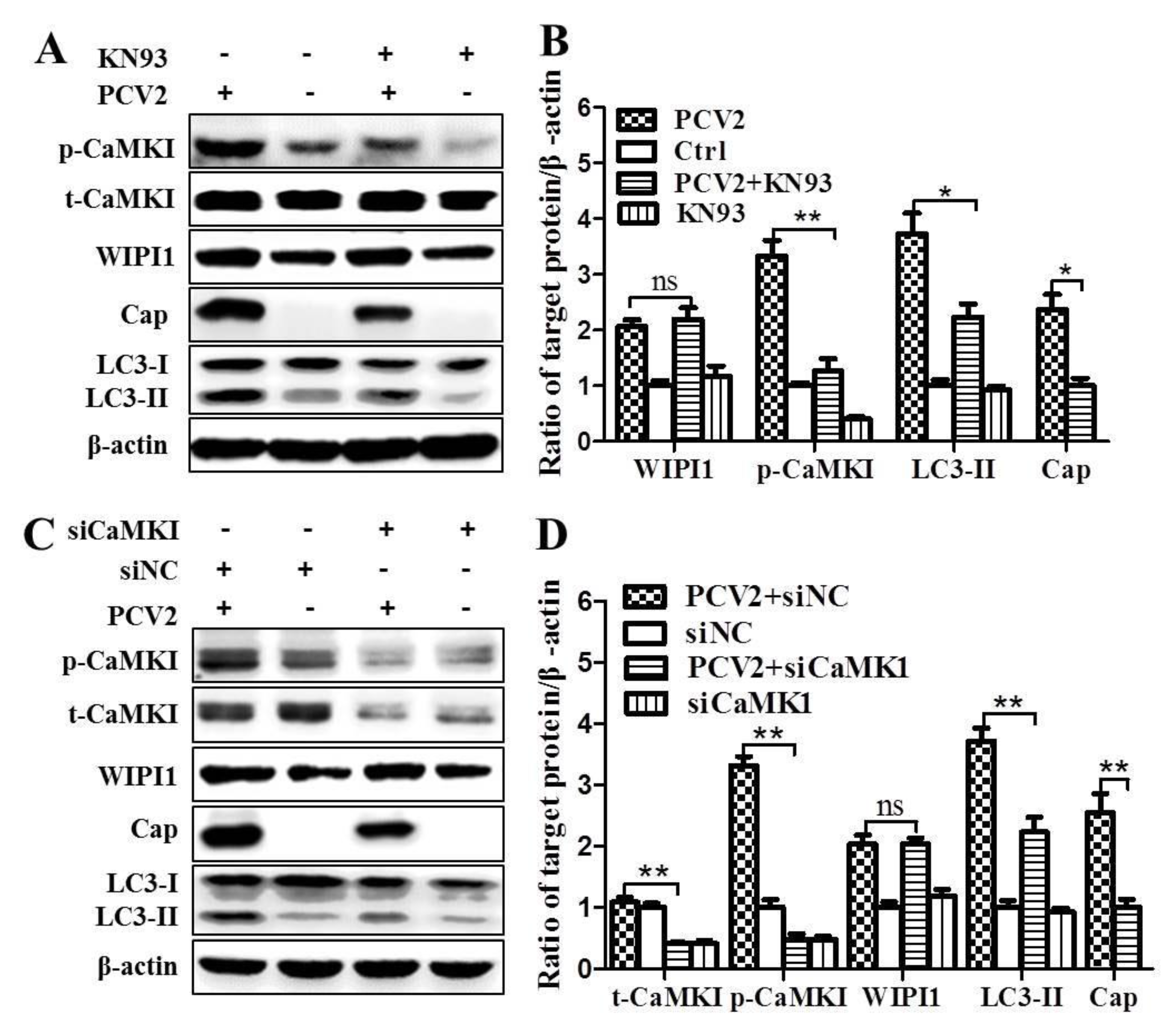
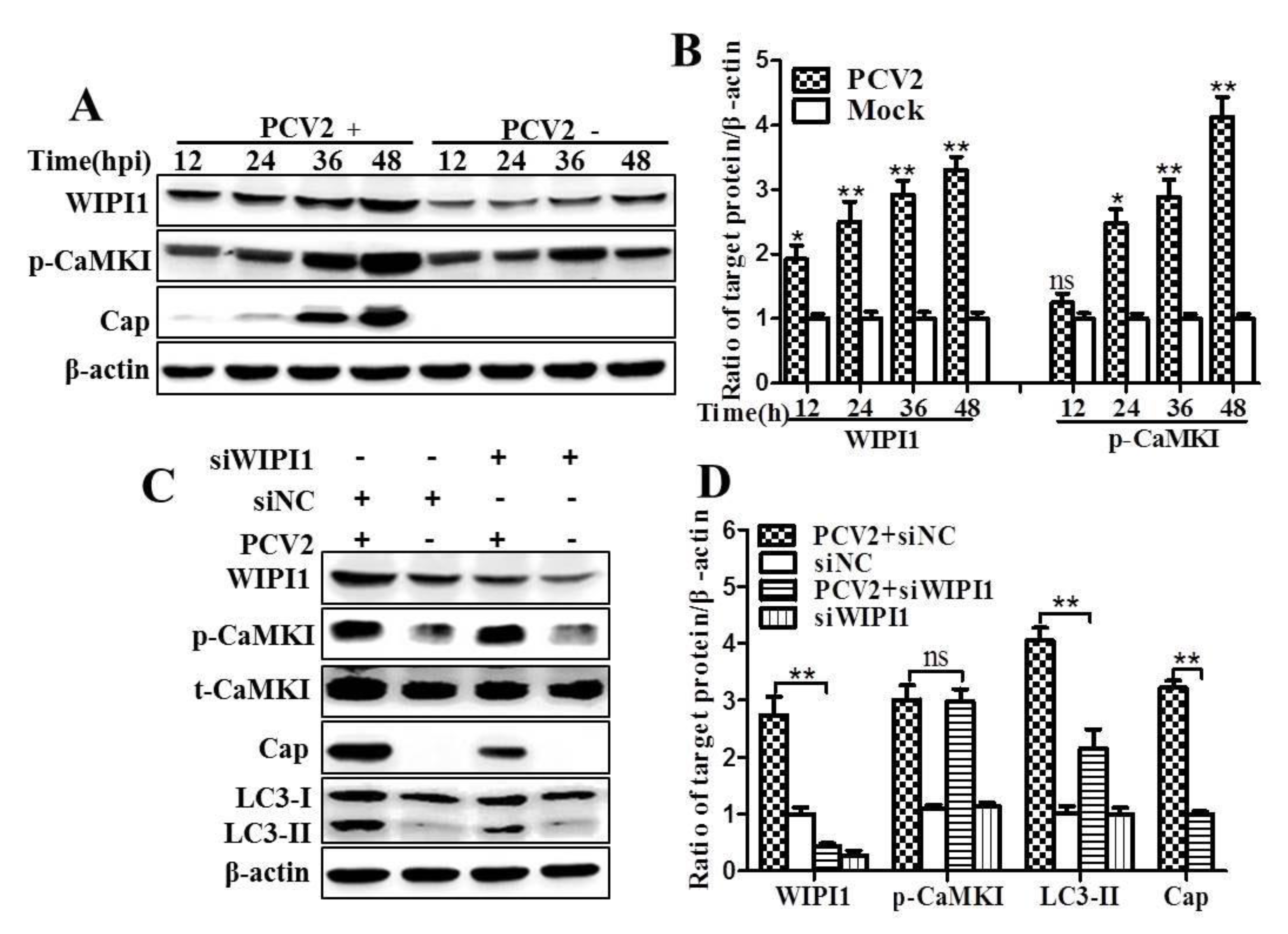
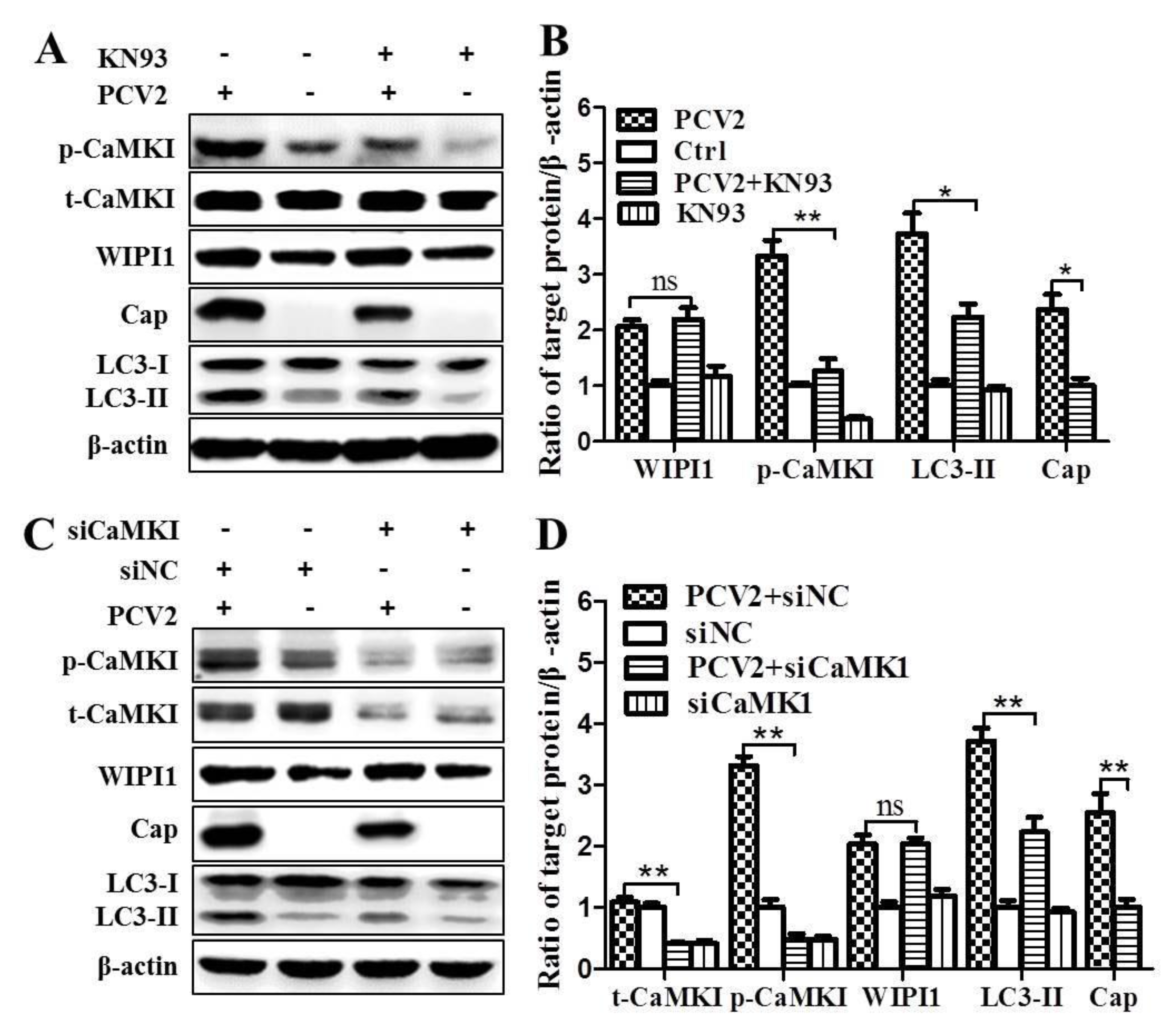
3.2. PCV2 Employs CaMKI/WIPI1 for Autophagy Induction Independent of AMPK
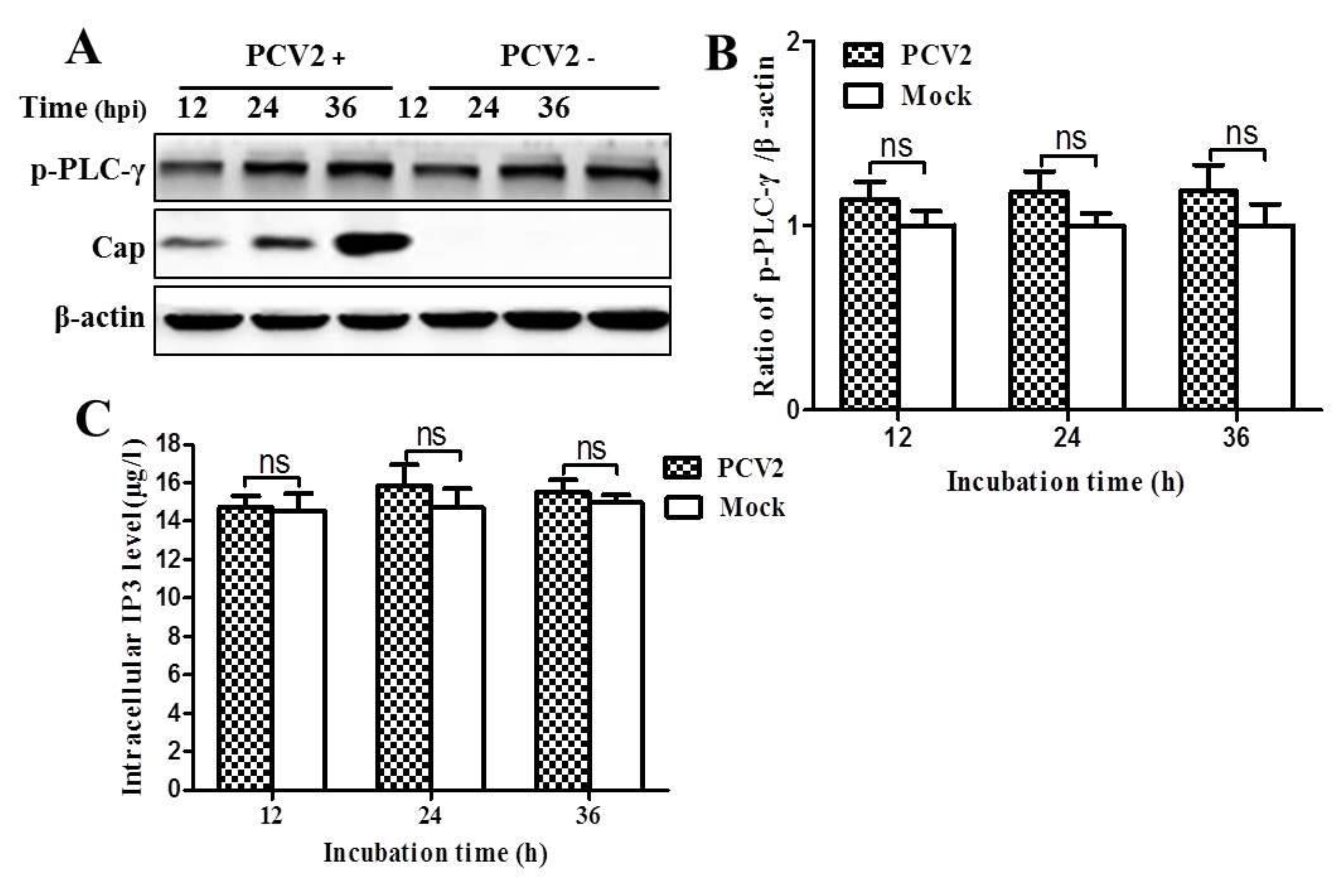
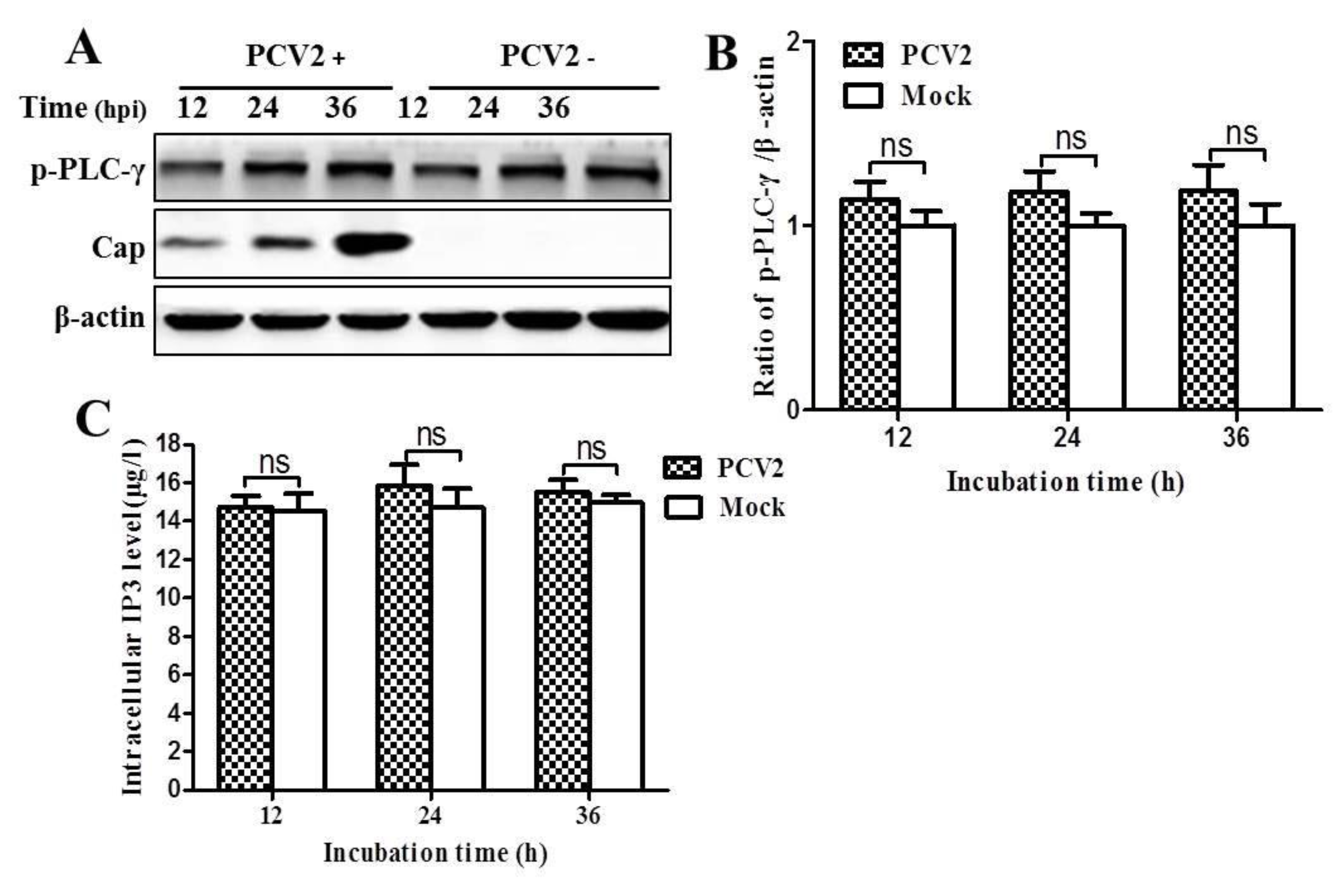
3.3. PCV2 Infection Increases Cytosolic Ca2+ Release from the ER via IP3R to Activate CaMKKβ
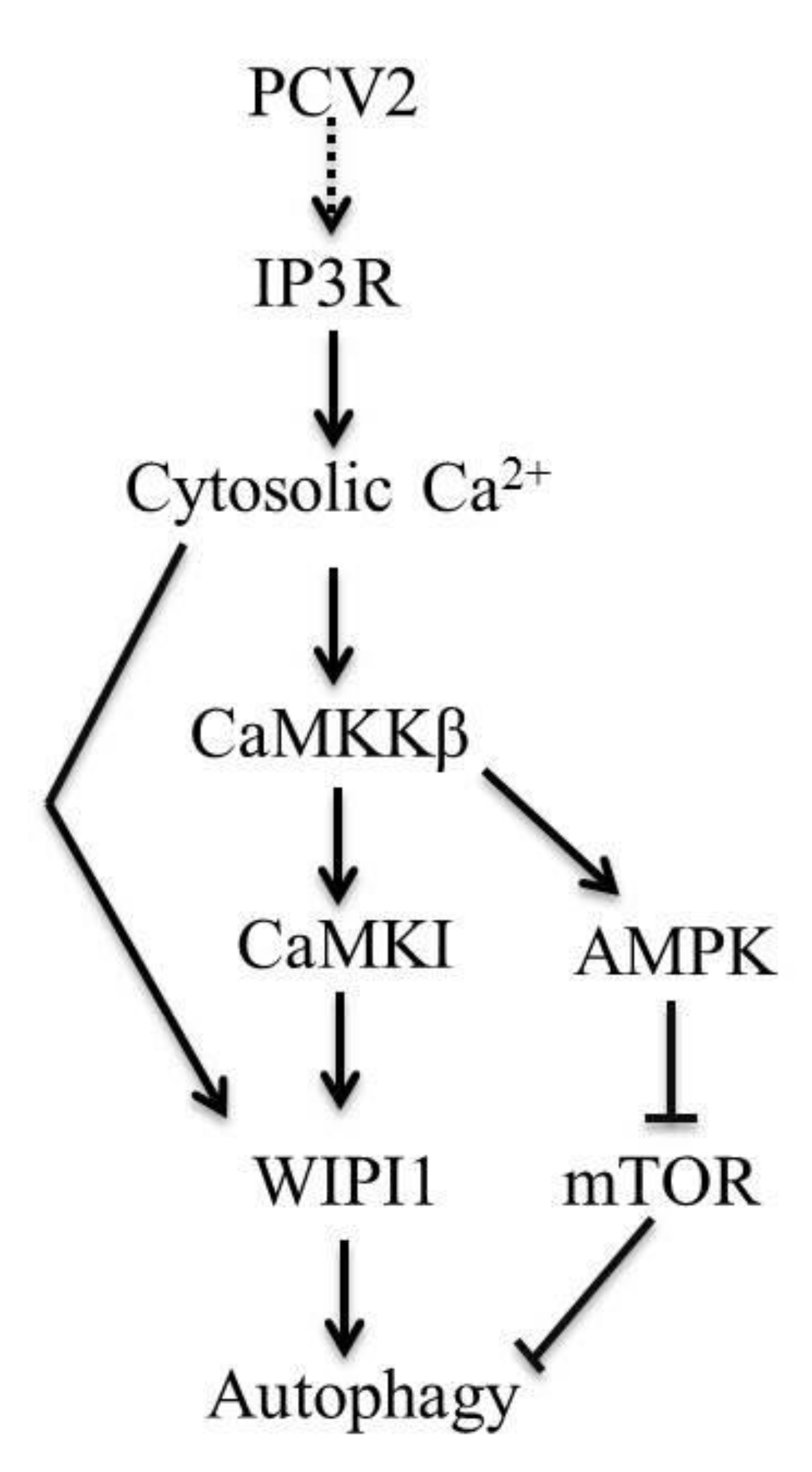
4. Discussion
4.1. AMPK Activation Is Due to Activation of CaMKKβ as a Result of Increased Cytosolic Ca2+ in PCV2-Infected Cells
4.2. CaMKI and WIPI1 Are Activated by CaMKKβ in PCV2-Infected Cells and Involved in Autophagy
4.3. CaMKKβ Is Involved in Positive Regulation of PCV2 Replication
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ellis, J.; Hassard, L.; Clark, E.; Harding, J.; Allan, G.; Willson, P.; Strokappe, J.; Martin, K.; McNeilly, F.; Meehan, B.; et al. Isolation of circovirus from lesions of pigs with postweaning multisystemic wasting syndrome. Can. Vet. J. 1998, 39, 44–51. [Google Scholar] [PubMed]
- Meng, X.J. Porcine circovirus type 2 (PCV2): Pathogenesis and interaction with the immune system. Ann. Rev. Anim. Biosci. 