
Roles of the Picornaviral 3C Proteinase in the Viral Life Cycle and Host Cells


Abstract
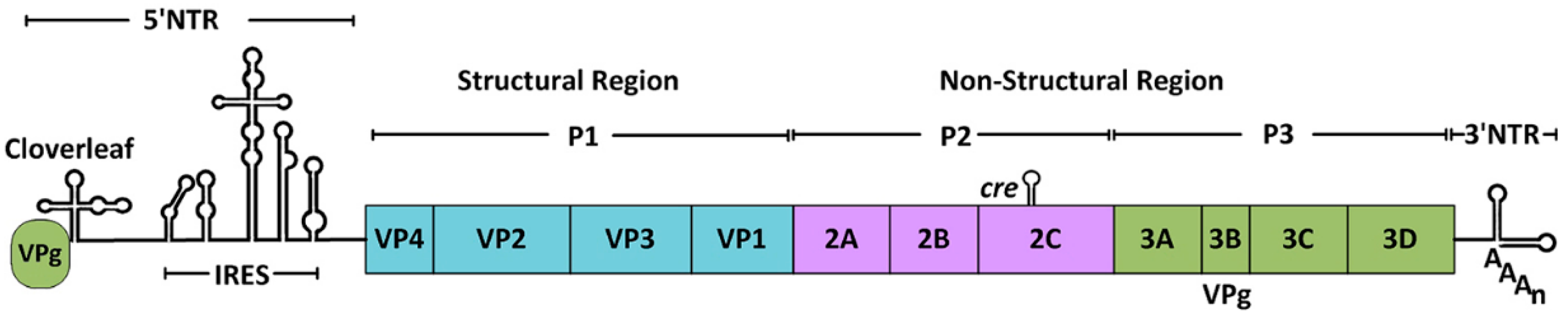
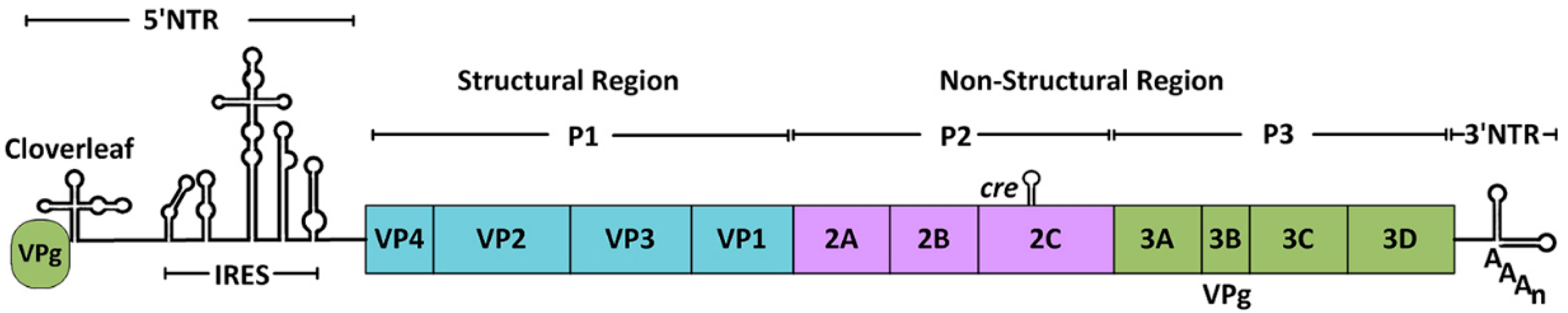
1. Introduction
2. The Mechanism of Proteolysis
3. The Role of 3Cpro in the Viral Life Cycle
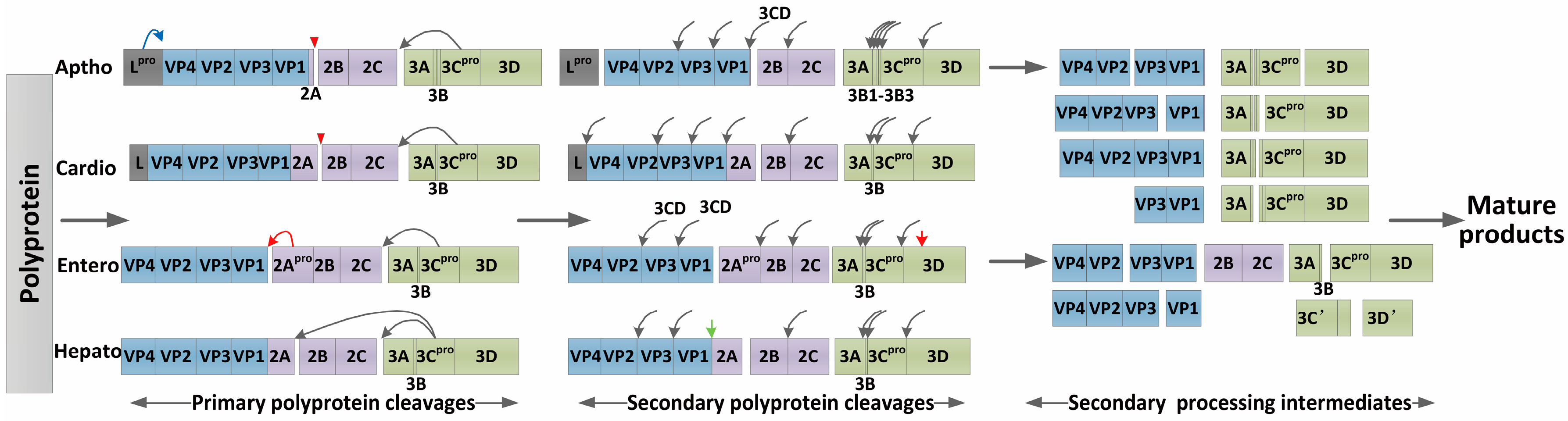
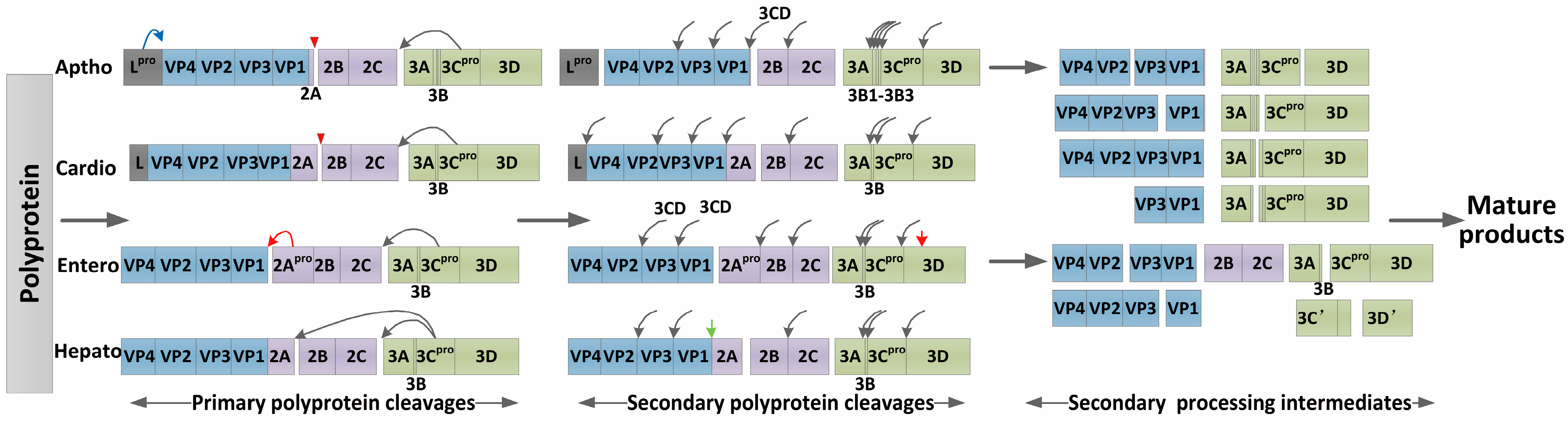
3.1. Processing of the Viral Polyprotein
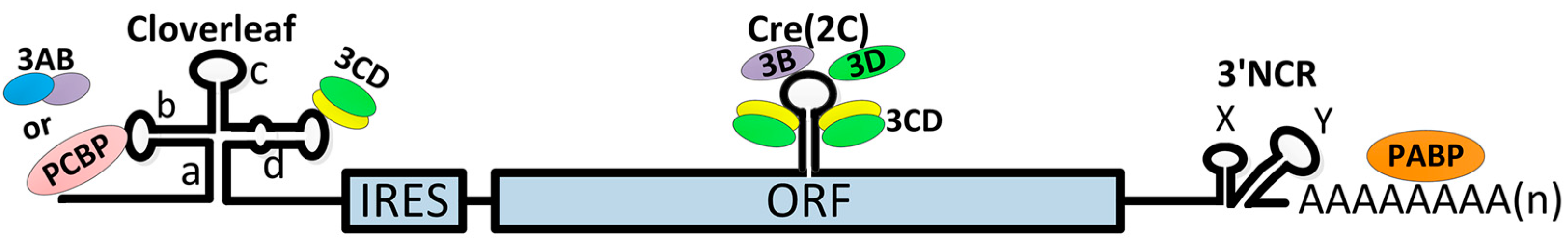
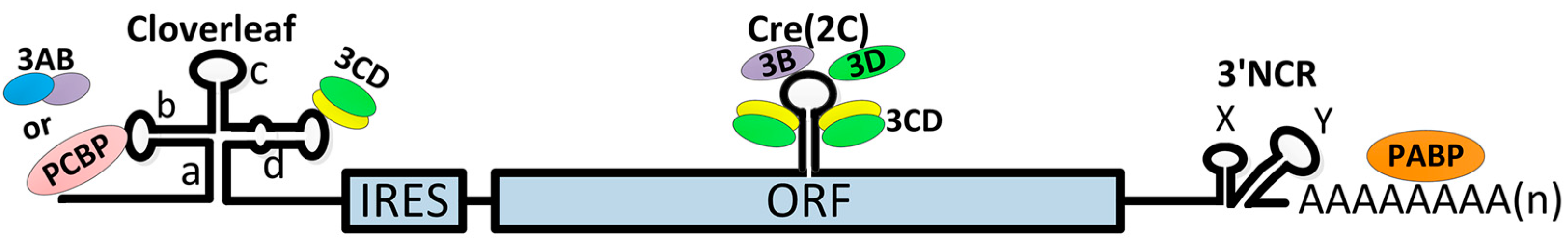
3.2. Initiation of Viral RNA Synthesis
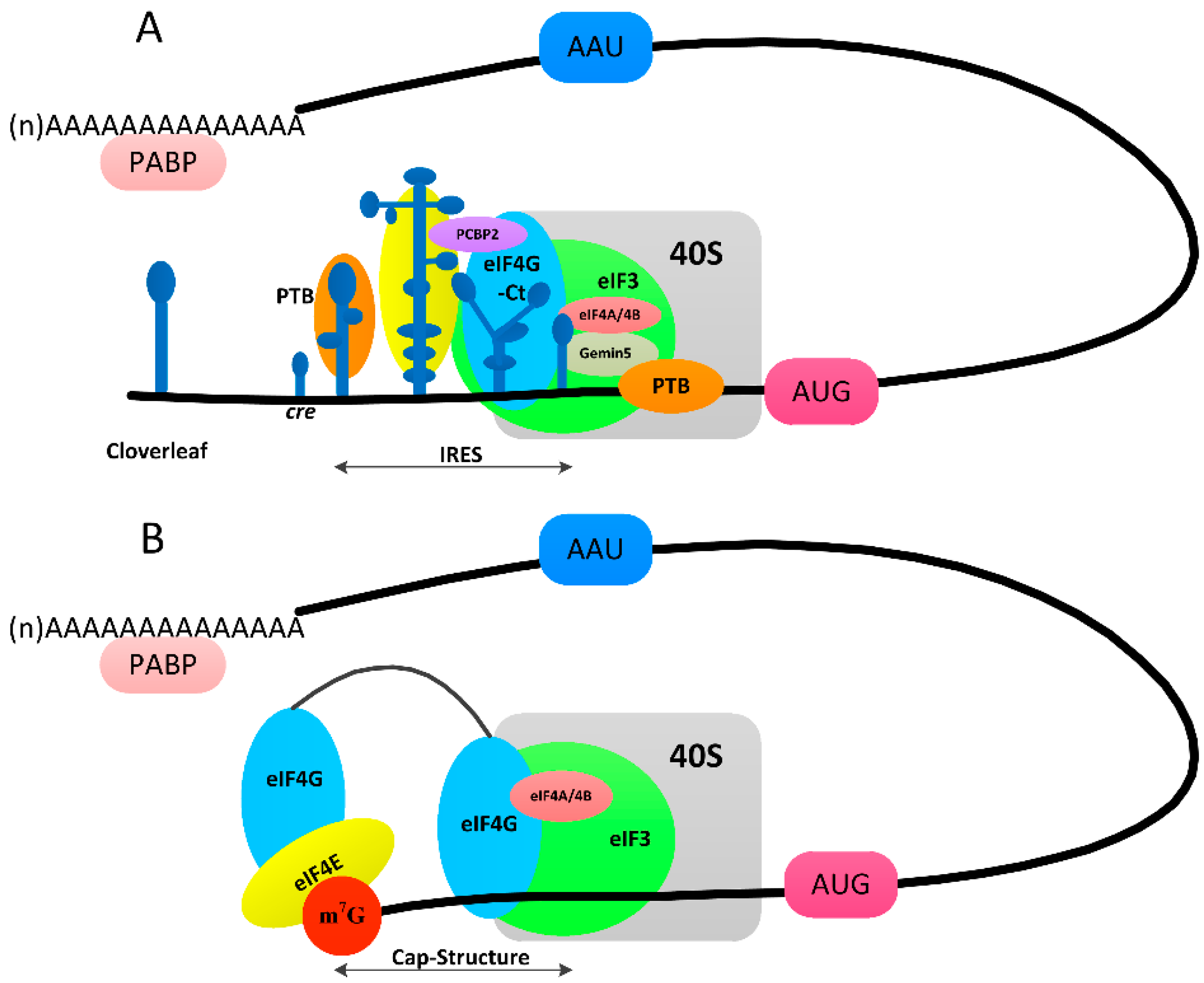
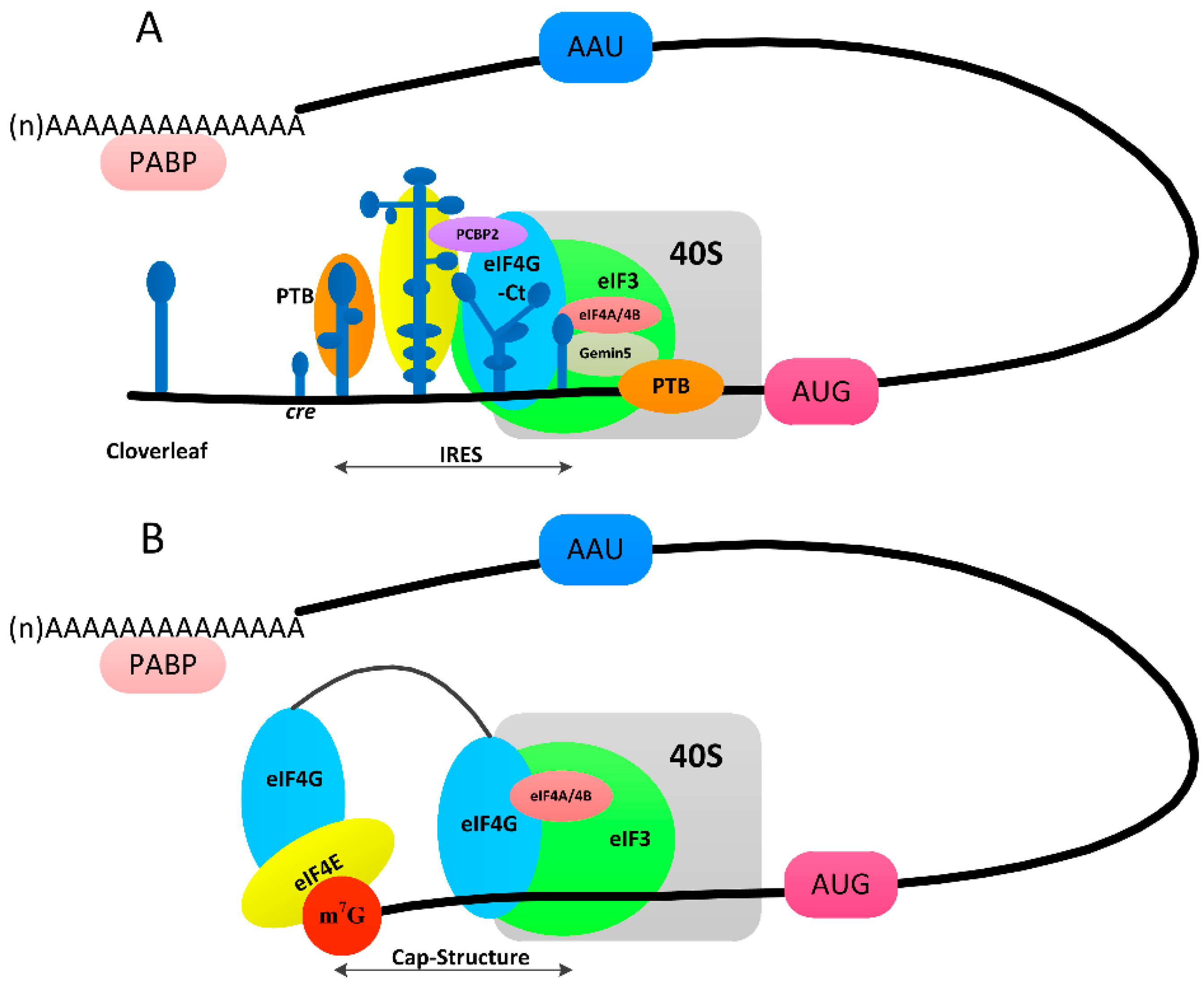
3.3. Switch from Translation to RNA Replication
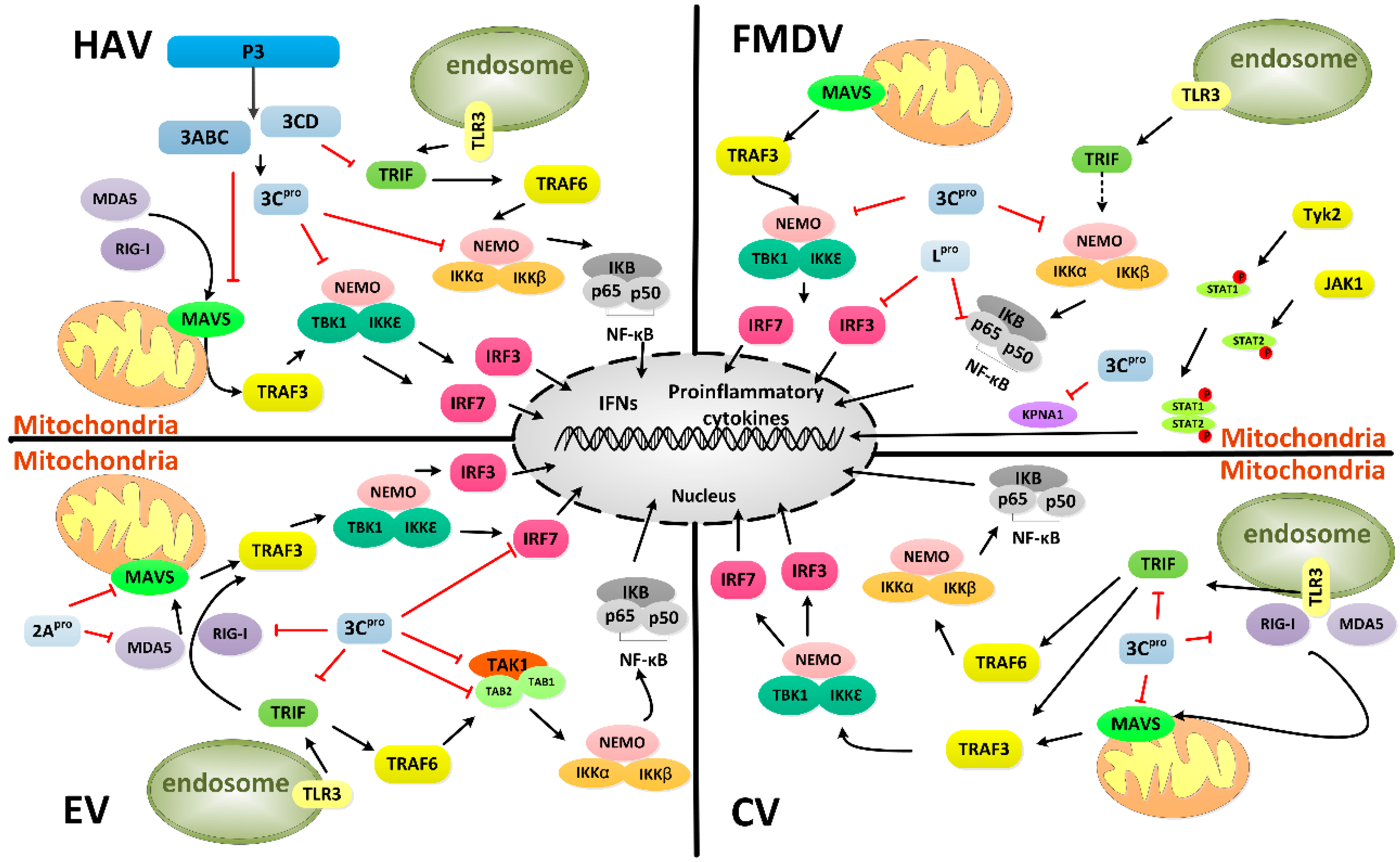
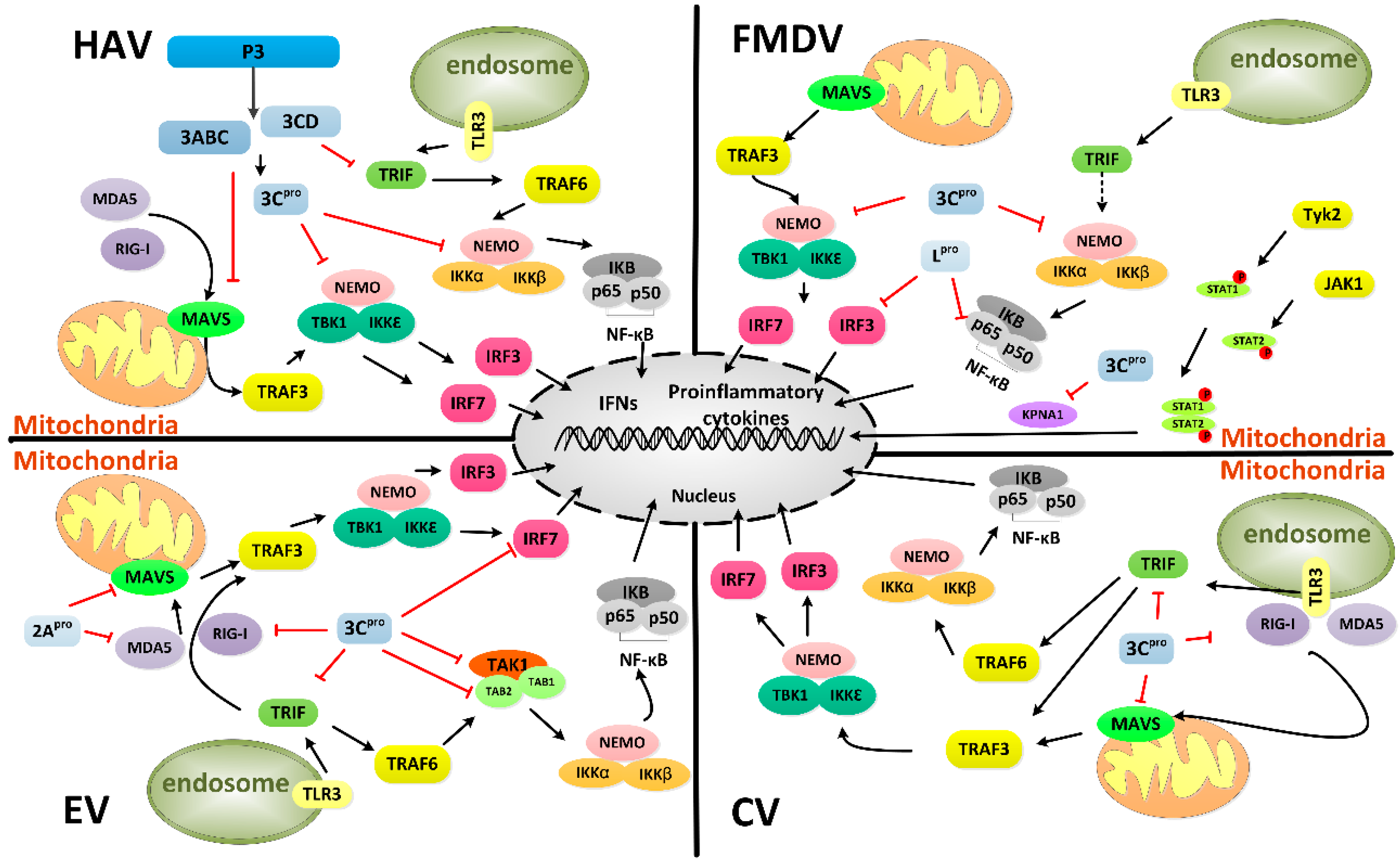
4. Host Cells Intervention by 3Cpro
4.1. Rapid Shut-off of Transcription in Host Cells
4.2. Rapid Inhibition of Protein Synthesis Initiation in Host Cells
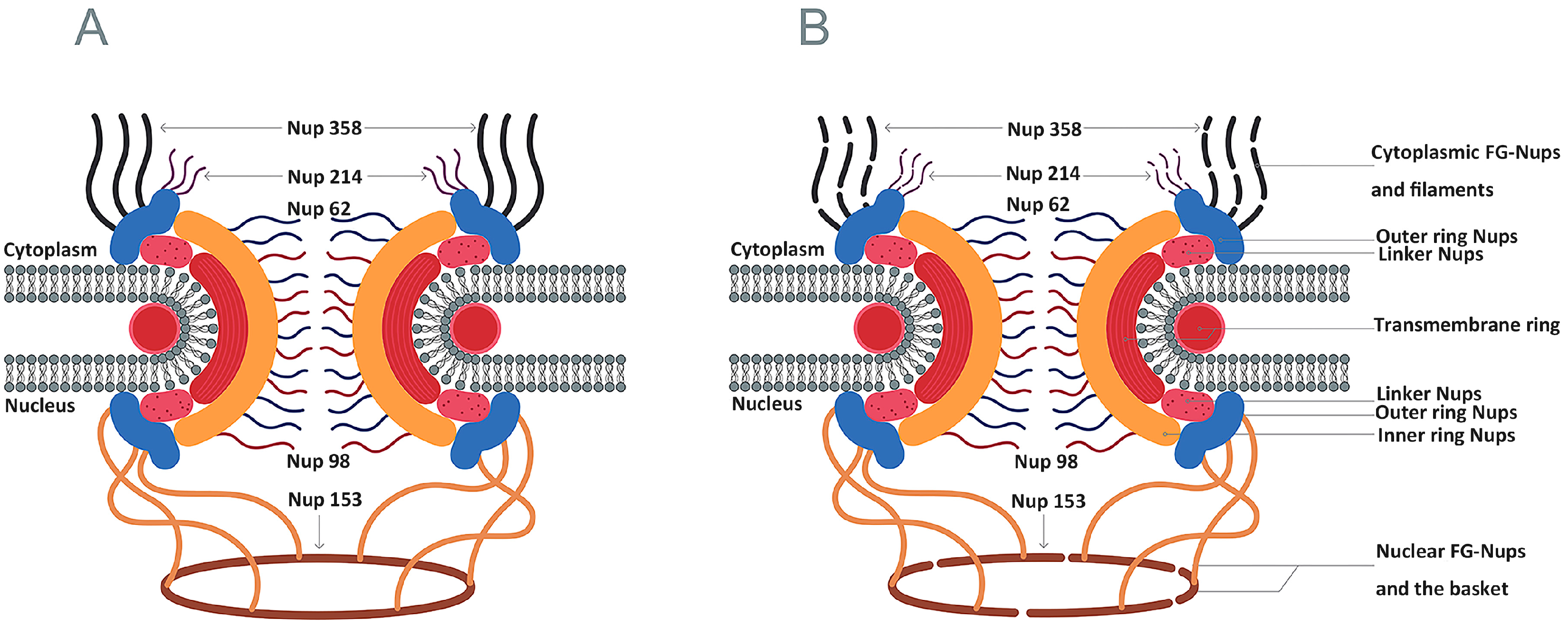
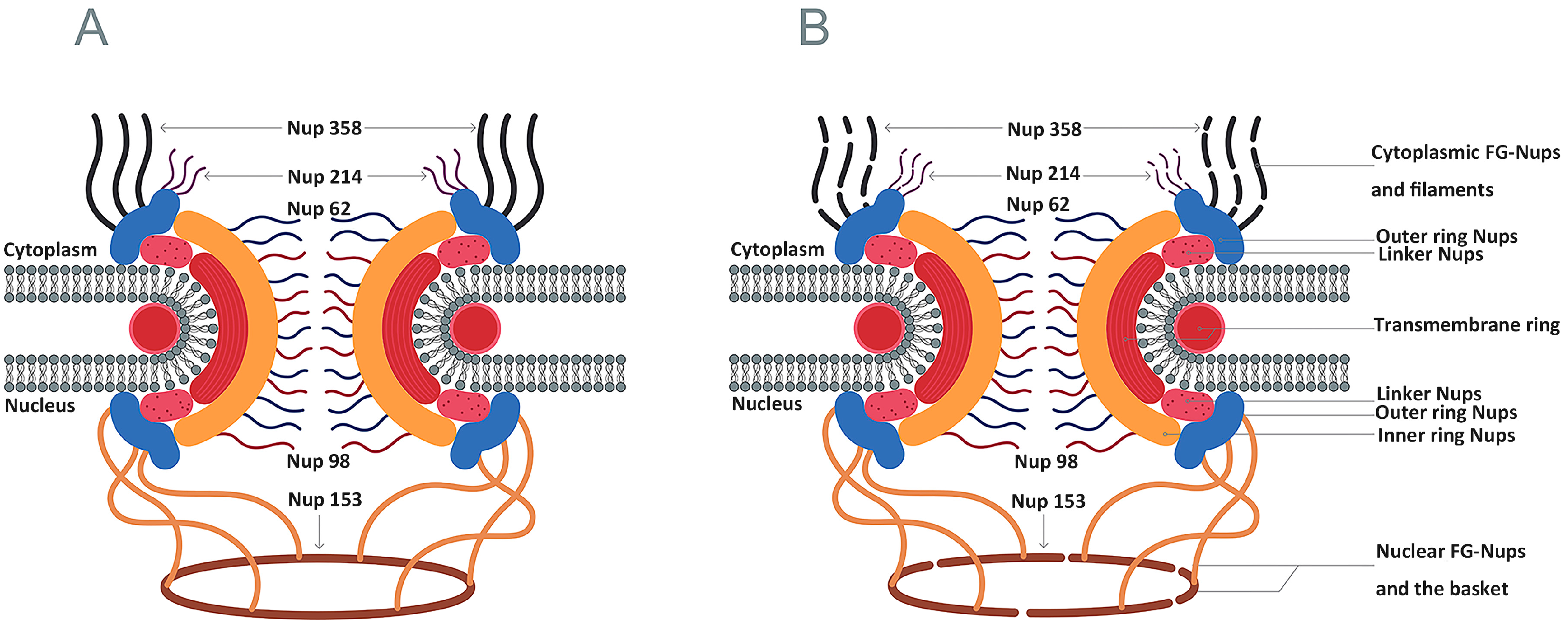
4.3. Inhibition of Nucleocytoplasmic Trafficking by the Nuclear Pore Complex
4.4. Induction of Cell Death
4.5. Other Functions
5. The Role of the Pathogenic Process
6. Treatment
6.1. Inhibitors of Picornaviral 3Cpro
6.2. Broad-Spectrum Inhibitors of 3Cpro and 3CLpro
6.3. Natural Medicine
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Adams, M.J.; King, A.M.Q.; Carstens, E.B. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2013). Arch. Virol. 2013, 158, 2023–2030. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; Academic Press/Elsevier: London, UK, 2012; pp. 855–880. [Google Scholar]
- Lewis, S.A.; Morgan, D.O.; Grubman, M.J. Expression, processing, and assembly of foot-and-mouth disease virus capsid structures in heterologous systems: Induction of a neutralizing antibody response in guinea pigs. J. Virol. 1991, 65, 6572–6580. [Google Scholar] [PubMed]
- Khan, A.; Sharif, S.; Shaukat, S.; Khan, S.; Zaidi, S. An outbreak of acute hemorrhagic conjunctivitis (AHC) caused by coxsackievirus A24 variant in pakistan. Virus Res. 2008, 137, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Sapkal, G.N.; Bondre, V.P.; Fulmali, P.V.; Patil, P.; Gopalkrishna, V.; Dadhania, V.; Ayachit, V.M.; Gangale, D.; Kushwaha, K.P.; Rathi, A.K.; et al. Enteroviruses in patients with acute encephalitis, Uttar Pradesh, India. Emerg. Infect. Dis. 2009, 15, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tan, X.J.; Wang, H.Y.; Yan, D.M.; Zhu, S.L.; Wang, D.Y.; Ji, F.; Wang, X.J.; Gao, Y.J.; Chen, L.; et al. An outbreak of hand, foot, and mouth disease associated with subgenotype C4 of human enterovirus 71 in Shandong, China. J. Clin. Virol. 2009, 44, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Turner, R.B.; Gwaltney, J.M.; Chi-Burris, K.; Gersten, M.; Hsyu, P.; Patick, A.K.; Smith, G.J.; Zalman, L.S. Phase II, randomized, double-blind, placebo-controlled studies of ruprintrivir nasal spray 2-percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrob. Agents Chemother. 2003, 47, 3907–3916. [Google Scholar] [CrossRef] [PubMed]
- Patick, A.K. Rhinovirus chemotherapy. Antiviral Res. 2006, 71, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.V.; Rieder, E.; Kim, D.W.; van Boom, J.H.; Wimmer, E. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J. Virol. 2000, 74, 10359–10370. [Google Scholar] [CrossRef] [PubMed]
- Birtley, J.R.; Knox, S.R.; Jaulent, A.M.; Brick, P.; Leatherbarrow, R.J.; Curry, S. Crystal structure of foot-and-mouth disease virus 3C protease. New insights into catalytic mechanism and cleavage specificity. J. Biol. Chem. 2005, 280, 11520–11527. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, E.M.; Cherney, M.M.; McKendrick, J.; Frormann, S.; Luo, C.; Malcolm, B.A.; Vederas, J.C.; James, M.N. Crystal structure of an inhibitor complex of the 3C proteinase from hepatitis A virus (HAV) and implications for the polyprotein processing in HAV. Virology 1999, 265, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Mosimann, S.C.; Cherney, M.M.; Sia, S.; Plotch, S.; James, M.N. Refined X-ray crystallographic structure of the poliovirus 3C gene product. J. Mol. Biol. 1997, 273, 1032–1047. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.A.; Smith, W.W.; Ferre, R.A.; Condon, B.; Budahazi, G.; Sisson, W.; Villafranca, J.E.; Janson, C.A.; McElroy, H.E.; Gribskov, C.L.; et al. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell 1994, 77, 761–771. [Google Scholar] [CrossRef]
- Matthews, D.A.; Dragovich, P.S.; Webber, S.E.; Fuhrman, S.A.; Patick, A.K.; Zalman, L.S.; Hendrickson, T.F.; Love, R.A.; Prins, T.J.; Marakovits, J.T.; et al. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Nalt. Acad. Sci. USA 1999, 96, 11000–11007. [Google Scholar] [CrossRef]
