
Remodeling of the Host Cell Plasma Membrane by HIV-1 Nef and Vpu: A Strategy to Ensure Viral Fitness and Persistence


Abstract
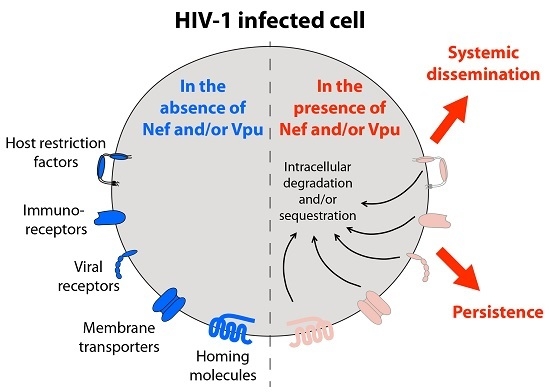
:
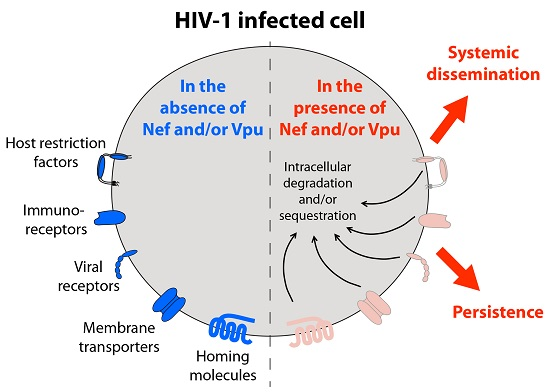
1. Introduction
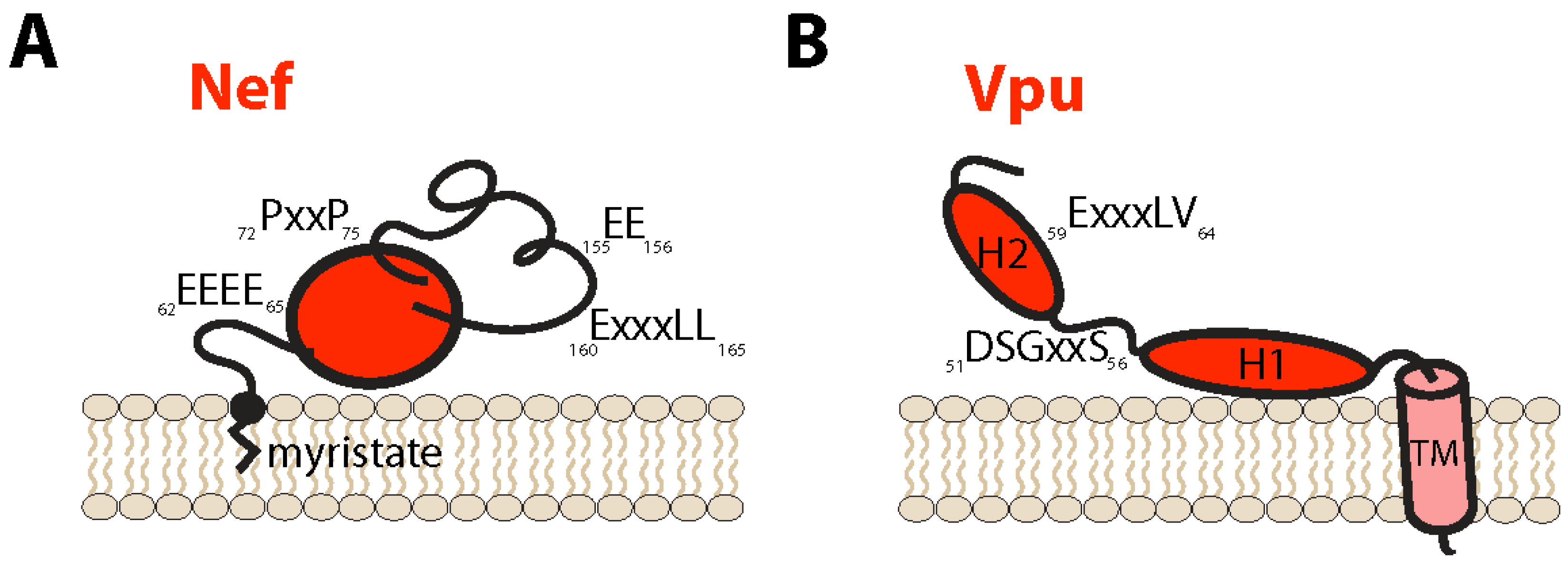
2. Negative Factor (Nef) Protein
3. Viral Protein U (Vpu)
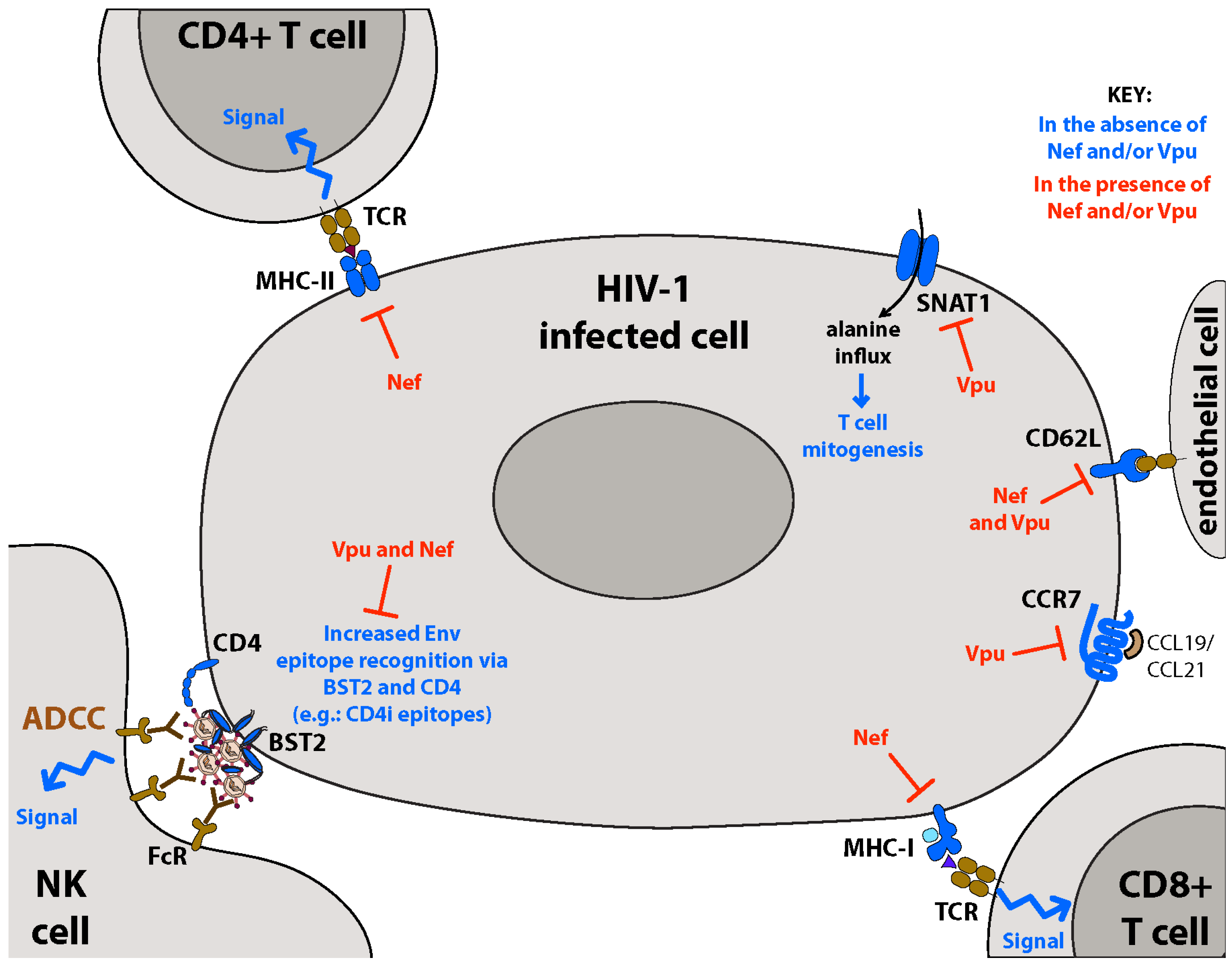
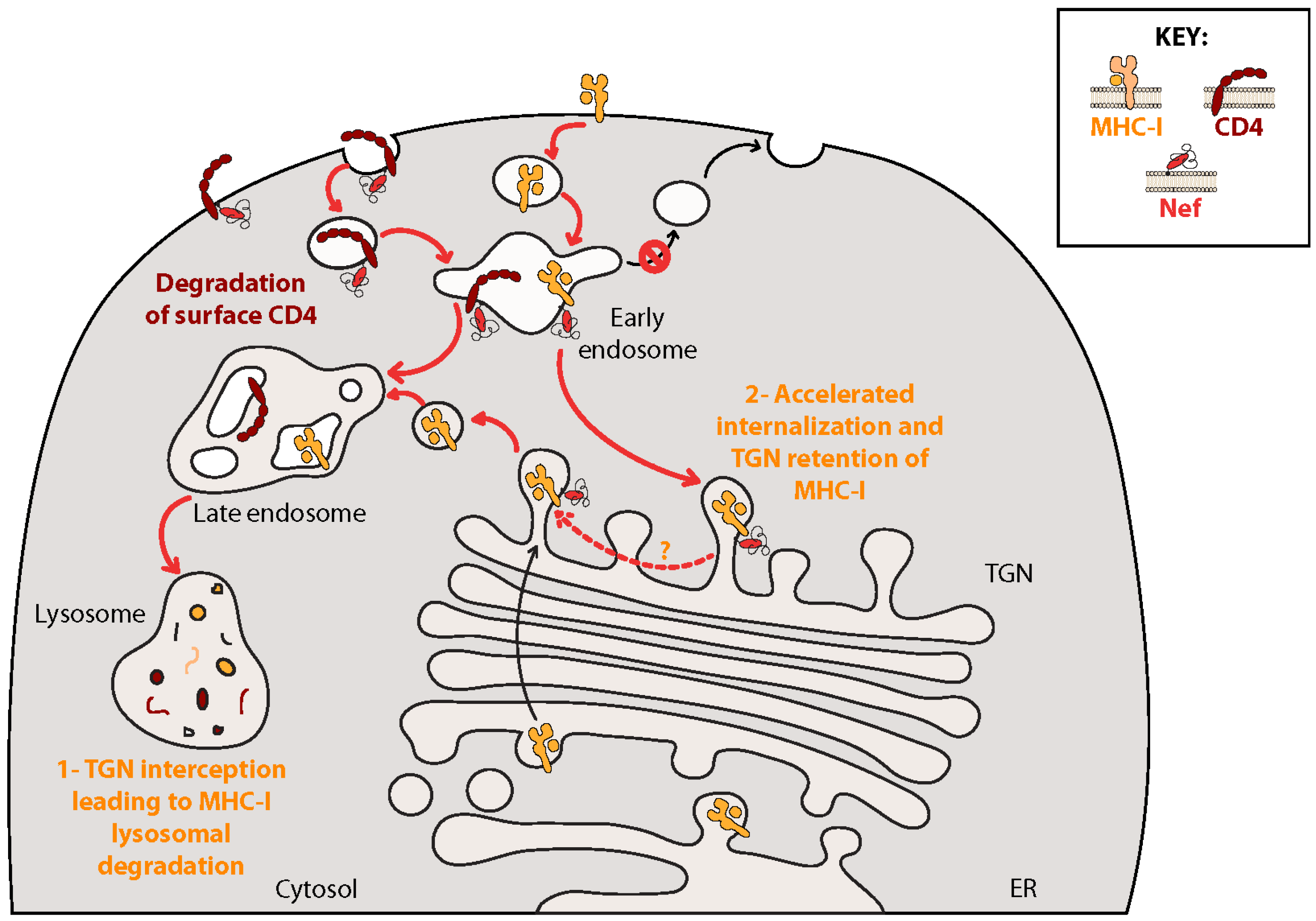
4. Downregulation of the CD4 Viral Receptor: Prevention of Superinfection, Enhancement of Viral Release and Protection from Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC)
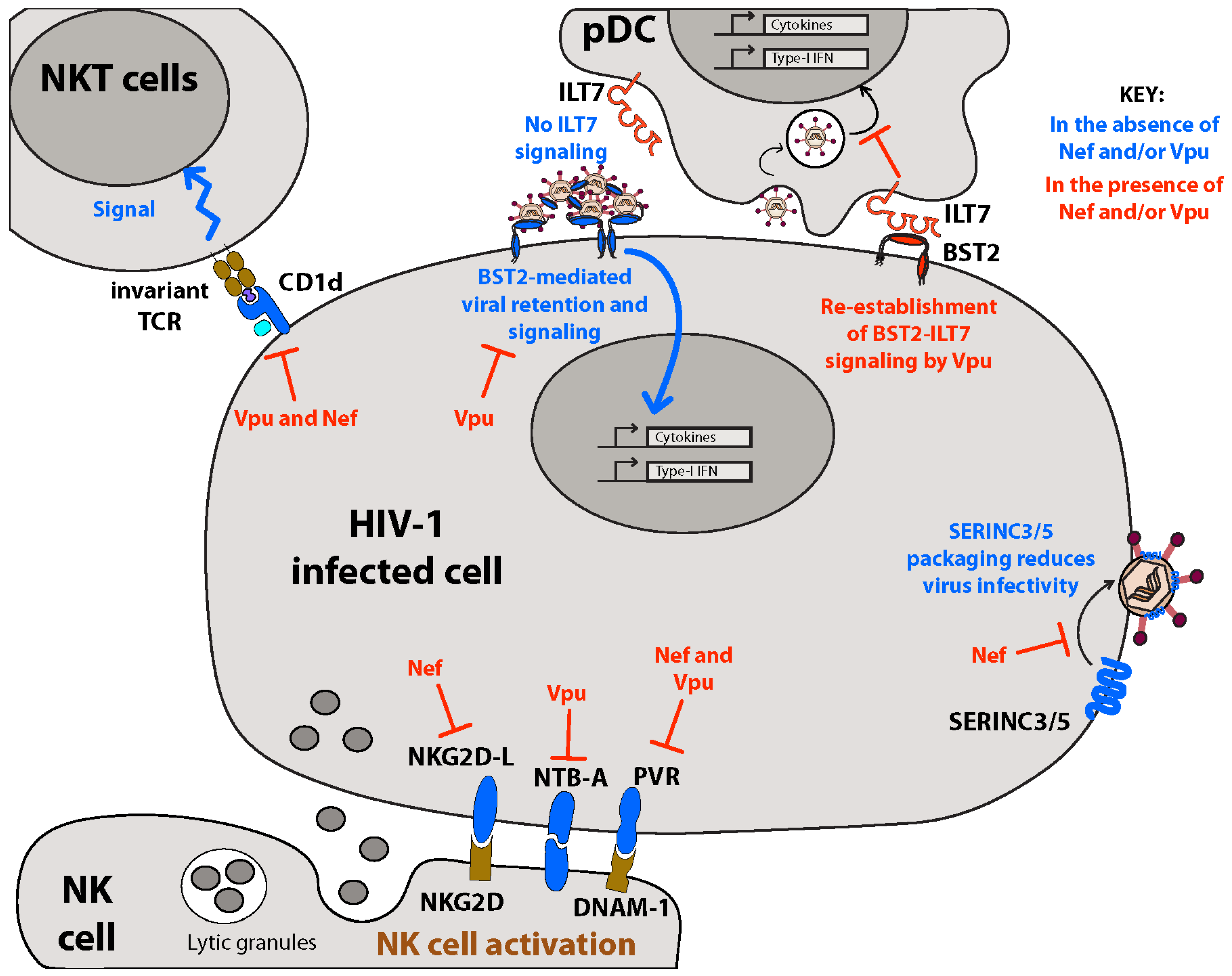
5. Membrane-Associated Restriction Factors: BST2 and SERINC3/5
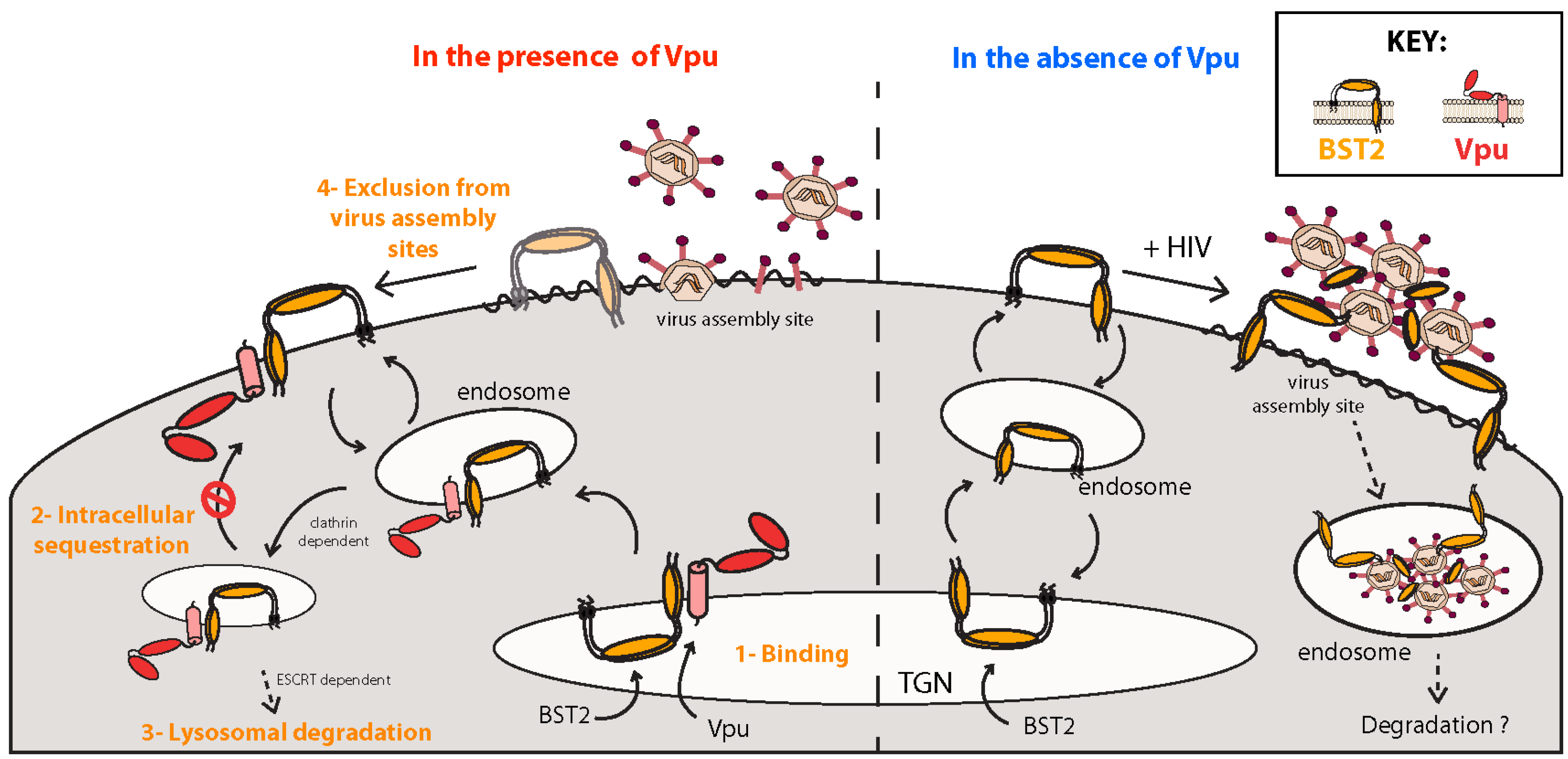
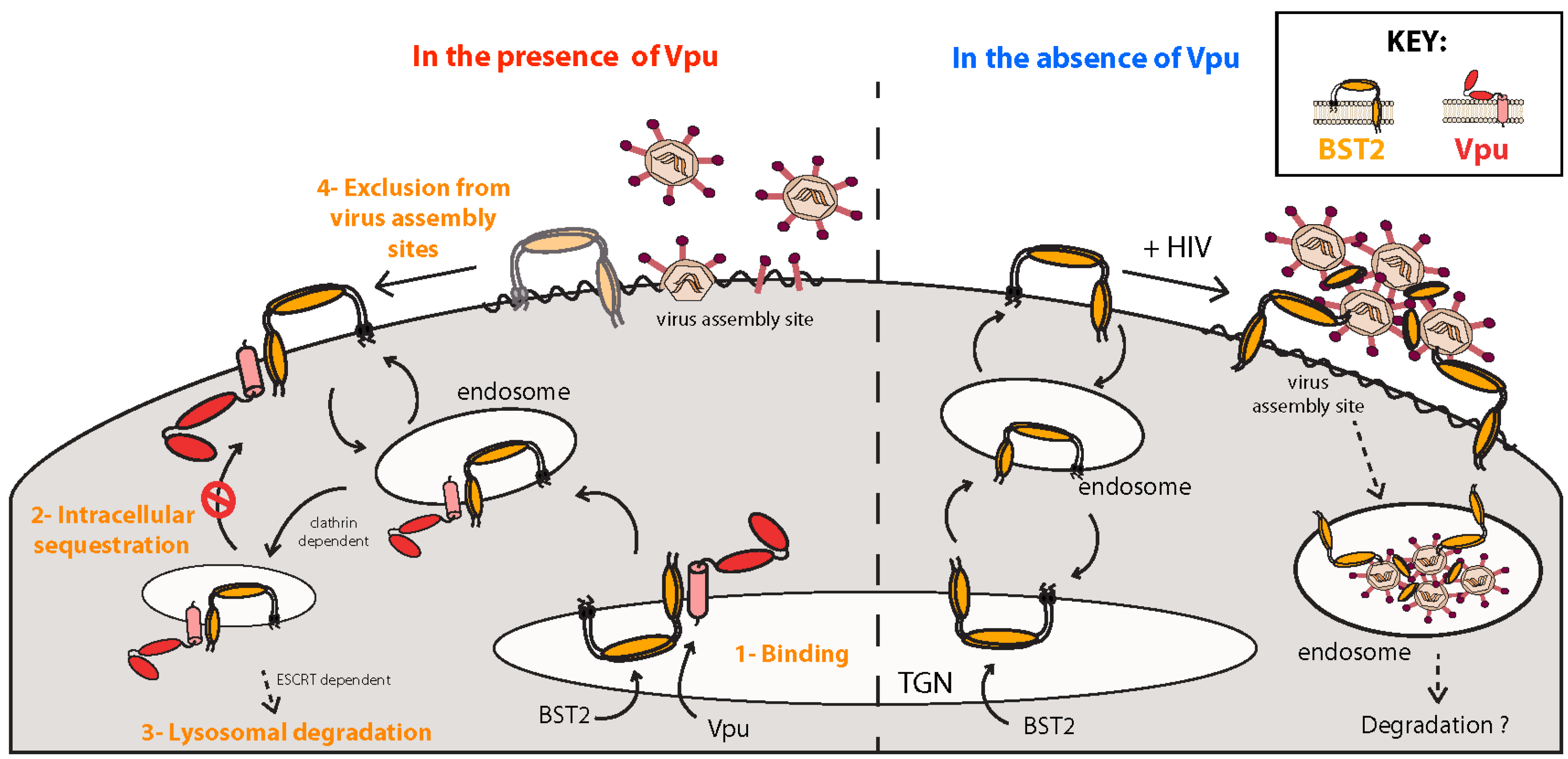
5.1. BST2, a Host Restriction Factor Counteracted by Vpu
5.1.1. Inhibition of Virus Particle Release
5.1.2. Mechanisms of Vpu-Mediated BST2 Antagonism
5.1.3. Sensor of Virus Assembly and Activator of NFκB-Dependent Proinflammatory Responses
5.1.4. Distorting IFN-I Homeostasis through Altered Membrane-Initiated Signaling Cascades
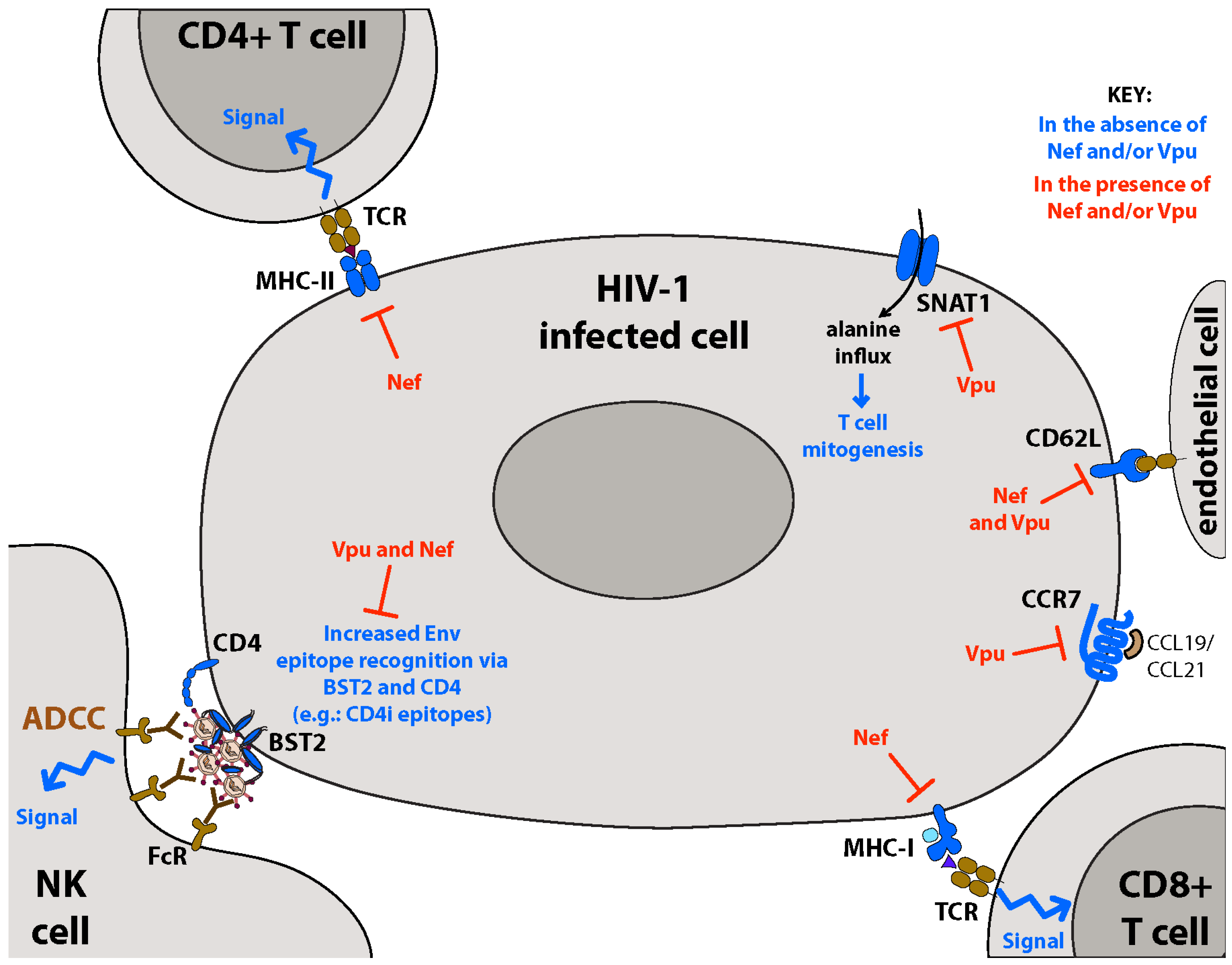
5.1.5. Vpu, BST2 and ADCC
5.2. SERINC3/5, Host Restriction Factors Counteracted by Nef
6. Tetraspanins: Virus Assembly Areas and the Control of Membrane Fusion
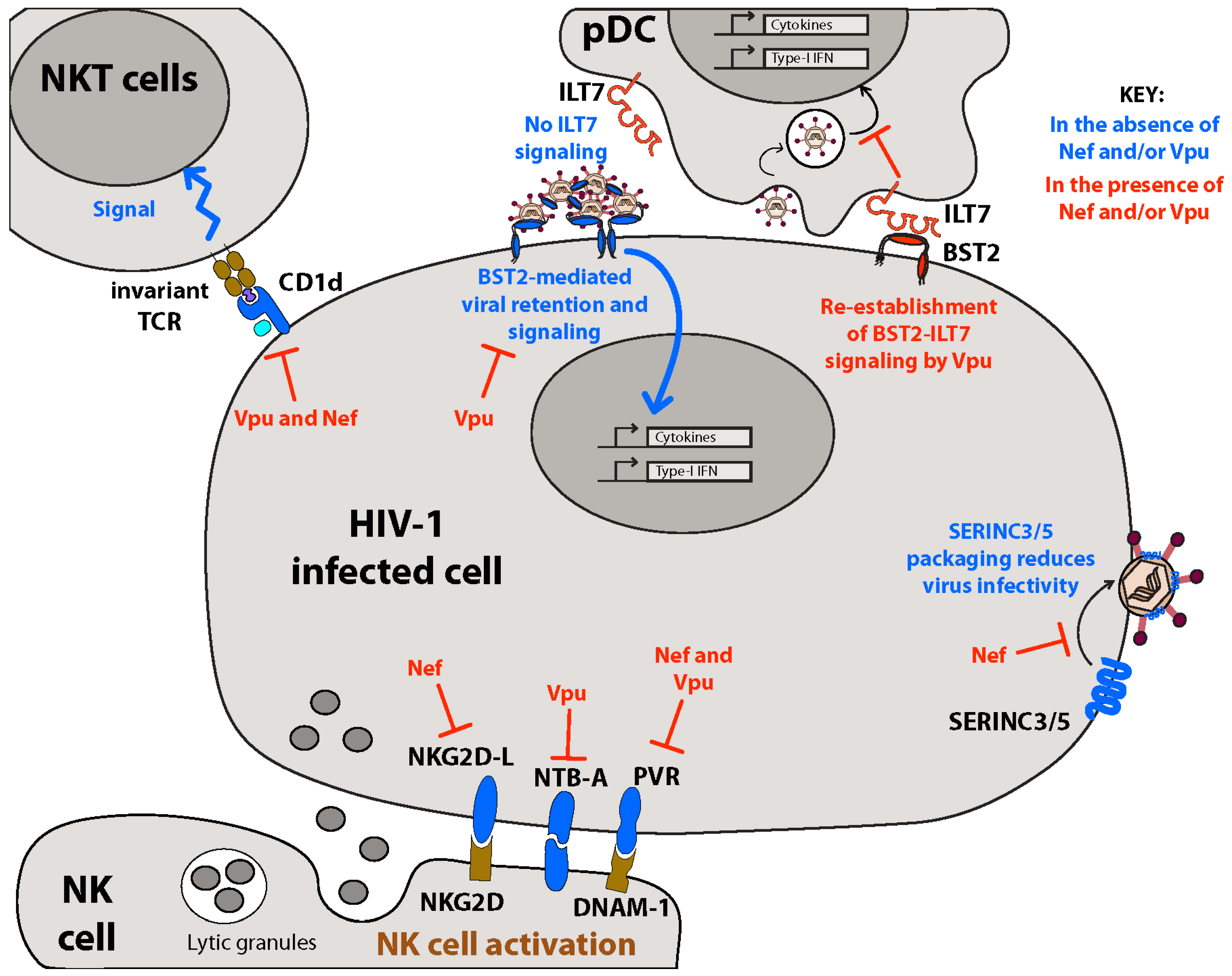
7. Inhibiting Immune Detection through Downregulation of Immunoreceptors: MHC-I/II, NKG2D-L, PVR, NTB-A, and CD1d
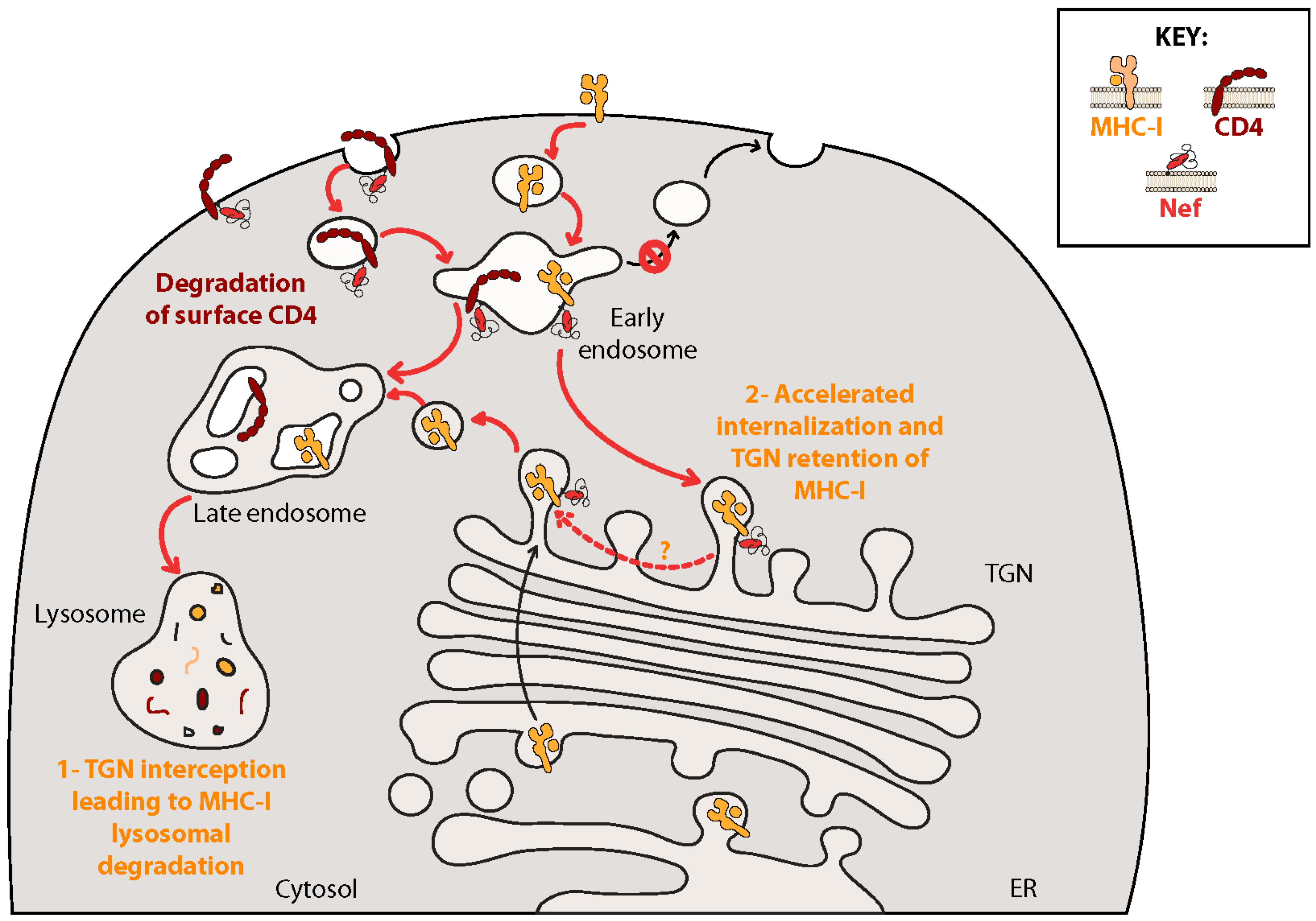
7.1. Nef Inhibits Adaptive Immune Responses: MHC-I/II
7.2. Inhibiting Innate Immunity: Downregulation of NK Cell Activating Receptors
8. Altered Cell Homing: Downregulation of CD62L and CCR7
9. Downregulation of Metabolic Transporters: SNAT1
10. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Goldberg, D.M.; Riordan, J.R. Role of membranes in disease. Clin. Physiol. Biochem. 1986, 4, 305–336. [Google Scholar] [PubMed]
- Ashrafuzzaman, M.; Tuszynski, J. Membrane-Related Diseases. In Membrane Biophysics; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2012; pp. 151–170. [Google Scholar]
- Maartens, G.; Celum, C.; Lewin, S.R. HIV infection: Epidemiology, pathogenesis, treatment, and prevention. Lancet 2014, 384, 258–271. [Google Scholar] [CrossRef]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 2008, 105, 7552–7557. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Keele, B.F.; Learn, G.H.; Giorgi, E.E.; Li, H.; Decker, J.M.; Wang, S.; Baalwa, J.; Kraus, M.H.; et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009, 206, 1273–1289. [Google Scholar] [CrossRef] [PubMed]
- Keele, B.F.; Li, H.; Learn, G.H.; Hraber, P.; Giorgi, E.E.; Grayson, T.; Sun, C.; Chen, Y.; Yeh, W.W.; Letvin, N.L.; et al. Low-dose rectal inoculation of rhesus macaques by SIVsmE660 or SIVmac251 recapitulates human mucosal infection by HIV-1. J. Exp. Med. 2009, 206, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.J.; Li, Q.; Abel, K.; Kim, E.Y.; Ma, Z.M.; Wietgrefe, S.; La Franco-Scheuch, L.; Compton, L.; Duan, L.; Shore, M.D.; et al. Propagation and dissemination of infection after vaginal transmission of simian immunodeficiency virus. J. Virol. 2005, 79, 9217–9227. [Google Scholar] [CrossRef] [PubMed]
- Haase, A.T. Early events in sexual transmission of HIV and SIV and opportunities for interventions. Annu. Rev. Med. 2011, 62, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Allan, J.S.; Coligan, J.E.; Lee, T.H.; McLane, M.F.; Kanki, P.J.; Groopman, J.E.; Essex, M. A new HTLV-III/LAV encoded antigen detected by antibodies from AIDS patients. Science 1985, 230, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Deacon, N.J.; Tsykin, A.; Solomon, A.; Smith, K.; Ludford-Menting, M.; Hooker, D.J.; McPhee, D.A.; Greenway, A.L.; Ellett, A.; Chatfield, C.; et al. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 1995, 270, 988–991. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, F.; Greenough, T.C.; Brettler, D.B.; Sullivan, J.L.; Desrosiers, R.C. Brief report: Absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N. Engl. J. Med. 1995, 332, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Kestler, H.W., 3rd; Ringler, D.J.; Mori, K.; Panicali, D.L.; Sehgal, P.K.; Daniel, M.D.; Desrosiers, R.C. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 1991, 65, 651–662. [Google Scholar] [CrossRef]