2013, 1, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K. Transcriptional analysis of porcine circovirus type 2. Virology 2003, 305, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K. Porcine circovirus: Transcription and DNA replication. Virus Res. 2012, 164, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Lekcharoensuk, P.; Morozov, I.; Paul, P.S.; Thangthumniyom, N.; Wajjawalku, W.; Meng, X.J. Epitope mapping of the major capsid protein of type 2 porcine circovirus (PCV2) by using chimeric PCV1 and PCV2. J. Virol. 2004, 78, 8135–8145. [Google Scholar] [CrossRef] [PubMed]
- Khayat, R.; Brunn, N.; Speir, J.A.; Hardham, J.M.; Ankenbauer, R.G.; Schneemann, A.; Johnson, J.E. The 2.3-angstrom structure of porcine circovirus 2. J. Virol. 2011, 85, 7856–7862. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Xu, F.; Li, J.; Shuai, J.; Li, X.; Fang, W. Porcine circovirus type 2 explores the autophagic machinery for replication in pk-15 cells. Virus Res. 2012, 163, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Walia, R.; Dardari, R.; Chaiyakul, M.; Czub, M. Porcine circovirus-2 capsid protein induces cell death in PK15 cells. Virology 2014, 468–470, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, I.; Kwang, J. Characterization of a previously unidentified viral protein in porcine circovirus type 2-infected cells and its role in virus-induced apoptosis. J. Virol. 2005, 79, 8262–8274. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Cao, J.; Zhou, N.; Jin, Y.; Wu, J.; Zhou, J. Identification and functional analysis of the novel orf4 protein encoded by porcine circovirus type 2. J. Virol. 2013, 87, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Guo, K.; Xu, H.; Wang, T.; Zhang, Y. Identification of putative ORF5 protein of porcine circovirus type 2 and functional analysis of GFP-fused ORF5 protein. PloS ONE 2015, 10, e0127859. [Google Scholar]
- Klausmann, S.; Sydler, T.; Summerfield, A.; Lewis, F.I.; Weilenmann, R.; Sidler, X.; Brugnera, E. T-cell reprogramming through targeted CD4-coreceptor and t-cell receptor expression on maturing thymocytes by latent circoviridae family member porcine circovirus type 2 cell infections in the thymus. Emerg. Microbes Infect. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Sydler, T.; Bragger, S.; Handke, M.; Hartnack, S.; Lewis, F.I.; Sidler, X.; Brugnera, E. Latent porcine circovirus type 2-infected domestic pigs: A potential infection model for the effective development of vaccines against latent or chronic virus induced diseases. Vaccine 2016, 34, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479–480, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Chiramel, A.I.; Brady, N.R.; Bartenschlager, R. Divergent roles of autophagy in virus infection. Cells 2013, 2, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Ghislat, G.; Knecht, E. Ca2+-sensor proteins in the autophagic and endocytic traffic. Curr. Protein Pept. Sci. 2013, 14, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Bultynck, G.; Parys, J.B. A dual role for Ca(2+) in autophagy regulation. Cell Calcium 2011, 50, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef] [PubMed]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Grotemeier, A.; Alers, S.; Pfisterer, S.G.; Paasch, F.; Daubrawa, M.; Dieterle, A.; Viollet, B.; Wesselborg, S.; Proikas-Cezanne, T.; Stork, B. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell. Signal. 2010, 22, 914–925. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, S.G.; Mauthe, M.; Codogno, P.; Proikas-Cezanne, T. Ca2+/calmodulin-dependent kinase (CaMK) signaling via CaMKI and AMP-activated protein kinase contributes to the regulation of WIPI-1 at the onset of autophagy. Mol. Pharmacol. 2011, 80, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Proikas-Cezanne, T.; Takacs, Z.; Donnes, P.; Kohlbacher, O. WIPI proteins: Essential PtdIns3P effectors at the nascent autophagosome. J. Cell Sci. 2015, 128, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; Proikas-Cezanne, T. Function of human WIPI proteins in autophagosomal rejuvenation of endomembranes? FEBS Lett. 2015, 589, 1546–1551. [Google Scholar] [CrossRef] [PubMed]