- Yin, J.; Bergmann, E.M.; Cherney, M.M.; Lall, M.S.; Jain, R.P.; Vederas, J.C.; James, M.N. Dual modes of modification of hepatitis a virus 3C protease by a serine-derived β-lactone: Selective crystallization and formation of a functional catalytic triad in the active site. J. Mol. Biol. 2005, 354, 854–871. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, T.R.; Roqué-Rosell, N.; Birtley, J.R.; Leatherbarrow, R.J.; Curry, S. Structural and mutagenic analysis of foot-and-mouth disease virus 3C protease reveals the role of the β-ribbon in proteolysis. J. Virol. 2007, 81, 115–124. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cui, S.; Wang, J.; Fan, T.; Qin, B.; Guo, L.; Lei, X.; Wang, J.; Wang, M.; Jin, Q. Crystal structure of human enterovirus 71 3C protease. J. Mol. Biol. 2011, 408, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, E.M.; Mosimann, S.C.; Chernaia, M.M.; Malcolm, B.A.; James, M.N. The refined crystal structure of the 3C gene product from hepatitis a virus: Specific proteinase activity and RNA recognition. J. Virol. 1997, 71, 2436–2448. [Google Scholar] [PubMed]
- Wang, J.; Fan, T.; Yao, X.; Wu, Z.; Guo, L.; Lei, X.; Wang, J.; Wang, M.; Jin, Q.; Cui, S. Crystal structures of enterovirus 71 3C protease complexed with rupintrivir reveal the roles of catalytically important residues. J. Virol. 2011, 85, 10021–10030. [Google Scholar] [CrossRef] [PubMed]
- Curry, S.; Roqué-Rosell, N.; Zunszain, P.A.; Leatherbarrow, R.J. Foot-and-mouth disease virus 3C protease: Recent structural and functional insights into an antiviral target. Int. J. Biochem. Cell Biol. 2007, 39, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Seipelt, J.; Guarné, A.; Bergmann, E.; James, M.; Sommergruber, W.; Fita, I.; Skern, T. The structures of picornaviral proteinases. Virus Res. 1999, 62, 159–168. [Google Scholar] [CrossRef]
- Guarné, A.; Hampoelz, B.; Glaser, W.; Carpena, X.; Tormo, J.; Fita, I.; Skern, T. Structural and biochemical features distinguish the foot-and-mouth disease virus leader proteinase from other papain-like enzymes. J. Mol. Biol. 2000, 302, 1227–1240. [Google Scholar] [CrossRef] [PubMed]
- Salas, E.M.; Ryan, M.D. Translation and protein process. In The Picornaviruses; Ehrenfeld, E., Domingo, E., Roos, R., Eds.; ASM Press: Washington, DC, USA, 2010; pp. 141–150. [Google Scholar]
- Palmenberg, A.; Neubauer, D.; Skern, T. Genome organization and encoded proteins. In The Picornaviruses; Ehrenfeld, E., Domingo, E., Roos, R., Eds.; ASM Press: Washington, DC, USA, 2010; pp. 3–17. [Google Scholar]
- Castelló, A.; Alvarez, E.; Carrasco, L. The multifaceted poliovirus 2A protease: Regulation of gene expression by picornavirus proteases. J. Biomed. Biotechnol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Amineva, S.P.; Aminev, A.G.; Palmenberg, A.C.; Gern, J.E. Rhinovirus 3C protease precursors 3CD and 3CD′ localize to the nuclei of infected cells. J. Gen. Virol. 2004, 85, 2969–2979. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.D.; Belsham, G.J.; King, A.M. Specificity of enzyme-substrate interactions in foot-and-mouth disease virus polyprotein processing. Virology 1989, 173, 35–45. [Google Scholar] [CrossRef]
- Ypma-Wong, M.F.; Dewalt, P.G.; Johnson, V.H.; Lamb, J.G.; Semler, B.L. Protein 3CD is the major poliovirus proteinase responsible for cleavage of the P1 capsid precursor. Virology 1988, 166, 265–270. [Google Scholar] [CrossRef]
- Guarné, A.; Tormo, J.; Kirchweger, R.; Pfistermueller, D.; Fita, I.; Skern, T. Structure of the foot-and-mouth disease virus leader protease: A papain-like fold adapted for self-processing and eIF4G recognition. EMBO J. 1998, 17, 7469–7479. [Google Scholar] [CrossRef] [PubMed]
- Spear, A.; Ogram, S.A.; Morasco, B.J.; Smerage, L.E.; Flanegan, J.B. Viral precursor protein P3 and its processed products perform discrete and essential functions in the poliovirus RNA replication complex. Virology 2015, 485, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Paul, A.V.; Wimmer, E.; Rieder, E. Functional dissection of a poliovirus cis-acting replication element (PV-cre(2C)): Analysis of single- and dual-cre viral genomes and proteins that bind specifically to PV-cre RNA. J. Virol. 2003, 77, 5152–5166. [Google Scholar] [CrossRef] [PubMed]
- Pathak, H.B.; Arnold, J.J.; Wiegand, P.N.; Hargittai, M.R.; Cameron, C.E. Picornavirus genome replication: Assembly and organization of the VPg uridylylation ribonucleoprotein (initiation) complex. J. Biol. Chem. 2007, 282, 16202–16213. [Google Scholar] [CrossRef] [PubMed]
- Pathak, H.B.; Oh, H.S.; Goodfellow, I.G.; Arnold, J.J.; Cameron, C.E. Picornavirus genome replication: Roles of precursor proteins and rate-limiting steps in oriI-dependent VPg uridylylation. J. Biol. Chem. 2008, 283, 30677–30688. [Google Scholar] [CrossRef] [PubMed]
- Kusov, Y.Y.; Morace, G.; Probst, C.; Gauss-Müller, V. Interaction of hepatitis A virus (HAV) precursor proteins 3AB and 3ABC with the 5′ and 3′ termini of the HAV RNA. Virus Res. 1997, 51, 151–157. [Google Scholar] [CrossRef]
- Paul, A.V.; Wimmer, E. Initiation of protein-primed picornavirus RNA synthesis. Virus Res. 2015, 206, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Andino, R.; Rieckhof, G.E.; Trono, D.; Baltimore, D. Substitutions in the protease (3Cpro) gene of poliovirus can suppress a mutation in the 5’noncoding region. J. Virol. 1990, 64, 607–612. [Google Scholar] [PubMed]
