
Viral Evasion of Natural Killer Cell Activation

Abstract
:1. Introduction
2. NKG2D-Mediated Evasion
2.1. Viral Protein-Based Inhibition of NKG2D Ligands
2.2. Viral MiRNA-Based Inhibition of NKG2D Ligands
2.3. Soluble NKG2D Ligands
2.4. Cytokine-Mediated Inhibition of NKG2D Expression
3. NCR-Mediated Evasion
4. Evasion of NK Cell Adaptive Responses
5. Evasion of NK Cell Activation by γ-Herpesvirus
6. Future Perspective
7. Open Questions
- (1)
- Different viruses employ distinct strategies for immune evasion, and it remains to be determined whether there are additional mechanisms in other viruses to evade NK cell activation and avoid destruction. Additionally, the molecular details of the innate and adaptive functions of NK cells remain limited. Further investigation may reveal additional novel strategies for viral evasion of NK cell immunity.
- (2)
- Although a large number of studies have confirmed viral evasion of adaptive and innate immune responses, the mechanisms of the viral evasion of the NK cell adaptive immune response still largely remain unknown. More studies are required to reveal whether viruses can avoid NK cell memory-like adaptive functions and, if so, how this process works.
- (3)
- It is worth investigating additional viral effects on NKG2D and ligands in NKT- and T cell-based immune responses. NKG2D is not only expressed on NK cells but also on NKT cells and T cells, and modulation of NKG2D and ligands may contribute broadly to viral evasion.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moretta, A.; Marcenaro, E.; Parolini, S.; Ferlazzo, G.; Moretta, L. NK cells at the interface between innate and adaptive immunity. Cell Death Differ. 2008, 15, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Cretney, E.; Kelly, J.M.; Westwood, J.A.; Street, S.E.; Yagita, H.; Takeda, K.; van Dommelen, S.L.; Degli-Esposti, M.A.; Hayakawa, Y. Activation of NK cell cytotoxicity. Mol. Immunol. 2005, 42, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Arnon, T.I.; Markel, G.; Mandelboim, O. Tumor and viral recognition by natural killer cells receptors. Semin. Cancer Biol. 2006, 16, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Watzl, C.; Long, E.O. Signal transduction during activation and inhibition of natural killer cells. Curr. Protoc. Immunol. 2010. [Google Scholar] [CrossRef]
- Orr, M.T.; Lanier, L.L. Natural killer cell education and tolerance. Cell 2010, 142, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.H.; Bouvier, M. MHC class I antigen presentation: Learning from viral evasion strategies. Nat. Rev. Immunol. 2009, 9, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Eagle, R.A.; Traherne, J.A.; Hair, J.R.; Jafferji, I.; Trowsdale, J. ULBP6/RAET1L is an additional human NKG2D ligand. Eur. J. Immunol. 2009, 39, 3207–3216. [Google Scholar] [CrossRef] [PubMed]