- Liu, L.X.; Heveker, N.; Fackler, O.T.; Arold, S.; Le Gall, S.; Janvier, K.; Peterlin, B.M.; Dumas, C.; Schwartz, O.; Benichou, S.; et al. Mutation of a conserved residue (D123) required for oligomerization of human immunodeficiency virus type 1 Nef protein abolishes interaction with human thioesterase and results in impairment of Nef biological functions. J. Virol. 2000, 74, 5310–5319. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Molloy, S.S.; Thomas, L.; Liu, G.; Xiang, Y.; Rybak, S.L.; Thomas, G. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell 1998, 94, 205–216. [Google Scholar] [CrossRef]
- Piguet, V.; Wan, L.; Borel, C.; Mangasarian, A.; Demaurex, N.; Thomas, G.; Trono, D. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat. Cell. Biol. 2000, 2, 163–167. [Google Scholar] [PubMed]
- Saksela, K.; Cheng, G.; Baltimore, D. Proline-rich (PxxP) motifs in HIV-1 Nef bind to SH3 domains of a subset of Src kinases and are required for the enhanced growth of Nef+ viruses but not for down-regulation of CD4. EMBO J. 1995, 14, 484–491. [Google Scholar] [PubMed]
- Piguet, V.; Gu, F.; Foti, M.; Demaurex, N.; Gruenberg, J.; Carpentier, J.L.; Trono, D. Nef-induced CD4 degradation: A diacidic-based motif in Nef functions as a lysosomal targeting signal through the binding of beta-COP in endosomes. Cell 1999, 97, 63–73. [Google Scholar] [CrossRef]
- Schwartz, S.; Felber, B.K.; Fenyo, E.M.; Pavlakis, G.N. Env and Vpu proteins of human immunodeficiency virus type 1 are produced from multiple bicistronic mRNAs. J. Virol. 1990, 64, 5448–5456. [Google Scholar] [PubMed]
- Pacyniak, E.; Gomez, M.L.; Gomez, L.M.; Mulcahy, E.R.; Jackson, M.; Hout, D.R.; Wisdom, B.J.; Stephens, E.B. Identification of a region within the cytoplasmic domain of the subtype B Vpu protein of human immunodeficiency virus type 1 (HIV-1) that is responsible for retention in the golgi complex and its absence in the Vpu protein from a subtype C HIV-1. AIDS Res. Hum. Retrovir. 2005, 21, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Henklein, P.; Boldyreff, B.; Wingender, E.; Strebel, K.; Porstmann, T. The human immunodeficiency virus type 1 encoded Vpu protein is phosphorylated by casein kinase-2 (CK-2) at positions Ser52 and Ser56 within a predicted alpha-helix-turn-alpha-helix-motif. J. Mol. Biol. 1994, 236, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef]
- Cohen, E.A.; Terwilliger, E.F.; Sodroski, J.G.; Haseltine, W.A. Identification of a protein encoded by the Vpu gene of HIV-1. Nature 1988, 334, 532–534. [Google Scholar] [CrossRef] [PubMed]
- Strebel, K.; Klimkait, T.; Martin, M.A. A novel gene of HIV-1, Vpu, and its 16-kilodalton product. Science 1988, 241, 1221–1223. [Google Scholar] [CrossRef] [PubMed]
- Hout, D.R.; Gomez, M.L.; Pacyniak, E.; Gomez, L.M.; Inbody, S.H.; Mulcahy, E.R.; Culley, N.; Pinson, D.M.; Powers, M.F.; Wong, S.W.; et al. Scrambling of the amino acids within the transmembrane domain of Vpu results in a simian-human immunodeficiency virus (SHIVTM) that is less pathogenic for pig-tailed macaques. Virology 2005, 339, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Misawa, N.; Fukuhara, M.; Iwami, S.; An, D.S.; Ito, M.; Koyanagi, Y. Vpu augments the initial burst phase of HIV-1 propagation and downregulates BST2 and CD4 in humanized mice. J. Virol. 2012, 86, 5000–5013. [Google Scholar] [CrossRef] [PubMed]