- Andino, R.; Rieckhof, G.E.; Baltimore, D. A functional ribonucleoprotein complex forms around the 5′ end of poliovirus RNA. Cell 1990, 63, 369–380. [Google Scholar] [CrossRef]
- Andino, R.; Rieckhof, G.E.; Achacoso, P.L.; Baltimore, D. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5’-end of viral RNA. EMBO J. 1993, 12, 3587–3598. [Google Scholar] [PubMed]
- Nayak, A.; Goodfellow, I.G.; Belsham, G.J. Factors required for the uridylylation of the foot-and-mouth disease virus 3B1, 3B2, and 3B3 peptides by the RNA-dependent RNA polymerase (3Dpol) in vitro. J. Virol. 2005, 79, 7698–7706. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nayak, A.; Goodfellow, I.G.; Woolaway, K.E.; Birtley, J.; Curry, S.; Belsham, G.J. Role of RNA structure and RNA binding activity of foot-and-mouth disease virus 3C protein in VPg uridylylation and virus replication. J. Virol. 2006, 80, 9865–9875. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Herold, J.; Andino, R. Poliovirus RNA replication requires genome circularization through a protein-protein bridge. Mol. Cell 2001, 7, 581–591. [Google Scholar] [CrossRef]
- Harris, K.S.; Xiang, W.; Alexander, L.; Lane, W.S.; Paul, A.V.; Wimmer, E. Interaction of poliovirus polypeptide 3CDpro with the 5’and 3’termini of the poliovirus genome. Identification of viral and cellular cofactors needed for efficient binding. J. Biol. Chem. 1994, 269, 27004–27014. [Google Scholar] [PubMed]
- Franco, D.; Pathak, H.B.; Cameron, C.E.; Rombaut, B.; Wimmer, E.; Paul, A.V. Stimulation of poliovirus synthesis in a HeLa cell-free in vitro translation-RNA replication system by viral protein 3CDpro. J. Virol. 2005, 79, 6358–6367. [Google Scholar] [CrossRef] [PubMed]
- Blair, W.S.; Parsley, T.B.; Bogerd, H.P.; Towner, J.S.; Semler, B.L.; Cullen, B.R. Utilization of a mammalian cell-based RNA binding assay to characterize the RNA binding properties of picornavirus 3C proteinases. RNA 1998, 4, 215–225. [Google Scholar] [PubMed]
- Moustafa, I.M.; Gohara, D.W.; Uchida, A.; Yennawar, N.; Cameron, C.E. Conformational ensemble of the poliovirus 3CD precursor observed by md simulations and confirmed by saxs: A strategy to expand the viral proteome? Viruses 2015, 7, 5962–5986. [Google Scholar] [CrossRef] [PubMed]
- Gamarnik, A.V.; Andino, R. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 1998, 12, 2293–2304. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.J.; Daijogo, S.; Semler, B.L. Inhibition of poliovirus-induced cleavage of cellular protein PCBP2 reduces the levels of viral RNA replication. J. Virol. 2014, 88, 3192–3201. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.M.; Boehr, D.D. Allosteric functional switch in poliovirus 3C protease. FASEB J. 2015, 29. [Google Scholar] [CrossRef]
- Banerjee, R.; Weidman, M.K.; Navarro, S.; Comai, L.; Dasgupta, A. Modifications of both selectivity factor and upstream binding factor contribute to poliovirus-mediated inhibition of RNA polymerase I transcription. J. Gen. Virol. 2005, 86, 2315–2322. [Google Scholar] [CrossRef] [PubMed]
- Kundu, P.; Raychaudhuri, S.; Tsai, W.; Dasgupta, A. Shutoff of RNA polymerase II transcription by poliovirus involves 3C protease-mediated cleavage of the TATA-binding protein at an alternative site: Incomplete shutoff of transcription interferes with efficient viral replication. J. Virol. 2005, 79, 9702–9713. [Google Scholar] [CrossRef] [PubMed]
- Yalamanchili, P.; Datta, U.; Dasgupta, A. Inhibition of host cell transcription by poliovirus: Cleavage of transcription factor CREB by poliovirus-encoded protease 3Cpro. J. Virol. 1997, 71, 1220–1226. [Google Scholar] [PubMed]
- Yalamanchili, P.; Weidman, K.; Dasgupta, A. Cleavage of transcriptional activator Oct-1 by poliovirus encoded protease 3Cpro. Virology 1997, 239, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Weidman, M.K.; Yalamanchili, P.; Ng, B.; Tsai, W.; Dasgupta, A. Poliovirus 3C protease-mediated degradation of transcriptional activator p53 requires a cellular activity. Virology 2001, 291, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Igo, M.; Yalamanchili, P.; Berk, A.J.; Dasgupta, A. DNA binding domain and subunit interactions of transcription factor IIIC revealed by dissection with poliovirus 3C protease. Mol. Cell. Biol. 1996, 16, 4163–4171. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Falk, M.M.; Grigera, P.R.; Bergmann, I.E.; Zibert, A.; Multhaup, G.; Beck, E. Foot-and-mouth disease virus protease 3C induces specific proteolytic cleavage of host cell histone H3. J. Virol. 1990, 64, 748–756. [Google Scholar] [PubMed]