- Tsuyuki, S.; Takabayashi, M.; Kawazu, M.; Kudo, K.; Watanabe, A.; Nagata, Y.; Kusama, Y.; Yoshida, K. Detection of WIPI1 mRNA as an indicator of autophagosome formation. Autophagy 2014, 10, 497–513. [Google Scholar] [CrossRef] [PubMed]
- Proikas-Cezanne, T.; Ruckerbauer, S.; Stierhof, Y.D.; Berg, C.; Nordheim, A. Human WIPI-1 puncta-formation: A novel assay to assess mammalian autophagy. FEBS Lett. 2007, 581, 3396–3404. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Frey, T.K.; Yang, J.J. Viral calciomics: Interplays between Ca2+ and virus. Cell Calcium 2009, 46, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Torres, J.; Liu, D.X. The emerging roles of viroporins in ER stress response and autophagy induction during virus infection. Viruses 2015, 7, 2834–2857. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Harry, B.L.; Zhou, Q.; Skeen-Gaar, R.R.; Ge, X.; Lee, E.S.; Mitani, S.; Xue, D. Hepatitis B virus x protein targets the Bcl-2 protein CED-9 to induce intracellular ca2+ increase and cell death in Caenorhabditis elegans. Proc. Nat. Acad. Sci. USA 2012, 109, 18465–18470. [Google Scholar] [CrossRef] [PubMed]