- Dave, V.P.; Hajjar, F.; Dieng, M.M.; Haddad, E.; Cohen, E.A. Efficient BST2 antagonism by Vpu is critical for early HIV-1 dissemination in humanized mice. Retrovirology 2013, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 1992, 66, 7193–7200. [Google Scholar] [PubMed]
- Guy, B.; Kieny, M.P.; Riviere, Y.; Le Peuch, C.; Dott, K.; Girard, M.; Montagnier, L.; Lecocq, J.P. HIV F/3' orf encodes a phosphorylated GTP-binding protein resembling an oncogene product. Nature 1987, 330, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Wildum, S.; Schindler, M.; Munch, J.; Kirchhoff, F. Contribution of Vpu, Env, and Nef to CD4 down-modulation and resistance of human immunodeficiency virus type 1-infected T cells to superinfection. J. Virol. 2006, 80, 8047–8059. [Google Scholar] [CrossRef] [PubMed]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein regulates the formation of intracellular gp160-CD4 complexes. J. Virol. 1992, 66, 226–234. [Google Scholar] [PubMed]
- Ross, T.M.; Oran, A.E.; Cullen, B.R. Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Curr. Biol. 1999, 9, 613–621. [Google Scholar] [CrossRef]
- Smalls-Mantey, A.; Connors, M.; Sattentau, Q.J. Comparative efficiency of HIV-1-infected T cell killing by NK cells, monocytes and neutrophils. PLoS ONE 2013, 8, e74858. [Google Scholar]
- Rolland, M.; Edlefsen, P.T.; Larsen, B.B.; Tovanabutra, S.; Sanders-Buell, E.; Hertz, T.; deCamp, A.C.; Carrico, C.; Menis, S.; Magaret, C.A.; et al. Increased HIV-1 vaccine efficacy against viruses with genetic signatures in Env V2. Nature 2012, 490, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.W.; Navis, M.; Isitman, G.; Wren, L.; Silvers, J.; Amin, J.; Kent, S.J.; Stratov, I. Activation of NK cells by ADCC antibodies and HIV disease progression. J. Acquir. Immune Defic. Syndr. 2011, 58, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Li, D.; He, X.; Zhao, Y.; Peng, H.; Ma, P.; Hong, K.; Liang, H.; Shao, Y. Impaired natural killer cell-induced antibody-dependent cell-mediated cytotoxicity is associated with human immunodeficiency virus-1 disease progression. Clin. Exp. Immunol. 2013, 171, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Gilbert, P.B.; McElrath, M.J.; Zolla-Pazner, S.; Tomaras, G.D.; Alam, S.M.; Evans, D.T.; Montefiori, D.C.; Karnasuta, C.; Sutthent, R.; et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 2012, 366, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Yates, N.L.; Liao, H.X.; Fong, Y.; Decamp, A.; Vandergrift, N.A.; Williams, W.T.; Alam, S.M.; Ferrari, G.; Yang, Z.Y.; Seaton, K.E.; et al. Vaccine-Induced Env V1-V2 IgG3 Correlates with Lower HIV-1 Infection Risk and Declines Soon After Vaccination. Sci. Transl. Med. 2014, 6, 228ra39. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Pollara, J.; Kozink, D.; Harms, T.; Drinker, M.; Freel, S.; Moody, M.A.; Alam, S.M.; Tomaras, G.D.; Ochsenbauer, C.; et al. An HIV-1 gp120 envelope human monoclonal antibody that recognizes a C1 conformational epitope mediates potent antibody-dependent cellular cytotoxicity (ADCC) activity and defines a common ADCC epitope in human HIV-1 serum. J. Virol. 2011, 85, 7029–7036. [Google Scholar] [CrossRef] [PubMed]
- Bonsignori, M.; Pollara, J.; Moody, M.A.; Alpert, M.D.; Chen, X.; Hwang, K.K.; Gilbert, P.B.; Huang, Y.; Gurley, T.C.; Kozink, D.M.; et al. Antibody-dependent cellular cytotoxicity-mediating antibodies from an HIV-1 vaccine efficacy trial target multiple epitopes and preferentially use the VH1 gene family. J. Virol. 2012, 86, 11521–11532. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, R.; Moore, J.; Accola, M.; Desjardin, E.; Robinson, J.; Sodroski, J. Involvement of the V1/V2 variable loop structure in the exposure of human immunodeficiency virus type 1 gp120 epitopes induced by receptor binding. J. Virol. 1995, 69, 5723–5733. [Google Scholar] [PubMed]