- Weng, K.F.; Li, M.L.; Hung, C.T.; Shih, S.R. Enterovirus 71 3C protease cleaves a novel target CstF-64 and inhibits cellular polyadenylation. PLoS Pathog. 2009, 5, e1000593. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Lozano, G.; Martínez-Salas, E. Structural insights into viral IRES-dependent translation mechanisms. Curr. Opin. Virol. 2015, 12, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Salas, E.; Pacheco, A.; Serrano, P.; Fernandez, N. New insights into internal ribosome entry site elements relevant for viral gene expression. J. Gen. Virol. 2008, 89, 611–626. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Salas, E.; Francisco-Velilla, R.; Fernandez-Chamorro, J.; Lozano, G.; Diaz-Toledano, R. Picornavirus IRES elements: RNA structure and host protein interactions. Virus Res. 2015, 206, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Belsham, G.J.; McInerney, G.M.; Ross-Smith, N. Foot-and-mouth disease virus 3C protease induces cleavage of translation initiation factors eIF4A and eIF4G within infected cells. J. Virol. 2000, 74, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Gradi, A.; Foeger, N.; Strong, R.; Svitkin, Y.V.; Sonenberg, N.; Skern, T.; Belsham, G.J. Cleavage of eukaryotic translation initiation factor 4GII within foot-and-mouth disease virus-infected cells: Identification of the L-protease cleavage site in vitro. J. Virol. 2004, 78, 3271–3278. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ross-Smith, N.; Proud, C.G.; Belsham, G.J. Cleavage of translation initiation factor 4AI (eIF4AI) but not eIF4AII by foot-and-mouth disease virus 3C protease: Identification of the eIF4AI cleavage site. FEBS Lett. 2001, 507, 1–5. [Google Scholar] [CrossRef]
- De Breyne, S.; Bonderoff, J.M.; Chumakov, K.M.; Lloyd, R.E.; Hellen, C.U. Cleavage of eukaryotic initiation factor eIF5B by enterovirus 3C proteases. Virology 2008, 378, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.; Daijogo, S.; Walter, B.L.; Nguyen, J.H.; Semler, B.L. Cellular protein modification by poliovirus: The two faces of poly (rC)-binding protein. J. Virol. 2007, 81, 8919–8932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Seitz, S.; Kusov, Y.; Zell, R.; Gauss-Müller, V. RNA interaction and cleavage of poly (C)-binding protein 2 by hepatitis A virus protease. Biochem. Biophys. Res. Commun. 2007, 364, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Kuyumcu-Martinez, N.M.; Van Eden, M.E.; Younan, P.; Lloyd, R.E. Cleavage of poly (A)-binding protein by poliovirus 3C protease inhibits host cell translation: A novel mechanism for host translation shutoff. Mol. Cell. Biol. 2004, 24, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Morace, G.; Gauss-Müller, V.; Kusov, Y. Poly(A) binding protein, C-terminally truncated by the hepatitis A virus proteinase 3C, inhibits viral translation. Nucleic Acids Res. 2007, 35, 5975–5984. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kobayashi, M.; Arias, C.; Garabedian, A.; Palmenberg, A.C.; Mohr, I. Site-specific cleavage of the host poly (A) binding protein by the encephalomyocarditis virus 3C proteinase stimulates viral replication. J. Virol. 2012, 86, 10686–10694. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fung, G.; Ng, C.S.; Zhang, J.; Shi, J.; Wong, J.; Piesik, P.; Han, L.; Chu, F.; Jagdeo, J.; Jan, E.; et al. Production of a dominant-negative fragment due to G3BP1 cleavage contributes to the disruption of mitochondria-associated protective stress granules during CVB3 infection. PLoS ONE 2013, 8, e79546. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, P.; Schafer, E.A.; Rieder, E. The nuclear protein Sam68 is cleaved by the FMDV 3C protease redistributing Sam68 to the cytoplasm during FMDV infection of host cells. Virology 2012, 425, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Flather, D.; Semler, B.L. Picornaviruses and nuclear functions: Targeting a cellular compartment distinct from the replication site of a positive-strand RNA virus. Front. Microbiol. 2015, 6, 594. [Google Scholar] [CrossRef] [PubMed]
- Watters, K.; Palmenberg, A.C. Differential processing of nuclear pore complex proteins by rhinovirus 2A proteases from different species and serotypes. J. Virol. 2011, 85, 10874–10883. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.J.; Younessi, P.; Fulcher, A.J.; McCuaig, R.; Thomas, B.J.; Bardin, P.G.; Jans, D.A.; Ghildyal, R. Rhinovirus 3C protease facilitates specific nucleoporin cleavage and mislocalisation of nuclear proteins in infected host cells. PLoS ONE 2013, 8, e71316. [Google Scholar] [CrossRef] [PubMed]
- Caly, L.; Ghildyal, R.; Jans, D.A. Respiratory virus modulation of host nucleocytoplasmic transport; target for therapeutic intervention? Front. Microbiol. 2015, 6, 848. [Google Scholar] [CrossRef] [PubMed]
- Park, N.; Katikaneni, P.; Skern, T.; Gustin, K.E. Differential targeting of nuclear pore complex proteins in poliovirus-infected cells. J. Virol. 2008, 82, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Park, N.; Skern, T.; Gustin, K.E. Specific cleavage of the nuclear pore complex protein Nup62 by a viral protease. J. Biol. Chem. 2010, 285, 28796–28805. [Google Scholar] [CrossRef] [PubMed]
- Ghildyal, R.; Jordan, B.; Li, D.; Dagher, H.; Bardin, P.G.; Gern, J.E.; Jans, D.A. Rhinovirus 3C protease can localize in the nucleus and alter active and passive nucleocytoplasmic transport. J. Virol. 2009, 83, 7349–7352. [Google Scholar] [CrossRef] [PubMed]