- Kadri, N.; Thanh, T.L.; Hoglund, P. Selection, tuning, and adaptation in mouse NK cell education. Immunol. Rev. 2015, 267, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Shifrin, N.; Raulet, D.H.; Ardolino, M. NK cell self tolerance, responsiveness and missing self recognition. Semin. Immunol. 2014, 26, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Pyzik, M.; Dumaine, A.; Charbonneau, B.; Fodil-Cornu, N.; Jonjic, S.; Vidal, S.M. Viral MHC class i-like molecule allows evasion of NK cell effector responses in vivo. J. Immunol. 2014, 193, 6061–6069. [Google Scholar] [CrossRef] [PubMed]
- Forbes, C.A.; Scalzo, A.A.; Degli-Esposti, M.A.; Coudert, J.D. Ly49C-dependent control of mcmv infection by NK cells is cis-regulated by MHC class I molecules. PLoS Pathog. 2014, 10, e1004161. [Google Scholar] [CrossRef] [PubMed]
- Kulpa, D.A.; Collins, K.L. The emerging role of HLA-C in HIV-1 infection. Immunology 2011, 134, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Fadda, L.; Korner, C.; Kumar, S.; van Teijlingen, N.H.; Piechocka-Trocha, A.; Carrington, M.; Altfeld, M. HLA-CW * 0102-restricted HIV-1 P24 epitope variants can modulate the binding of the inhibitory KIR2DL2 receptor and primary NK cell function. PLoS Pathog. 2012, 8, e1002805. [Google Scholar] [CrossRef] [PubMed]
- Heatley, S.L.; Pietra, G.; Lin, J.; Widjaja, J.M.; Harpur, C.M.; Lester, S.; Rossjohn, J.; Szer, J.; Schwarer, A.; Bradstock, K.; et al. Polymorphism in human cytomegalovirus UL40 impacts on recognition of human leukocyte antigen-E (HLA-E) by natural killer cells. J. Biol. Chem. 2013, 288, 8679–8690. [Google Scholar] [CrossRef] [PubMed]
- Holzemer, A.; Thobakgale, C.F.; Jimenez Cruz, C.A.; Garcia-Beltran, W.F.; Carlson, J.M.; van Teijlingen, N.H.; Mann, J.K.; Jaggernath, M.; Kang, S.G.; Korner, C.; et al. Selection of an HLA-C * 03:04-restricted HIV-1 P24 gag sequence variant is associated with viral escape from KIR2DL3+ natural killer cells: Data from an observational cohort in South Africa. PLoS Med. 2015, 12, e1001900. [Google Scholar] [CrossRef] [PubMed]
- Champsaur, M.; Lanier, L.L. Effect of NKG2D ligand expression on host immune responses. Immunol. Rev. 2010, 235, 267–285. [Google Scholar] [CrossRef] [PubMed]
- Campbell, T.M.; McSharry, B.P.; Steain, M.; Slobedman, B.; Abendroth, A. Varicella-zoster virus and herpes simplex virus 1 differentially modulate NKG2D ligand expression during productive infection. J. Virol. 2015, 89, 7932–7943. [Google Scholar] [CrossRef] [PubMed]
- Cosman, D.; Müllberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. Ulbps, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Rolle, A.; Mousavi-Jazi, M.; Eriksson, M.; Odeberg, J.; Soderberg-Naucler, C.; Cosman, D.; Karre, K.; Cerboni, C. Effects of human cytomegalovirus infection on ligands for the activating NKG2D receptor of NK cells: Up-regulation of UL16-binding protein (ULBP)1 and ULBP2 is counteracted by the viral UL16 protein. J. Immunol. 2003, 171, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Chalupny, N.J.; Rein-Weston, A.; Dosch, S.; Cosman, D. Down-regulation of the NKG2D ligand mica by the human cytomegalovirus glycoprotein UL142. Biochem. Biophys. Res. Commun. 2006, 346, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Bennett, N.J.; Ashiru, O.; Morgan, F.J.; Pang, Y.; Okecha, G.; Eagle, R.A.; Trowsdale, J.; Sissons, J.G.; Wills, M.R. Intracellular sequestration of the NKG2D ligand ULBP3 by human cytomegalovirus. J. Immunol. 2010, 185, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Seidel, E.; Le, V.T.; Bar-On, Y.; Tsukerman, P.; Enk, J.; Yamin, R.; Stein, N.; Schmiedel, D.; Oiknine Djian, E.; Weisblum, Y.; et al. Dynamic co-evolution of host and pathogen: HCMV downregulates the prevalent allele MICA * 008 to escape elimination by NK cells. Cell Rep. 2015, 10, 968–982. [Google Scholar] [CrossRef] [PubMed]