- Veillette, M.; Coutu, M.; Richard, J.; Batraville, L.A.; Dagher, O.; Bernard, N.; Tremblay, C.; Kaufmann, D.E.; Roger, M.; Finzi, A. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J. Virol. 2015, 89, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Pazgier, M.; Sajadi, M.M.; Kamin-Lewis, R.; Al-Darmarki, S.; Flinko, R.; Lovo, E.; Wu, X.; Robinson, J.E.; Seaman, M.S.; et al. Diverse specificity and effector function among human antibodies to HIV-1 envelope glycoprotein epitopes exposed by CD4 binding. Proc. Natl. Acad. Sci. USA 2013, 110, E69–E78. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.N.; Lukhele, S.; Hajjar, F.; Routy, J.P.; Cohen, E.A. HIV Nef and Vpu protect HIV-infected CD4+ T cells from antibody-mediated cell lysis through down-modulation of CD4 and BST2. Retrovirology 2014, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Veillette, M.; Desormeaux, A.; Medjahed, H.; Gharsallah, N.E.; Coutu, M.; Baalwa, J.; Guan, Y.; Lewis, G.; Ferrari, G.; Hahn, B.H.; et al. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J. Virol. 2014, 88, 2633–2644. [Google Scholar] [CrossRef] [PubMed]
- Grzesiek, S.; Stahl, S.J.; Wingfield, P.T.; Bax, A. The CD4 determinant for downregulation by HIV-1 Nef directly binds to Nef. Mapping of the Nef binding surface by NMR. Biochemistry 1996, 35, 10256–10261. [Google Scholar] [CrossRef] [PubMed]
- Preusser, A.; Briese, L.; Baur, A.S.; Willbold, D. Direct in vitro binding of full-length human immunodeficiency virus type 1 Nef protein to CD4 cytoplasmic domain. J. Virol. 2001, 75, 3960–3964. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, R.; Lindwasser, O.W.; Smith, W.J.; Hurley, J.H.; Bonifacino, J.S. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J. Virol. 2007, 81, 3877–3890. [Google Scholar] [CrossRef] [PubMed]
- Lindwasser, O.W.; Smith, W.J.; Chaudhuri, R.; Yang, P.; Hurley, J.H.; Bonifacino, J.S. A diacidic motif in human immunodeficiency virus type 1 Nef is a novel determinant of binding to AP-2. J. Virol. 2008, 82, 1166–1174. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, M.R.; Wonderlich, E.R.; Roeth, J.F.; Leonard, J.A.; Collins, K.L. HIV-1 Nef targets MHC-I and CD4 for degradation via a final common beta-COP-dependent pathway in T cells. PLoS Pathog. 2008, 4, e1000131. [Google Scholar] [CrossRef] [PubMed]
- DaSilva, L.L.; Sougrat, R.; Burgos, P.V.; Janvier, K.; Mattera, R.; Bonifacino, J.S. Human immunodeficiency virus type 1 Nef protein targets CD4 to the multivesicular body pathway. J. Virol. 2009, 83, 6578–6590. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Anton, L.C.; Bacik, I.; Cox, J.H.; Bour, S.; Bennink, J.R.; Orlowski, M.; Strebel, K.; Yewdell, J.W. CD4 glycoprotein degradation induced by human immunodeficiency virus type 1 Vpu protein requires the function of proteasomes and the ubiquitin-conjugating pathway. J. Virol. 1998, 72, 2280–2288. [Google Scholar] [PubMed]
- Binette, J.; Dube, M.; Mercier, J.; Halawani, D.; Latterich, M.; Cohen, E.A. Requirements for the selective degradation of CD4 receptor molecules by the human immunodeficiency virus type 1 Vpu protein in the endoplasmic reticulum. Retrovirology 2007, 4, 75. [Google Scholar] [CrossRef] [PubMed]
- Magadan, J.G.; Perez-Victoria, F.J.; Sougrat, R.; Ye, Y.; Strebel, K.; Bonifacino, J.S. Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog. 2010, 6, e1000869. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, P.; Desfarges, S.; Bartha, I.; Joos, B.; Zangger, N.; Munoz, M.; Gunthard, H. F.; Beerenwinkel, N.; Telenti, A.; Ciuffi, A. 24 hours in the life of HIV-1 in a T cell line. PLoS Pathog. 2013, 9, e1003161. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.J.; Friborg, J.; Checroune, F.; Gratton, S.; Boisvert, F.; Sekaly, R.P.; Cohen, E.A. Degradation of CD4 induced by human immunodeficiency virus type 1 Vpu protein: A predicted alpha-helix structure in the proximal cytoplasmic region of CD4 contributes to Vpu sensitivity. Virology 1995, 209, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Bour, S.; Schubert, U.; Strebel, K. The human immunodeficiency virus type 1 Vpu protein specifically binds to the cytoplasmic domain of CD4: Implications for the mechanism of degradation. J. Virol. 1995, 69, 1510–1520. [Google Scholar] [PubMed]