- Alber, F.; Dokudovskaya, S.; Veenhoff, L.M.; Zhang, W.; Kipper, J.; Devos, D.; Suprapto, A.; Karni-Schmidt, O.; Williams, R.; Chait, B.T. The molecular architecture of the nuclear pore complex. Nature 2007, 450, 695–701. [Google Scholar] [PubMed]
- Grossman, E.; Medalia, O.; Zwerger, M. Functional architecture of the nuclear pore complex. Annu. Rev. Biophys. 2012, 41, 557–584. [Google Scholar] [CrossRef] [PubMed]
- Porter, F.W.; Palmenberg, A.C. Leader-induced phosphorylation of nucleoporins correlates with nuclear trafficking inhibition by cardioviruses. J. Virol. 2009, 83, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Ricour, C.; Delhaye, S.; Hato, S.V.; Olenyik, T.D.; Michel, B.; van Kuppeveld, F.J.; Gustin, K.E.; Michiels, T. Inhibition of mrna export and dimerization of interferon regulatory factor 3 by theiler’s virus leader protein. J. Gen. Virol. 2009, 90, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Barco, A.; Feduchi, E.; Carrasco, L. Poliovirus protease 3Cpro kills cells by apoptosis. Virology 2000, 266, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Hsu, T.A.; Chen, T.C.; Chang, S.C.; Lee, J.C.; Chen, C.C.; Stollar, V.; Shih, S.R. The 3C protease activity of enterovirus 71 induces human neural cell apoptosis. Virology 2002, 293, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Chau, D.H.; Yuan, J.; Zhang, H.; Cheung, P.; Lim, T.; Liu, Z.; Sall, A.; Yang, D. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 2007, 12, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Shubin, A.V.; Demidyuk, I.V.; Lunina, N.A.; Komissarov, A.A.; Roschina, M.P.; Leonova, O.G.; Kostrov, S.V. Protease 3C of hepatitis A virus induces vacuolization of lysosomal/endosomal organelles and caspase-independent cell death. BMC Cell Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Amero, C.D.; Arnold, J.J.; Moustafa, I.M.; Cameron, C.E.; Foster, M.P. Identification of the oriI-binding site of poliovirus 3C protein by nuclear magnetic resonance spectroscopy. J. Virol. 2008, 82, 4363–4370. [Google Scholar] [CrossRef] [PubMed]
- Joachims, M.; Harris, K.S.; Etchison, D. Poliovirus protease 3C mediates cleavage of microtubule-associated protein 4. Virology 1995, 211, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Mogensen, M.M.; Powell, P.P.; Curry, S.; Wileman, T. Foot-and-mouth disease virus 3C protease induces fragmentation of the Golgi compartment and blocks intra-Golgi transport. J. Virol. 2013, 87, 11721–11729. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Autophagy: An emerging immunological paradigm. J. Immunol. 2012, 189, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Pålsson-McDermott, E.M.; O’Neill, L.A. Building an immune system from nine domains. Biochem. Soc. Trans. 2007, 35, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.; O’neill, L.A. The interleukin-1 receptor/toll-like receptor superfamily: Signal generators for pro-inflammatory interleukins and microbial products. J. Leukoc. Biol. 2000, 67, 508–514. [Google Scholar] [PubMed]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, G.; Saha, S.K.; Guo, B.; He, J.Q.; Shahangian, A.; Zarnegar, B.; Perry, A.; Cheng, G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 2006, 439, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Häcker, H.; Redecke, V.; Blagoev, B.; Kratchmarova, I.; Hsu, L.C.; Wang, G.G.; Kamps, M.P.; Raz, E.; Wagner, H.; Häcker, G.; et al. Specificity in toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 2006, 439, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Matsumoto, M.; Funami, K.; Akazawa, T.; Seya, T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 2003, 4, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.M.; Maniatis, T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Barton, G.M. Toll-like receptors: Key players in antiviral immunity. Curr. Opin. Virol. 2011, 1, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Liu, X.; Ma, Y.; Sun, Z.; Yang, Y.; Jin, Q.; He, B.; Wang, J. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 2010, 84, 8051–8061. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xi, X.; Lei, X.; Zhang, X.; Cui, S.; Wang, J.; Jin, Q.; Zhao, Z. Enterovirus 71 protease 2A pro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathol. 2013, 9, e1003231. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Sun, Z.; Liu, X.; Jin, Q.; He, B.; Wang, J. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by toll-like receptor 3. J. Virol. 2011, 85, 8811–8818. [Google Scholar] [CrossRef] [PubMed]