- Scherbik, S.V.; Brinton, M.A. Virus-induced Ca2+ influx extends survival of West Nile virus-infected cells. J. Virol. 2010, 84, 8721–8731. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Hyser, J.M.; Utama, B.; Estes, M.K. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-beta signaling is required for rotavirus replication. Proc. Nat. Acad. Sci. USA 2012, 109, E3405–E3413. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Dai, L.; Han, H.; Zhang, S. PCV2 induces apoptosis and modulates calcium homeostasis in piglet lymphocytes in vitro. Res. Vet. Sci. 2012, 93, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Zhou, Y.; Xu, F.; Shuai, J.; Li, X.; Fang, W. Porcine circovirus type 2 induces autophagy via the AMPK/ERK/TSC2/mTOR signaling pathway in PK-15 cells. J. Virol. 2012, 86, 12003–12012. [Google Scholar] [CrossRef] [PubMed]
- Mank, M.; Santos, A.F.; Direnberger, S.; Mrsic-Flogel, T.D.; Hofer, S.B.; Stein, V.; Hendel, T.; Reiff, D.F.; Levelt, C.; Borst, A.; et al. A genetically encoded calcium indicator for chronic in vivo two-photon imaging. Nat. Methods 2008, 5, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.C.; Ho, M.Y.; Liang, S.M.; Liang, C.M. Recombinant protein rVP1 upregulates BECN1-independent autophagy, MAPK1/3 phosphorylation and MMP9 activity via WIPI1/WIPI2 to promote macrophage migration. Autophagy 2013, 9, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Kadamur, G.; Ross, E.M. Mammalian phospholipase c. Ann. Rev. Hysiol. 