- Bour, S.; Strebel, K. The HIV-1 Vpu protein: A multifunctional enhancer of viral particle release. Microbes Infect. 2003, 5, 1029–1039. [Google Scholar] [CrossRef]
- Neil, S.J.; Eastman, S.W.; Jouvenet, N.; Bieniasz, P.D. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006, 2, e39. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, R.J.; Talbot, K.J.; Callahan, M.; Harper, W.; Panganiban, A.T. Cell type-dependence for Vpu function. J. Med. Primatol. 1994, 23, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Varthakavi, V.; Smith, R.M.; Bour, S.P.; Strebel, K.; Spearman, P. Viral protein U counteracts a human host cell restriction that inhibits HIV-1 particle production. Proc. Natl. Acad. Sci. USA 2003, 100, 15154–15159. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Sandrin, V.; Sundquist, W.I.; Bieniasz, P.D. An interferon-alpha-induced tethering mechanism inhibits HIV-1 and Ebola virus particle release but is counteracted by the HIV-1 Vpu protein. Cell Host Microbe 2007, 2, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.F.; Iwabu, Y.; Tokunaga, K. Structural Basis for the Antiviral Activity of BST-2/Tetherin and Its Viral Antagonism. Front. Microbiol. 2011, 2, 250. [Google Scholar] [CrossRef] [PubMed]
- Hinz, A.; Miguet, N.; Natrajan, G.; Usami, Y.; Yamanaka, H.; Renesto, P.; Hartlieb, B.; McCarthy, A.A.; Simorre, J.P.; Gottlinger, H.; et al. Structural basis of HIV-1 tethering to membranes by the BST-2/tetherin ectodomain. Cell Host Microbe 2010, 7, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Schubert, H.L.; Zhai, Q.; Sandrin, V.; Eckert, D.M.; Garcia-Maya, M.; Saul, L.; Sundquist, W.I.; Steiner, R.A.; Hill, C.P. Structural and functional studies on the extracellular domain of BST2/tetherin in reduced and oxidized conformations. Proc. Natl. Acad. Sci. USA 2010, 107, 17951–17956. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Bieniasz, P.D. Mechanism of HIV-1 virion entrapment by tetherin. PLoS Pathog. 2013, 9, e1003483. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Rocha, S.; Mangeat, B.; Blanchet, F.; Uji, I.H.; Hofkens, J.; Piguet, V. Quantitative multicolor super-resolution microscopy reveals tetherin HIV-1 interaction. PLoS Pathog. 2011, 7, e1002456. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, E.; Andrew, A.J.; Kao, S.; Strebel, K. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc. Natl. Acad. Sci. USA 2009, 106, 2868–2873. [Google Scholar] [CrossRef] [PubMed]
- Dube, M.; Roy, B.B.; Guiot-Guillain, P.; Binette, J.; Mercier, J.; Chiasson, A.; Cohen, E.A. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010, 6, e1000856. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu Binds Directly to Tetherin and Displaces It from Nascent Virions. PLoS Pathog. 2013, 9, e1003299. [Google Scholar] [CrossRef] [PubMed]
- Vigan, R.; Neil, S.J. Determinants of tetherin antagonism in the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein. J. Virol. 2010, 84, 12958–12970. [Google Scholar] [CrossRef] [PubMed]