- Papon, L.; Oteiza, A.; Imaizumi, T.; Kato, H.; Brocchi, E.; Lawson, T.G.; Akira, S.; Mechti, N. The viral RNA recognition sensor RIG-I is degraded during encephalomyocarditis virus (EMCV) infection. Virology 2009, 393, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Barral, P.M.; Morrison, J.M.; Drahos, J.; Gupta, P.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 2007, 81, 3677–3684. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.I.; Weng, K.F.; Shih, S.R. Viral and host factors that contribute to pathogenicity of enterovirus 71. Future Microbiol. 2012, 7, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Xiao, X.; Xue, Q.; Jin, Q.; He, B.; Wang, J. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J. Virol. 2013, 87, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Han, N.; Xiao, X.; Jin, Q.; He, B.; Wang, J. Enterovirus 71 3C inhibits cytokine expression through cleavage of the TAK1/TAB1/TAB2/TAB3 complex. J. Virol. 2014, 88, 9830–9841. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Z.; Zhao, X.; Yu, R.; Zhang, X.; Wu, S.; Liu, J.; Chi, X.; Song, X.; Fu, L.; et al. Enterovirus 71 inhibits cellular type I interferon signaling by downregulating jak1 protein expression. Viral Immunol. 2014, 27, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mao, D.; Roswit, W.T.; Jin, X.; Patel, A.C.; Patel, D.A.; Agapov, E.; Wang, Z.; Tidwell, R.M.; Atkinson, J.J.; et al. PARP9-DTX3L ubiquitin ligase targets host histone H2BJ and viral 3C protease to enhance interferon signaling and control viral infection. Nat. Immunol. 2015, 16, 1215–1227. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, Y.; Qu, L.; Chen, Z.; Yi, M.; Li, K.; Lemon, S.M. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. USA 2007, 104, 7253–7258. [Google Scholar] [CrossRef] [PubMed]
- Qu, L. Evasion of RIG-I/MDA5 and TLR3-Mediated Innate Immunity by Hepatitis A Virus. Ph.D. Thesis, University of Texas Medical Branch, Galveston, TX, USA, 2010. [Google Scholar]
- Qu, L.; Feng, Z.; Yamane, D.; Liang, Y.; Lanford, R.E.; Li, K.; Lemon, S.M. Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease-polymerase processing intermediate, 3CD. PLoS Pathog. 2011, 7, e1002169. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, L.; Wei, D.; Zhang, H.; Luo, R.; Chen, H.; Li, K.; Xiao, S. Hepatitis A virus 3C protease cleaves NEMO to impair induction of β interferon. J. Virol. 2014, 88, 10252–10258. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, L.; Li, K.; Zhong, H.; Fan, J.; Ouyang, C.; Zhang, H.; Duan, E.; Luo, R.; Zhang, Z.; et al. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J. Virol. 2012, 86, 9311–9322. [Google Scholar] [CrossRef] [PubMed]
- De Los Santos, T.; Diaz-San Segundo, F.; Grubman, M.J. Degradation of nuclear factor κB during foot-and-mouth disease virus infection. J. Virol. 2007, 81, 12803–12815. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fang, L.; Luo, R.; Ye, R.; Fang, Y.; Xie, L.; Chen, H.; Xiao, S. Foot-and-mouth disease virus leader proteinase inhibits dsRNA-induced type I interferon transcription by decreasing interferon regulatory factor 3/7 in protein levels. Biochem. Biophys. Res. Commun. 2010, 399, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Bi, J.; Liu, J.; Liu, X.; Wu, X.; Jiang, P.; Yoo, D.; Zhang, Y.; Wu, J.; Wan, R.; et al. 3Cpro of foot-and-mouth disease virus antagonizes the interferon signaling pathway by blocking STAT1/STAT2 nuclear translocation. J. Virol. 2014, 88, 4908–4920. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morosky, S.A.; Delorme-Axford, E.; Dybdahl-Sissoko, N.; Oberste, M.S.; Wang, T.; Coyne, C.B. The Coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011, 7, e1001311. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; He, X.; Zheng, Z.; Zhang, Z.; Wei, C.; Guan, K.; Hou, L.; Zhang, B.; Zhu, L.; Cao, Y.; et al. Downregulation of MicroRNA miR-526a by enterovirus inhibits RIG-I-Dependent innate immune response. J. Virol. 2014, 88, 11356–11368. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Fung, G.; Shi, J.; Deng, H.; Hou, J.; Wang, C.; Hong, A.; Zhang, J.; Jia, W.; Luo, H. Cytoplasmic translocation, aggregation, and cleavage of TDP-43 by enteroviral proteases modulate viral pathogenesis. Cell Death Differ. 2015, 22, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S.; Zhou, R.; Webber, S.E.; Prins, T.J.; Kwok, A.K.; Okano, K.; Fuhrman, S.A.; Zalman, L.S.; Maldonado, F.C.; Brown, E.L.; et al. Structure-based design of ketone-containing, tripeptidyl human rhinovirus 3C protease inhibitors. Bioorg. Med. Chem. Lett. 2000, 10, 45–48. [Google Scholar] [CrossRef]
- Shepherd, T.A.; Cox, G.A.; McKinney, E.; Tang, J.; Wakulchik, M.; Zimmerman, R.E.; Villarreal, E.C. Small peptidic aldehyde inhibitors of human rhinovirus 3C protease. Bioorg. Med. Chem. Lett. 1996, 6, 2893–2896. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, B.; Zhai, Y.; Yin, Z.; Sun, Y.; Rao, Z. Peptidyl aldehyde NK-1.8 k suppresses enterovirus 71 and enterovirus 68 infection by targeting protease 3C. Antimicrob. Agents Chemother. 2015, 59, 2636–2646. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Hanzlik, R.P. Structure-activity relationships for inhibition of papain by peptide Michael acceptors. J. Med. Chem. 1992, 35, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.S.; Venkatraman, S.; Furness, K.; Nimkar, S.; Shepherd, T.A.; Wang, Q.M.; Aubé, J.; Hanzlik, R.P. Synthesis and evaluation of peptidyl Michael acceptors that inactivate human rhinovirus 3C protease and inhibit virus replication. J. Med. Chem. 1998, 41, 2579–2587. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S.; Webber, S.E.; Babine, R.E.; Fuhrman, S.A.; Patick, A.K.; Matthews, D.A.; Lee, C.A.; Reich, S.H.; Prins, T.J.; Marakovits, J.T.; et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 1. Michael acceptor structure-activity studies. J. Med. Chem. 1998, 41, 2806–2818. [Google Scholar] [CrossRef] [PubMed]