- Fielding, C.A.; Aicheler, R.; Stanton, R.J.; Wang, E.C.; Han, S.; Seirafian, S.; Davies, J.; McSharry, B.P.; Weekes, M.P.; Antrobus, P.R.; et al. Two novel human cytomegalovirus NK cell evasion functions target MICA for lysosomal degradation. PLoS Pathog. 2014, 10, e1004058. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.L.; Hudson, A.W. The human herpesvirus-7 (HHV-7) U21 immunoevasin subverts NK-mediated cytoxicity through modulation of MICA and MICB. PLoS Pathog. 2011, 7, e1002362. [Google Scholar] [CrossRef] [PubMed]
- Rancan, C.; Schirrmann, L.; Huls, C.; Zeidler, R.; Moosmann, A. Latent membrane protein LMP2A impairs recognition of ebv-infected cells by CD8+ T cells. PLoS Pathog. 2015, 11, e1004906. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Boname, J.M.; Field, S.; Nejentsev, S.; Salio, M.; Cerundolo, V.; Wills, M.; Lehner, P.J. Down-regulation of NKG2D and NKp80 ligands by kaposi’s sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 1656–1661. [Google Scholar] [CrossRef] [PubMed]
- McSharry, B.P.; Burgert, H.G.; Owen, D.P.; Stanton, R.J.; Prod’homme, V.; Sester, M.; Koebernick, K.; Groh, V.; Spies, T.; Cox, S.; et al. Adenovirus E3/19K promotes evasion of NK cell recognition by intracellular sequestration of the NKG2D ligands major histocompatibility complex class I chain-related proteins A and B. J. Virol. 2008, 82, 4585–4594. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, X.J.; Shi, K.Q.; Chen, Y.P.; Ren, Y.F.; Song, Y.J.; Li, G.; Xue, Y.F.; Fang, Y.X.; Deng, Z.J.; et al. Hepatitis B surface antigen inhibits MICA and MICB expression via induction of cellular miRNAs in hepatocellular carcinoma cells. Carcinogenesis 2014, 35, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Cerboni, C.; Neri, F.; Casartelli, N.; Zingoni, A.; Cosman, D.; Rossi, P.; Santoni, A.; Doria, M. Human immunodeficiency virus 1 Nef protein downmodulates the ligands of the activating receptor NKG2D and inhibits natural killer cell-mediated cytotoxicity. J. Gen. Virol. 2007, 88, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Matusali, G.; Potesta, M.; Santoni, A.; Cerboni, C.; Doria, M. The human immunodeficiency virus type 1 Nef and Vpu proteins downregulate the natural killer cell-activating ligand PVR. J. Virol. 2012, 86, 4496–4504. [Google Scholar] [CrossRef] [PubMed]
- Sowrirajan, B.; Barker, E. The natural killer cell cytotoxic function is modulated by HIV-1 accessory proteins. Viruses 2011, 3, 1091–1111. [Google Scholar] [CrossRef] [PubMed]