- Skasko, M.; Wang, Y.; Tian, Y.; Tokarev, A.; Munguia, J.; Ruiz, A.; Stephens, E.B.; Opella, S.J.; Guatelli, J. HIV-1 Vpu protein antagonizes innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J. Biol. Chem. 2012, 287, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Janvier, K.; Pelchen-Matthews, A.; Renaud, J.B.; Caillet, M.; Marsh, M.; Berlioz-Torrent, C. The ESCRT-0 component HRS is required for HIV-1 Vpu-mediated BST-2/tetherin down-regulation. PLoS Pathog. 2011, 7, e1001265. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Fruh, K.; Moses, A.V. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a {beta}TrCP-dependent mechanism. J. Virol. 2009, 83, 7931–7947. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via beta-TrCP and endo-lysosomal trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Tokarev, A.A.; Munguia, J.; Guatelli, J.C. Serine-threonine ubiquitination mediates downregulation of BST-2/tetherin and relief of restricted virion release by HIV-1 Vpu. J. Virol. 2011, 85, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Gustin, J.K.; Douglas, J.L.; Bai, Y.; Moses, A.V. Ubiquitination of BST-2 protein by HIV-1 Vpu protein does not require lysine, serine, or threonine residues within the BST-2 cytoplasmic domain. J. Biol. Chem. 2012, 287, 14837–14850. [Google Scholar] [CrossRef] [PubMed]
- Tervo, H.M.; Homann, S.; Ambiel, I.; Fritz, J.V.; Fackler, O.T.; Keppler, O.T. beta-TrCP is dispensable for Vpu's ability to overcome the CD317/Tetherin-imposed restriction to HIV-1 release. Retrovirology 2011, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Fritz, J.V.; Bitzegeio, J.; Fackler, O.T.; Keppler, O.T. HIV-1 Vpu blocks recycling and biosynthetic transport of the intrinsic immunity factor CD317/tetherin to overcome the virion release restriction. MBio 2011, 2, e00036–11. [Google Scholar] [CrossRef] [PubMed]
- Kueck, T.; Neil, S.J. A cytoplasmic tail determinant in HIV-1 Vpu mediates targeting of tetherin for endosomal degradation and counteracts interferon-induced restriction. PLoS Pathog. 2012, 8, e1002609. [Google Scholar] [CrossRef] [PubMed]
- Kueck, T.; Foster, T.L.; Weinelt, J.; Sumner, J.C.; Pickering, S.; Neil, S.J.D. Serine Phosphorylation of HIV-1 Vpu and Its Binding to Tetherin Regulates Interaction with Clathrin Adaptors. PLoS Pathog. 2015, 11, e1005141. [Google Scholar] [CrossRef] [PubMed]
- Galao, R.P.; Le Tortorec, A.; Pickering, S.; Kueck, T.; Neil, S.J. Innate sensing of HIV-1 assembly by Tetherin induces NFkappaB-dependent proinflammatory responses. Cell Host Microbe 2012, 12, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Weinelt, J.; Neil, S.J. Differential sensitivities of tetherin isoforms to counteraction by primate lentiviruses. J. Virol. 2014, 88, 5845–5858. [Google Scholar] [CrossRef] [PubMed]
- Dube, M.; Paquay, C.; Roy, B.B.; Bego, M.G.; Mercier, J.; Cohen, E.A. HIV-1 Vpu antagonizes BST-2 by interfering mainly with the trafficking of newly synthesized BST-2 to the cell surface. Traffic 2011, 12, 1714–1729. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.; Kwan, W.; Guatelli, J. Role of the endocytic pathway in the counteraction of BST-2 by human lentiviral pathogens. J. Virol. 2011, 85, 9834–9846. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Weber, E.; Tokarev, A.; Lewinski, M.; Rizk, M.; Suarez, M.; Guatelli, J.; Xiong, Y. Structural basis of HIV-1 Vpu-mediated BST2 antagonism via hijacking of the clathrin adaptor protein complex 1. Elife 2014, 3, e02362. [Google Scholar] [CrossRef] [PubMed]
- Galao, R.P.; Pickering, S.; Curnock, R.; Neil, S.J. Retroviral retention activates a Syk-dependent HemITAM in human tetherin. Cell Host Microbe 2014, 16, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Cocka, L.J.; Bates, P. Identification of alternatively translated Tetherin isoforms with differing antiviral and signaling activities. PLoS Pathog. 2012, 8, e1002931. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Takeuchi, J.S.; Ren, F.; Matsuda, K.; Sato, K.; Kimura, Y.; Misawa, N.; Yoshikawa, R.; Nakano, Y.; Yamada, E.; et al. Characterization of red-capped mangabey tetherin: Implication for the co-evolution of primates and their lentiviruses. Sci. Rep. 2014, 4, 5529. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Trinchieri, G.; Liu, Y.J. Plasmacytoid dendritic cells in immunity. Nat. Immunol. 2004, 5, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Lepelley, A.; Louis, S.; Sourisseau, M.; Law, H.K.; Pothlichet, J.; Schilte, C.; Chaperot, L.; Plumas, J.; Randall, R.E.; Si-Tahar, M.; et al. Innate sensing of HIV-infected cells. PLoS Pathog. 2011, 7, e1001284. [Google Scholar] [CrossRef] [PubMed]