2013, 75, 127–154. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Chisari, F.V. Viruses and the autophagy machinery. Cell Cycle 2010, 9, 1295–1307. [Google Scholar] [CrossRef] [PubMed]
- Mankouri, J.; Harris, M. Viruses and the fuel sensor: The emerging link between AMPK and virus replication. Rev. Med. Virol. 2011, 21, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Carling, D.; Viollet, B. Beyond energy homeostasis: The expanding role of AMP-activated protein kinase in regulating metabolism. Cell Metab. 2015, 21, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Wayman, G.A.; Tokumitsu, H.; Davare, M.A.; Soderling, T.R. Analysis of CaM-kinase signaling in cells. Cell Calcium 2011, 50, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Racioppi, L.; Means, A.R. Calcium/calmodulin-dependent protein kinase kinase 2: Roles in signaling and pathophysiology. J. Biol. Chem. 2012, 287, 31658–31665. [Google Scholar] [CrossRef] [PubMed]
- Moser, T.S.; Schieffer, D.; Cherry, S. AMP-activated kinase restricts Rift Valley fever virus infection by inhibiting fatty acid synthesis. PLoS Pathog. 2012, 8, e1002661. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.; Lo, K.W.; Ko, C.W.; Young, L.S.; Dawson, C.W. Inhibition of the LKB1-AMPK pathway by the Epstein-Barr virus-encoded LMP1 promotes proliferation and transformation of human nasopharyngeal epithelial cells. J. Pathol. 2013, 230, 336–346. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, Y.; Qi, B.; Zhou, Y.; Jiang, X.; Zhang, X.; Li, X.; Fang, W. Porcine Circovirus Type 2 Activates CaMMKβ to Initiate Autophagy in PK-15 Cells by Increasing Cytosolic Calcium. Viruses 2016, 8, 135. https://doi.org/10.3390/v8050135
Gu Y, Qi B, Zhou Y, Jiang X, Zhang X, Li X, Fang W. Porcine Circovirus Type 2 Activates CaMMKβ to Initiate Autophagy in PK-15 Cells by Increasing Cytosolic Calcium. Viruses. 2016; 8(5):135. https://doi.org/10.3390/v8050135
Chicago/Turabian StyleGu, Yuanxing, Baozhu Qi, Yingshan Zhou, Xiaowu Jiang, Xian Zhang, Xiaoliang Li, and Weihuan Fang. 2016. "Porcine Circovirus Type 2 Activates CaMMKβ to Initiate Autophagy in PK-15 Cells by Increasing Cytosolic Calcium" Viruses 8, no. 5: 135. https://doi.org/10.3390/v8050135
APA StyleGu, Y., Qi, B., Zhou, Y., Jiang, X., Zhang, X., Li, X., & Fang, W. (2016). Porcine Circovirus Type 2 Activates CaMMKβ to Initiate Autophagy in PK-15 Cells by Increasing Cytosolic Calcium. Viruses, 8(5), 135. https://doi.org/10.3390/v8050135