- Bolduan, S.; Hubel, P.; Reif, T.; Lodermeyer, V.; Hohne, K.; Fritz, J.V.; Sauter, D.; Kirchhoff, F.; Fackler, O.T.; Schindler, M.; et al. HIV-1 Vpu affects the anterograde transport and the glycosylation pattern of NTB-A. Virology 2013, 440, 190–203. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shah, A.H.; Sowrirajan, B.; Davis, Z.B.; Ward, J.P.; Campbell, E.M.; Planelles, V.; Barker, E. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe 2010, 8, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Bose, S.K.; Meyer, K.; Ray, R. Hepatitis c virus impairs natural killer cell-mediated augmentation of complement synthesis. J. Virol. 2014, 88, 2564–2571. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.C.; Lim, J.B.; Park, J.H.; Lee, J.M. Cell-to-cell contact with hepatitis C virus-infected cells reduces functional capacity of natural killer cells. J. Virol. 2011, 85, 12557–12569. [Google Scholar] [CrossRef] [PubMed]
- Jensen, H.; Andresen, L.; Nielsen, J.; Christensen, J.P.; Skov, S. Vesicular stomatitis virus infection promotes immune evasion by preventing NKG2D-ligand surface expression. PLoS ONE 2011, 6, e23023. [Google Scholar] [CrossRef] [PubMed]
- Muntasell, A.; Magri, G.; Pende, D.; Angulo, A.; Lopez-Botet, M. Inhibition of NKG2D expression in NK cells by cytokines secreted in response to human cytomegalovirus infection. Blood 2010, 115, 5170–5179. [Google Scholar] [CrossRef] [PubMed]
- Sene, D.; Levasseur, F.; Abel, M.; Lambert, M.; Camous, X.; Hernandez, C.; Pene, V.; Rosenberg, A.R.; Jouvin-Marche, E.; Marche, P.N.; et al. Hepatitis C virus (HCV) evades NKG2D-dependent NK cell responses through NS5A-mediated imbalance of inflammatory cytokines. PLoS Pathog. 2010, 6, e1001184. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Fu, B.; Gao, Y.; Liao, X.; Sun, R.; Tian, Z.; Wei, H. TGF-beta1 down-regulation of NKG2D/DAP10 and 2B4/sap expression on human NK cells contributes to hbv persistence. PLoS Pathog. 2012, 8, e1002594. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, S.; Lambert, M.; Zucman, D.; Choukem, S.P.; Tognarelli, S.; Pages, C.; Lebbe, C.; Caillat-Zucman, S. Human herpesvirus 8 (HHV8) sequentially shapes the NK cell repertoire during the course of asymptomatic infection and kaposi sarcoma. PLoS Pathog. 2012, 8, e1002486. [Google Scholar] [CrossRef] [PubMed]
- Stern-Ginossar, N.; Elefant, N.; Zimmermann, A.; Wolf, D.G.; Saleh, N.; Biton, M.; Horwitz, E.; Prokocimer, Z.; Prichard, M.; Hahn, G.; et al. Host immune system gene targeting by a viral miRNA. Science 2007, 317, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Stern-Ginossar, N.; Sarid, R.; Mandelboim, O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 2009, 5, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Bauman, Y.; Nachmani, D.; Vitenshtein, A.; Tsukerman, P.; Drayman, N.; Stern-Ginossar, N.; Lankry, D.; Gruda, R.; Mandelboim, O. An identical miRNA of the human JC and BK polyoma viruses targets the stress-induced ligand ULBP3 to escape immune elimination. Cell Host Microbe 2011, 9, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.A.; Trossman, D.S.; Yokoyama, W.M.; Carayannopoulos, L.N. Zoonotic orthopoxviruses encode a high-affinity antagonist of NKG2D. J. Exp. Med. 2007, 204, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Matusali, G.; Tchidjou, H.K.; Pontrelli, G.; Bernardi, S.; D’Ettorre, G.; Vullo, V.; Buonomini, A.R.; Andreoni, M.; Santoni, A.; Cerboni, C.; et al. Soluble ligands for the NKG2D receptor are released during HIV-1 infection and impair NKG2D expression and cytotoxicity of NK cells. FASEB J. 2013, 27, 2440–2450. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Zocher, G.; Steinle, A.; Stehle, T. Structure of the HCMV UL16-MICB complex elucidates select binding of a viral immunoevasin to diverse NKG2D ligands. PLoS Pathog. 2010, 6, e1000723. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. Viruses and microRNAs. Nat. Genet. 2006, 38, S25–S30. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and host interactions. Ann. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Kishikawa, T.; Otsuka, M.; Ohno, M.; Yoshikawa, T.; Sato, M.; Koike, K. Development of a screening method to identify regulators of mica shedding. Biochem. Biophys. Res. Commun. 2015, 465, 764–768. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.; Steinle, A.; Watzl, C.; Mandelboim, O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. 2013, 34, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Arnon, T.I.; Achdout, H.; Levi, O.; Markel, G.; Saleh, N.; Katz, G.; Gazit, R.; Gonen-Gross, T.; Hanna, J.; Nahari, E.; et al. Inhibition of the NKp30 activating receptor by PP65 of human cytomegalovirus. Nat. Immunol. 2005, 6, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Jarahian, M.; Fiedler, M.; Cohnen, A.; Djandji, D.; Hammerling, G.J.; Gati, C.; Cerwenka, A.; Turner, P.C.; Moyer, R.W.; Watzl, C.; et al. Modulation of NKp30- and NKp46-mediated natural killer cell responses by poxviral hemagglutinin. PLoS Pathog. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Tu, W.; Liu, Y.; Qin, G.; Zheng, J.; Chan, P.L.; Lam, K.T.; Peiris, J.S.; Lau, Y.L. Inhibition of human natural killer cell activity by influenza virions and hemagglutinin. J. Virol. 2010, 84, 4148–4157. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, Y.; Seidel, E.; Tsukerman, P.; Mandelboim, M.; Mandelboim, O. Influenza virus uses its neuraminidase protein to evade the recognition of two activating NK cell receptors. J. Infect. Dis. 2014, 210, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, Y.; Glasner, A.; Meningher, T.; Achdout, H.; Gur, C.; Lankry, D.; Vitenshtein, A.; Meyers, A.F.A.; Mandelboim, M.; Mandelboim, O. Neuraminidase-mediated, NKp46-dependent immune-evasion mechanism of influenza viruses. Cell Rep. 2013, 3, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Madrid, A.S.; Ganem, D. Kaposi’s sarcoma-associated herpesvirus ORF54/dUTPase downregulates a ligand for the NK activating receptor NKp44. J. Virol. 2012, 86, 8693–8704. [Google Scholar] [CrossRef] [PubMed]
- Fausther-Bovendo, H.; Sol-Foulon, N.; Candotti, D.; Agut, H.; Schwartz, O.; Debre, P.; Vieillard, V. HIV escape from natural killer cytotoxicity: Nef inhibits NKp44l expression on CD4+ T cells. AIDS 2009, 23, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Holder, K.A.; Stapleton, S.N.; Gallant, M.E.; Russell, R.S.; Grant, M.D. Hepatitis c virus-infected cells downregulate NKp30 and inhibit ex vivo NK cell functions. J. Immunol. 2013, 191, 3308–3318. [Google Scholar] [CrossRef] [PubMed]