- Fonteneau, J.F.; Larsson, M.; Beignon, A.S.; McKenna, K.; Dasilva, I.; Amara, A.; Liu, Y.J.; Lifson, J.D.; Littman, D.R.; Bhardwaj, N. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J. Virol. 2004, 78, 5223–5232. [Google Scholar] [CrossRef] [PubMed]
- Beignon, A.S.; McKenna, K.; Skoberne, M.; Manches, O.; DaSilva, I.; Kavanagh, D.G.; Larsson, M.; Gorelick, R.J.; Lifson, J.D.; Bhardwaj, N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Investig. 2005, 115, 3265–3275. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Bover, L.; Cho, M.; Wen, X.; Hanabuchi, S.; Bao, M.; Rosen, D.B.; Wang, Y.H.; Shaw, J.L.; Du, Q.; et al. Regulation of TLR7/9 responses in plasmacytoid dendritic cells by BST2 and ILT7 receptor interaction. J. Exp. Med. 2009, 206, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Bego, M.G.; Cote, E.; Aschman, N.; Mercier, J.; Weissenhorn, W.; Cohen, E.A. Vpu Exploits the Cross-Talk between BST2 and the ILT7 Receptor to Suppress Anti-HIV-1 Responses by Plasmacytoid Dendritic Cells. PLoS Pathog. 2015, 11, e1005024. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.F.; Heyer, L.N.; von Bredow, B.; Weisgrau, K.L.; Moldt, B.; Burton, D.R.; Rakasz, E.G.; Evans, D.T. Tetherin antagonism by Vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 6425–6430. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.A.; Hamlin, R.E.; Monroe, A.; Moldt, B.; Hotta, M.T.; Rodriguez Caprio, G.; Fierer, D.S.; Simon, V.; Chen, B.K. HIV-1 Vpu antagonism of tetherin inhibits antibody-dependent cellular cytotoxic responses by natural killer cells. J. Virol. 2014, 88, 6031–6046. [Google Scholar] [CrossRef] [PubMed]
- Chowers, M.Y.; Spina, C.A.; Kwoh, T.J.; Fitch, N.J.; Richman, D.D.; Guatelli, J.C. Optimal infectivity in vitro of human immunodeficiency virus type 1 requires an intact nef gene. J. Virol. 1994, 68, 2906–2914. [Google Scholar] [PubMed]
- Carl, S.; Greenough, T.C.; Krumbiegel, M.; Greenberg, M.; Skowronski, J.; Sullivan, J.L.; Kirchhoff, F. Modulation of different human immunodeficiency virus type 1 Nef functions during progression to AIDS. J. Virol. 2001, 75, 3657–3665. [Google Scholar] [CrossRef] [PubMed]
- Aiken, C.; Trono, D. Nef stimulates human immunodeficiency virus type 1 proviral DNA synthesis. J. Virol. 1995, 69, 5048–5056. [Google Scholar] [PubMed]
- Pizzato, M.; Helander, A.; Popova, E.; Calistri, A.; Zamborlini, A.; Palu, G.; Gottlinger, H.G. Dynamin 2 is required for the enhancement of HIV-1 infectivity by Nef. Proc. Natl. Acad. Sci. USA 2007, 104, 6812–6817. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Douglas, J.L.; Livingston, R.L.; Garcia, J.V. Infectivity enhancement by HIV-1 Nef is dependent on the pathway of virus entry: Implications for HIV-based gene transfer systems. Virology 1998, 241, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Pizzato, M.; Popova, E.; Gottlinger, H.G. Nef can enhance the infectivity of receptor-pseudotyped human immunodeficiency virus type 1 particles. J. Virol. 2008, 82, 10811–10819. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.D.; Warmerdam, M.T.; Page, K.A.; Feinberg, M.B.; Greene, W.C. Expression of the human immunodeficiency virus type 1 (HIV-1) nef gene during HIV-1 production increases progeny particle infectivity independently of gp160 or viral entry. J. Virol. 1995, 69, 579–584. [Google Scholar] [PubMed]