- Patick, A.K.; Binford, S.L.; Brothers, M.A.; Jackson, R.L.; Ford, C.E.; Diem, M.D.; Maldonado, F.; Dragovich, P.S.; Zhou, R.; Prins, T.J.; et al. In vitro antiviral activity of AG7088, a potent inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 1999, 43, 2444–2450. [Google Scholar] [PubMed]
- Tan, J.; George, S.; Kusov, Y.; Perbandt, M.; Anemüller, S.; Mesters, J.R.; Norder, H.; Coutard, B.; Lacroix, C.; Leyssen, P.; et al. 3C protease of enterovirus 68: Structure-based design of Michael acceptor inhibitors and their broad-spectrum antiviral effects against picornaviruses. J. Virol. 2013, 87, 4339–4351. [Google Scholar] [CrossRef] [PubMed]
- Van der Linden, L.; Ulferts, R.; Nabuurs, S.B.; Kusov, Y.; Liu, H.; George, S.; Lacroix, C.; Goris, N.; Lefebvre, D.; Lanke, K.H.; et al. Application of a cell-based protease assay for testing inhibitors of picornavirus 3C proteases. Antivir. Res. 2014, 103, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Kaplan, G.G.; Neyts, J.; Jochmans, D. Rapid and convenient assays to assess potential inhibitory activity on in vitro hepatitis A replication. Antivir. Res. 2013, 98, 325–331. [Google Scholar] [CrossRef] [PubMed]
- De Palma, A.M.; Pürstinger, G.; Wimmer, E.; Patick, A.K.; Andries, K.; Rombaut, B.; de Clercq, E.; Neyts, J. Potential use of antiviral agents in polio eradication. Emerg. Infect. Dis. 2008, 14, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Baxter, A.; Chambers, M.; Edfeldt, F.; Edman, K.; Freeman, A.; Johansson, C.; King, S.; Morley, A.; Petersen, J.; Rawlins, P.; et al. Non-covalent inhibitors of rhinovirus 3C protease. Bioorg. Med. Chem. Lett. 2011, 21, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.K.; Cho, J.H.; Jeong, P.; Lee, Y.; Lim, J.J.; Park, K.R.; Eom, S.H.; Kim, Y.C. Benserazide, the first allosteric inhibitor of Coxsackievirus B3 3C protease. FEBS Lett. 2015, 589, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.K.; Yun, S.H.; Ju, E.S.; Kim, B.K.; Lee, Y.J.; Yoo, D.K.; Kim, Y.C.; Jeon, E.S. Soluble Coxsackievirus B3 3C protease inhibitor prevents cardiomyopathy in an experimental chronic myocarditis murine model. Virus Res. 2015, 199, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.K.; Kim, J.H.; Kim, N.R.; Lee, W.G.; Lee, S.D.; Yun, S.H.; Jeon, E.S.; Kim, Y.C. Development of anti-coxsackievirus agents targeting 3C protease. Bioorg. Med. Chem. Lett. 2012, 22, 6952–6956. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Y.; Zhao, X.; Cui, Z.; Wang, M.; Wang, Y.; Li, L.; Sun, Q.; Yang, X.; Zeng, D.; Liu, Y.; et al. Cyanohydrin as an Anchoring Group for potent and selective inhibitors of enterovirus 71 3C protease. J. Med. Chem. 2015, 58, 9414–9420. [Google Scholar] [CrossRef] [PubMed]
- Ramajayam, R.; Tan, K.P.; Liang, P.H. Recent development of 3C and 3CL protease inhibitors for anti-coronavirus and anti-picornavirus drug discovery. Biochem. Soc. Trans. 2011, 39, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Shie, J.J.; Fang, J.M.; Kuo, T.H.; Kuo, C.J.; Liang, P.H.; Huang, H.J.; Wu, Y.T.; Jan, J.T.; Cheng, Y.S.; Wong, C.H. Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic α, β-unsaturated esters. Bioorg. Med. Chem. 2005, 13, 5240–5252. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Chen, S.J.; Hsu, M.F.; Wu, J.D.; Tseng, C.T.; Liu, Y.F.; Chen, H.C.; Kuo, C.W.; Wu, C.S.; Chang, L.W.; et al. Synthesis, crystal structure, structure-activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J. Med. Chem. 2006, 49, 4971–4980. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.J.; Liu, H.G.; Lo, Y.K.; Seong, C.M.; Lee, K.I.; Jung, Y.S.; Liang, P.H. Individual and common inhibitors of coronavirus and picornavirus main proteases. FEBS Lett. 2009, 583, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Prior, A.M.; Kim, Y.; Weerasekara, S.; Moroze, M.; Alliston, K.R.; Uy, R.A.; Groutas, W.C.; Chang, K.O.; Hua, D.H. Design, synthesis, and bioevaluation of viral 3C and 3C-like protease inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 6317–6320. [Google Scholar] [CrossRef] [PubMed]
- Mandadapu, S.R.; Weerawarna, P.M.; Prior, A.M.; Uy, R.A.; Aravapalli, S.; Alliston, K.R.; Lushington, G.H.; Kim, Y.; Hua, D.H.; Chang, K.O.; et al. Macrocyclic inhibitors of 3C and 3C-like proteases of picornavirus, norovirus, and coronavirus. Bioorg. Med. Chem. Lett. 2013, 23, 3709–3712. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Cordingley, M.G.; Ball, R.G.; Smith, J.L.; Dombrowski, A.W.; Goetz, M.A. Structure of stereochemistry of thysanone: A novel human rhinovirus 3C-protease inhibitor from Thysanophora penicilloides. Tetrahedron Lett. 1991, 32, 5279–5282. [Google Scholar] [CrossRef]