- Glasner, A.; Zurunic, A.; Meningher, T.; Lenac Rovis, T.; Tsukerman, P.; Bar-On, Y.; Yamin, R.; Meyers, A.F.; Mandeboim, M.; Jonjic, S.; et al. Elucidating the mechanisms of influenza virus recognition by NCR1. PLoS ONE 2012, 7, e36837. [Google Scholar] [CrossRef] [PubMed]
- Mandelboim, O.; Lieberman, N.; Lev, M.; Paul, L.; Arnon, T.I.; Bushkin, Y.; Davis, D.M.; Strominger, J.L.; Yewdell, J.W.; Porgador, A. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 2001, 409, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.D.; Alder, M.N. The evolution of adaptive immune systems. Cell 2006, 124, 815–822. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, J.G.; Goodarzi, M.; Drayton, D.L.; von Andrian, U.H. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat. Immunol. 2006, 7, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Min-Oo, G.; Kamimura, Y.; Hendricks, D.W.; Nabekura, T.; Lanier, L.L. Natural killer cells: Walking three paths down memory lane. Trends Immunol. 2013, 34, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Beilke, J.N.; Lanier, L.L. Adaptive immune features of natural killer cells. Nature 2009, 457, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.T.; Sun, J.C.; Hesslein, D.G.; Arase, H.; Phillips, J.H.; Takai, T.; Lanier, L.L. Ly49H signaling through DAP10 is essential for optimal natural killer cell responses to mouse cytomegalovirus infection. J. Exp. Med. 2009, 206, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Paust, S.; Gill, H.S.; Wang, B.-Z.; Flynn, M.P.; Moseman, E.A.; Senman, B.; Szczepanik, M.; Telenti, A.; Askenase, P.W.; Compans, R.W.; et al. Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat. Immunol. 2010, 11, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhang, T.; Hwang, I.; Kim, A.; Nitschke, L.; Kim, M.; Scott, J.M.; Kamimura, Y.; Lanier, L.L.; Kim, S. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 2015, 42, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Schlums, H.; Cichocki, F.; Tesi, B.; Theorell, J.; Beziat, V.; Holmes, T.D.; Han, H.; Chiang, S.C.; Foley, B.; Mattsson, K.; et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 2015, 42, 443–456. [Google Scholar] [CrossRef] [PubMed]
- French, A.R.; Pingel, J.T.; Wagner, M.; Bubic, I.; Yang, L.; Kim, S.; Koszinowski, U.; Jonjic, S.; Yokoyama, W.M. Escape of mutant double-stranded DNA virus from innate immune control. Immunity 2004, 20, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Voigt, V.; Forbes, C.A.; Tonkin, J.N.; Degli-Esposti, M.A.; Smith, H.R.; Yokoyama, W.M.; Scalzo, A.A. Murine cytomegalovirus M157 mutation and variation leads to immune evasion of natural killer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 13483–13488. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.E.; Johnson, L.R.; Kang, H.H.; Sun, J.C. BNIP3- and BNIP3l-mediated mitophagy promotes the generation of natural killer cell memory. Immunity 2015, 43, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.A.; Fehniger, T.A. Memory NK cells take out the (mitochondrial) garbage. Immunity 2015, 43, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.T. Viruses and the autophagy pathway. Virology 2015, 479–480, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.M.; Jung, J.U. Modulation of the autophagy pathway by human tumor viruses. Semin. Cancer Biol. 2013, 23, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Damania, B. Oncogenic gamma-herpesviruses: Comparison of viral proteins involved in tumorigenesis. Nat. Rev. Microbiol. 2004, 2, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Usherwood, E.J. Immune escape of gamma-herpesviruses from adaptive immunity. Rev. Med. Virol. 2014, 24, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Fukazawa, H.; Murakami, Y.; Takahashi, N.; Yamagoe, S.; Uehara, Y. Gamma-herpesviruses and cellular signaling in aids-associated malignancies. Cancer Sci. 2007, 98, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Pappworth, I.Y.; Wang, E.C.; Rowe, M. The switch from latent to productive infection in epstein-barr virus-infected B cells is associated with sensitization to NK cell killing. J. Virol. 2007, 81, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.R.; Quinn, L.L.; Rowe, M.; Zuo, J. Induction of the lytic cycle sensitizes epstein-barr virus-infected b cells to NK cell killing that is counteracted by virus-mediated NK cell evasion mechanisms in the late lytic cycle. J. Virol. 2015, 90, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Vosshenrich, C.A.; Di Santo, J.P. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat. Rev. Immunol. 2007, 7, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Di Santo, J.P. Humanized immune system (his) mice as a tool to study human NK cell development. Curr. Top. Microbiol. Immunol. 2008, 324, 109–124. [Google Scholar] [PubMed]
- Dorner, M.; Horwitz, J.A.; Robbins, J.B.; Barry, W.T.; Feng, Q.; Mu, K.; Jones, C.T.; Schoggins, J.W.; Catanese, M.T.; Burton, D.R.; et al. A genetically humanized mouse model for hepatitis C virus infection. Nature 2011, 474, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Imadome, K.; Takei, M. Modeling EBV infection and pathogenesis in new-generation humanized mice. Exp. Mol. Med. 2015, 47, e135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kovalev, G.I.; Su, L. HIV-1 infection and pathogenesis in a novel humanized mouse model. Blood 2007, 109, 2978–2981. [Google Scholar] [CrossRef] [PubMed]
- Slavuljica, I.; Busche, A.; Babic, M.; Mitrovic, M.; Gasparovic, I.; Cekinovic, D.; Markova Car, E.; Pernjak Pugel, E.; Cikovic, A.; Lisnic, V.J.; et al. Recombinant mouse cytomegalovirus expressing a ligand for the NKG2D receptor is attenuated and has improved vaccine properties. J. Clin. Investig. 2010, 120, 4532–4545. [Google Scholar] [CrossRef] [PubMed]
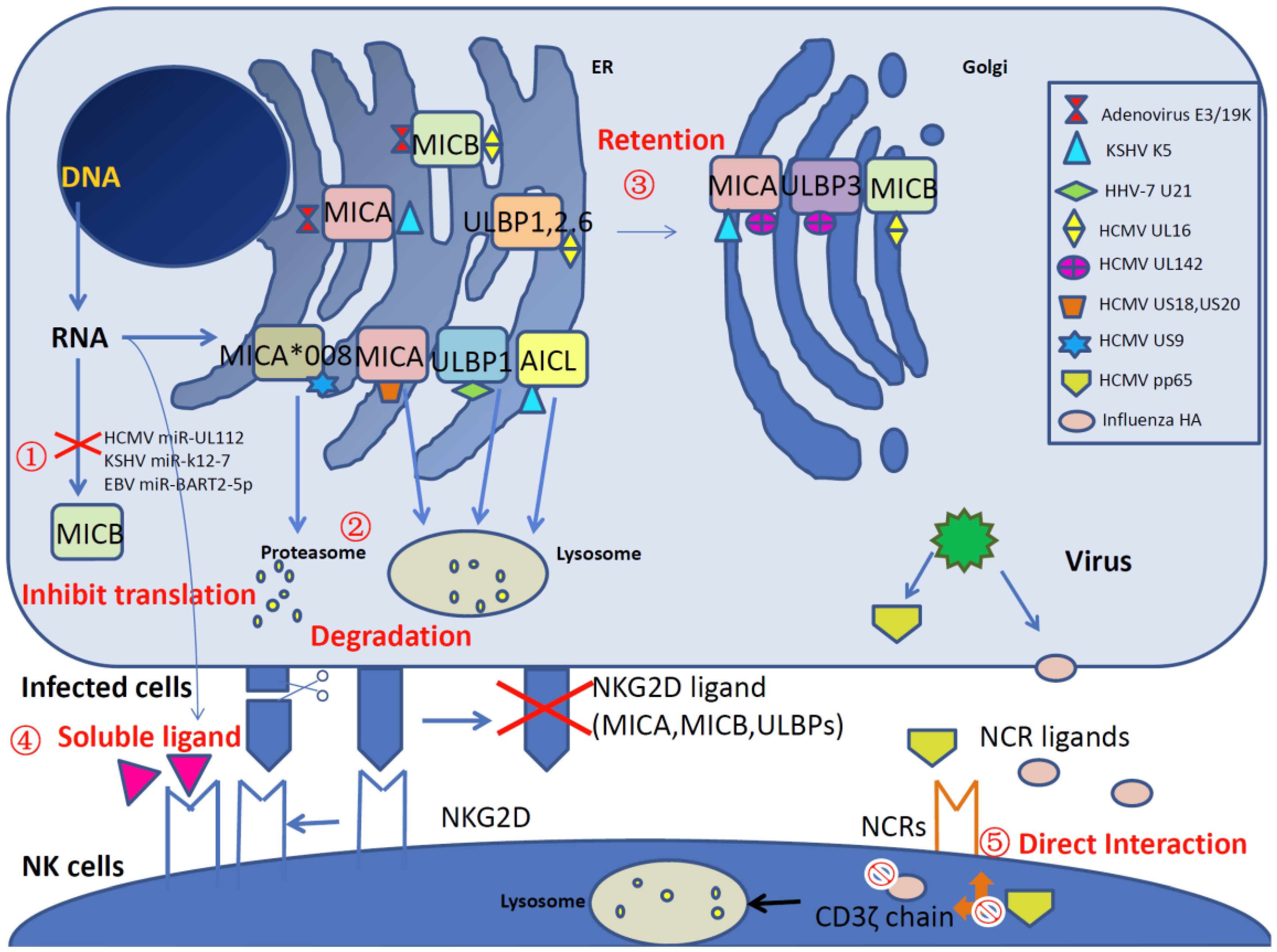

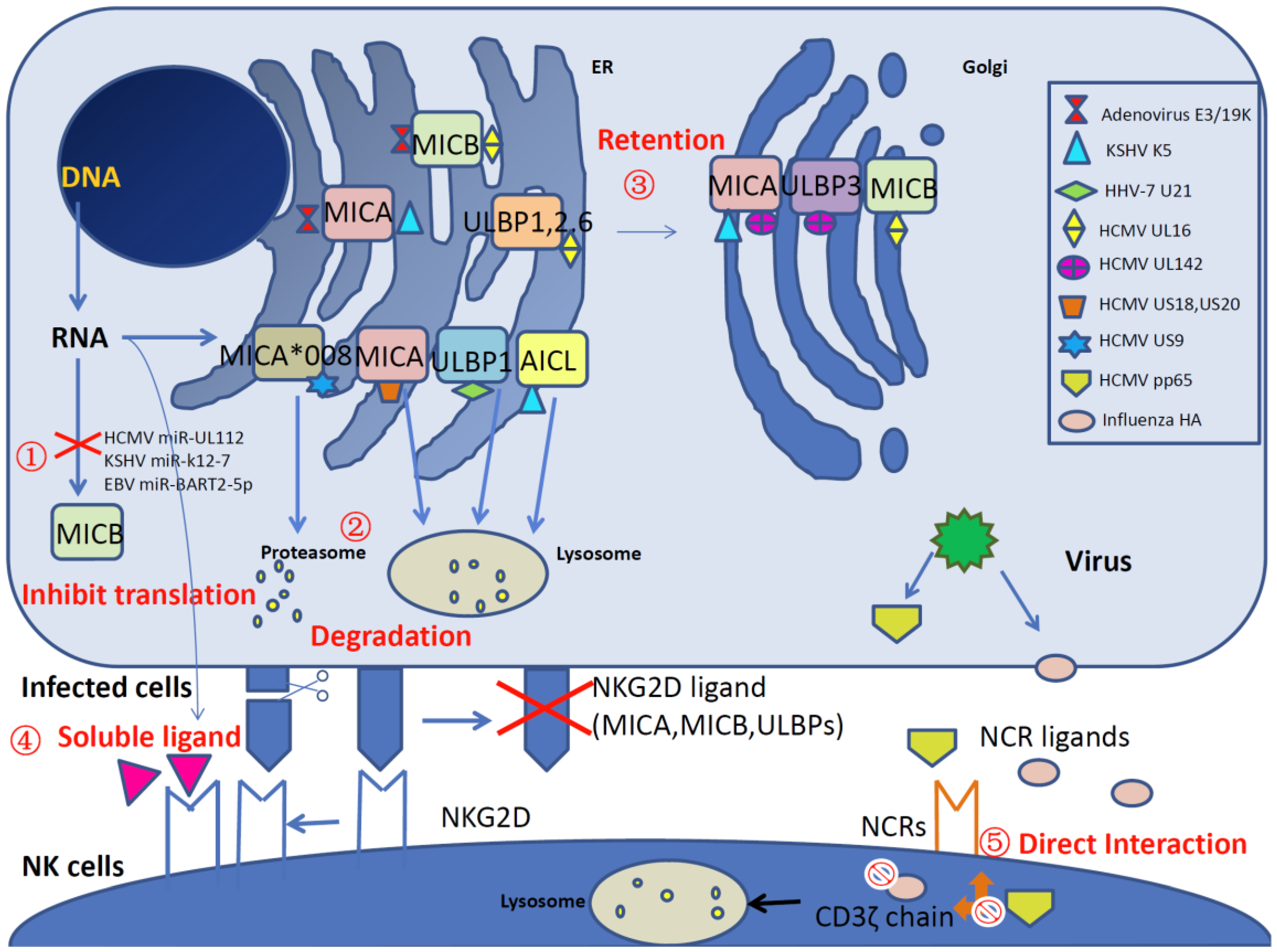{kind=link}
| Virus | Viral Product | Mechanisms | References |
|---|---|---|---|
| Viral proteins | |||
| HSV | ? | Decreases MICA, ULBP2, ULBP3 and ULBP1 on the cell surface | [17] |
| VZV | ? | Reduces ULBP2 and ULBP3 on the cell surface | [17] |
| HCMV | UL16 | Retains ULBP1, ULBP2, ULBP6 and MICB in the ER/cis-Golgi | [7,18,19] |
| UL142 | Retains ULBP3 and MICA in the cis-Golgi apparatus | [20,21] | |
| US9 | Induces MICA*008 proteasomal degradation | [22] | |
| US18, US20 | Induces MICA lysosomal degradation | [23] | |
| HHV-7 | U21 | Redirects ULBP1 to lysosomal degradation | [24] |
| Downregulates expression of MICA and MICB | |||
| EBV | LMP2A | Reduces the expression of MICA and ULBP4 | [25] |
| KSHV | K5 | Redistributes MICA to an intracellular compartment | [26] |
| Induces AICL endolysosomal degradation | [26] | ||
| Adenovirus | E3/19K | Retains MICA and MICB in the ER | [27] |
| HBV | HBsAg | Downregulates MICA and MICB by inducing human miRNAs | [28] |
| HIV | Nef | Downregulates the cell surface abundance of MICA, ULBP1 and ULBP2 | [29] |
| Vpu, Nef | Downregulates the expression of NTB-A and PVR | [30,31,32,33] | |
| HCV | NS2, NS5B | Downregulates MICA and MICB expression | [34] |
| ? | Downregulates NKG2D expression via cell-to-cell interaction | [35] | |
| VSV | ? | Suppresses MICA, MICB and ULBP2 expression | [36] |
| Cytokines and secretory molecules | |||
| HCMV | ? | Inhibits NKG2D/DAP10 expression through type I IFN and IL-12 | [37] |
| HCV | NS5A | Downregulates NKG2D expression through inducing IL-10-TGFβ | [38] |
| HBV | ? | Reduces NKG2D/DAP10 and 2B4/SAP expression through TGFβ | [39] |
| KSHV | ? | Downregulates NKG2D expression through PGE2 | [40] |
| Viral miRNA | |||
| HCMV | miR-UL112 | Inhibits MICB mRNA translation | [41] |
| EBV | miR-BART2-5p | Inhibits MICB mRNA translation | [42] |
| KSHV | miR-k12-7 | Inhibits MICB mRNA translation | [42] |
| JCV, BKV | 3p* miRNA | Inhibits ULBP3 mRNA translation | [43] |
| Soluble receptor and ligands | |||
| Zoonotic orthopoxviruses | OMCP | Secretes soluble NKG2D ligand | [44] |
| HIV | ? | Releases soluble NKG2D ligands via proteolytic shedding | [45] |
| Virus | Viral Product | Mechanisms | References |
|---|---|---|---|
| HCMV | pp65 | Inhibits the dissociation of NKp30 and CD3ζ chain | [51] |
| Poxvirus | HA | Inhibits NKp30-triggered activation | [52] |
| Influenza Virus | HA | Inhibits NKp46 through lysosomal degradation of CD3ζ chains | [53] |
| NA | Inhibits NKp44 and NKp46 recognition via the removal of sialic acid residues | [54,55] | |
| KSHV | ORF54/dUTPase | Inhibits the NKp44 ligand by interfering with intracellular trafficking | [56] |
| HIV | Nef | Inhibits the NKp44 ligand through intracellular retention | [57] |
| HCV | ? | Downregulates NKp30 expression in NK cells | [35,58] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Li, X.; Kuang, E. Viral Evasion of Natural Killer Cell Activation. Viruses 2016, 8, 95. https://doi.org/10.3390/v8040095
Ma Y, Li X, Kuang E. Viral Evasion of Natural Killer Cell Activation. Viruses. 2016; 8(4):95. https://doi.org/10.3390/v8040095
Chicago/Turabian StyleMa, Yi, Xiaojuan Li, and Ersheng Kuang. 2016. "Viral Evasion of Natural Killer Cell Activation" Viruses 8, no. 4: 95. https://doi.org/10.3390/v8040095
APA StyleMa, Y., Li, X., & Kuang, E. (2016). Viral Evasion of Natural Killer Cell Activation. Viruses, 8(4), 95. https://doi.org/10.3390/v8040095