- Schünemann, K.; Furkert, D.P.; Choi, E.C.; Connelly, S.; Fraser, J.D.; Sperry, J.; Brimble, M.A. Synthesis of the 2-methylene analogue of the HRV 3C protease inhibitor thysanone (2-carbathysanone). Org. Biomol. Chem. 2014, 12, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Young Jeong, J.; Sperry, J.; Taylor, J.A.; Brimble, M.A. Synthesis and evaluation of 9-deoxy analogues of (−)-thysanone, an inhibitor of HRV 3C protease. Eur. J. Med. Chem. 2014, 87, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Chang, Y.C.; Hsiao, N.W.; Hsieh, J.L.; Wang, C.Y.; Kung, S.H.; Tsai, F.J.; Lan, Y.C.; Lin, C.W. Fisetin and rutin as 3C protease inhibitors of enterovirus A71. J. Virol. Methods 2012, 182, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.C.; et al. Phosphatidylserine vesicles enable efficient en Bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Roosien, J.; Belsham, G.J.; Ryan, M.D.; King, A.M.; Vlak, J.M. Synthesis of foot-and-mouth disease virus capsid proteins in insect cells using baculovirus expression vectors. J. Gen. Virol. 1990, 71, 1703–1711. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification of RNA Polymerases | Host Cell Protein | Virus | Functions | Cleavage Site | Reference |
|---|---|---|---|---|---|
| I | TAF110 | PV | Regulates of transcriptional initiation | IQLQ265…ACAQ805G | [49] |
| II | TBP | PV | Involved in three RNA polymerase-mediated transcriptions and responsible for DNA binding | AAAVQQ104STSQQA | [50] |
| CREB-1 | PV | Binds to DNA elements for induction by cAMP | GQYIAITQ172GGAIQL | [51] | |
| Oct-1 | PV | Binds to the octamer sequence ATGCAAAT and activates the promoters of genes for some small nuclear RNAs | KLGFTQ329GDVGLA (Speculated) | [52] | |
| P53 | PV | Functions as a typical sequence specific transcription activator and a tumor suppressor | Not clear | [53] | |
| III | TFIIIC | PV | Binds to the B-box internal promoter element to process tRNA transcription | TSQPPVPQ732GEAEED | [54] |
| Other factors | CstF64 | EV 71 | Polyadenylation of host mRNA | MQASMQ251GGVPAPGQMP | [56] |
| Histone 3 | FMDV | DNA binding and involved in the structure of chromatin in eukaryotic cells | GKAPRKQL120ATKAAR | [55] |
| Host Cell Protein | Virus | Functions | Cleavage Site | Reference |
|---|---|---|---|---|
| eIF4A I | FMDV | Binds capped mRNA to the 40S ribosomal subunit and unwinds double-stranded RNA | CIGGTNVRAE143VQKLQMEA | [63] |
| eIF4G I | FMDV | Brings mRNA to the 40S ribosome in translation initiation | RRSQQGPRKE712PRKIIATVL | [61] |
| eIF5B | PV | Positions the initiation methionine tRNA on the start codon of the mRNA | LCAAVEVMEQ478GVPEKEET | [64] |
| CV | ||||
| HRV | ||||
| G3BP1 | CV | RNA-binding protein that interacts with Ras-GAP | EAGEQ325GDIEP | [70] |
| PCBP2 | HAV | Translational activation and control of gene expression | IGRQ306GAKI (Speculated) | [66] |
| PV | AMQQ253SHFP…IGRQ306GAK | [65] | ||
| PABP | PV | Involved in poly (A) shortening and translation initiation | Not clear | [67] |
| HAV | Not clear | [68] | ||
| EMCV | VRPPAAIQ437GVQAGA | [69] | ||
| Sam68 | FMDV | Involved in cellular differentiation and proliferation | C-terminal portion | [71] |
| Genus | Virus | Viral Protein | Host Protein | Reference |
|---|---|---|---|---|
| Enterovirus | Rhinovirus | 2Apro | Nup 62 | [77] |
| 3Cpro/3CD | Nup 153, Nup 214, Nup 358 | [78] | ||
| 3Cpro | Nup 62 | [78] | ||
| Poliovirus | 2Apro | Nup 62, Nup 98, Nup 153 | [76] | |
| Cardiovirus | Encephalomyocarditis virus | Leader protein | Nup 62, Nup 153, Nup 214 | [81] |
| Theiler’s murine encephalomyelitis virus | Leader protein | Nup 62, Nup 98 | [82] |
| Virus | Host Factors | Cleavage Site | Reference |
|---|---|---|---|
| FMDV | NEMO | LSSPLALPSQ383RRSPPEEPPD | [116] |
| EV71 | IRF7 | GDLLLQAVQQ189SCLADHLLTA | [108] |
| TRIF | TEGSAGPQ312SLPLPILEP | [104] | |
| TAK1 | KNQAKQQ360SESGRL | [109] | |
| TAB2 | SISDGQLQ113GGQSNSEL | [109] | |
| CVB3 | MAVS | SYPMPVQETQ148APESPGENSEQ | [120] |
| TRIF | Q190, Q653, Q659, Q671, Q702 (potential) | [120] | |
| HAV | NEMO | METVPVLKAQ304ADIYKADFQA | [115] |
| TRIF | SDWSQ190GCSLR…EQSQ554HLDGER | [114] | |
| MAVS | LASQ428VDSP…YKSE463GTFG (potential) | [113] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, D.; Chen, S.; Cheng, A.; Wang, M. Roles of the Picornaviral 3C Proteinase in the Viral Life Cycle and Host Cells. Viruses 2016, 8, 82. https://doi.org/10.3390/v8030082
Sun D, Chen S, Cheng A, Wang M. Roles of the Picornaviral 3C Proteinase in the Viral Life Cycle and Host Cells. Viruses. 2016; 8(3):82. https://doi.org/10.3390/v8030082
Chicago/Turabian StyleSun, Di, Shun Chen, Anchun Cheng, and Mingshu Wang. 2016. "Roles of the Picornaviral 3C Proteinase in the Viral Life Cycle and Host Cells" Viruses 8, no. 3: 82. https://doi.org/10.3390/v8030082
APA StyleSun, D., Chen, S., Cheng, A., & Wang, M. (2016). Roles of the Picornaviral 3C Proteinase in the Viral Life Cycle and Host Cells. Viruses, 8(3), 82. https://doi.org/10.3390/v8030082