- Chazal, N.; Singer, G.; Aiken, C.; Hammarskjold, M.L.; Rekosh, D. Human immunodeficiency virus type 1 particles pseudotyped with envelope proteins that fuse at low pH no longer require Nef for optimal infectivity. J. Virol. 2001, 75, 4014–4018. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Cohen, F.S.; Melikyan, G.B. The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J. Membr. Biol. 2004, 199, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Claas, C.; Stipp, C.S.; Hemler, M.E. Evaluation of prototype transmembrane 4 superfamily protein complexes and their relation to lipid rafts. J. Biol. Chem. 2001, 276, 7974–7984. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Mitar, I.; Sattentau, Q.J. Adhesion molecule interactions facilitate human immunodeficiency virus type 1-induced virological synapse formation between T cells. J. Virol. 2007, 81, 13916–13921. [Google Scholar] [CrossRef] [PubMed]
- Nydegger, S.; Khurana, S.; Krementsov, D.N.; Foti, M.; Thali, M. Mapping of tetraspanin-enriched microdomains that can function as gateways for HIV-1. J. Cell Biol. 2006, 173, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Hogue, I.B.; Grover, J.R.; Soheilian, F.; Nagashima, K.; Ono, A. Gag induces the coalescence of clustered lipid rafts and tetraspanin-enriched microdomains at HIV-1 assembly sites on the plasma membrane. J. Virol. 2011, 85, 9749–9766. [Google Scholar] [CrossRef] [PubMed]
- Symeonides, M.; Lambele, M.; Roy, N.H.; Thali, M. Evidence showing that tetraspanins inhibit HIV-1-induced cell-cell fusion at a post-hemifusion stage. Viruses 2014, 6, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Tachibana, I.; Miyado, K.; Kobayashi, M.; Miyazaki, T.; Funakoshi, T.; Kimura, H.; Yamane, H.; Saito, Y.; Goto, H.; et al. Tetraspanins CD9 and CD81 function to prevent the fusion of mononuclear phagocytes. J. Cell. Biol. 2003, 161, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Huerta, L.; Lopez-Balderas, N.; Rivera-Toledo, E.; Sandoval, G.; Gomez-Icazbalceta, G.; Villarreal, C.; Lamoyi, E.; Larralde, C. HIV-envelope-dependent cell-cell fusion: Quantitative studies. ScientificWorldJournal 2009, 9, 746–763. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Krementsov, D.N.; Khurana, S.; Roy, N.H.; Thali, M. Formation of syncytia is repressed by tetraspanins in human immunodeficiency virus type 1-producing cells. J. Virol. 2009, 83, 7467–7474. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Alonso, M.; Yanez-Mo, M.; Barreiro, O.; Alvarez, S.; Munoz-Fernandez, M.A.; Valenzuela-Fernandez, A.; Sanchez-Madrid, F. Tetraspanins CD9 and CD81 modulate HIV-1-induced membrane fusion. J. Immunol. 2006, 177, 5129–5137. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Aoki, J.; Misawa, N.; Daikoku, E.; Sano, K.; Tanaka, Y.; Koyanagi, Y. Modulation of human immunodeficiency virus type 1 infectivity through incorporation of tetraspanin proteins. J. Virol. 2008, 82, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Lambele, M.; Koppensteiner, H.; Symeonides, M.; Roy, N.H.; Chan, J.; Schindler, M.; Thali, M. Vpu Is the Main Determinant for Tetraspanin Downregulation in HIV-1-Infected Cells. J. Virol. 2015, 89, 3247–3255. [Google Scholar] [CrossRef] [PubMed]
- Haller, C.; Muller, B.; Fritz, J.V.; Lamas-Murua, M.; Stolp, B.; Pujol, F.M.; Keppler, O.T.; Fackler, O.T. HIV-1 Nef and Vpu are functionally redundant broad-spectrum modulators of cell surface receptors, including tetraspanins. J. Virol. 2014, 88, 14241–14257. [Google Scholar] [CrossRef] [PubMed]
- Matheson, N.J.; Sumner, J.; Wals, K.; Rapiteanu, R.; Weekes, M.P.; Vigan, R.; Weinelt, J.; Schindler, M.; Antrobus, R.; Costa, A.S.; et al. Cell Surface Proteomic Map of HIV Infection Reveals Antagonism of Amino Acid Metabolism by Vpu and Nef. Cell Host Microbe 2015, 18, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, O.; Marechal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.L.; Chen, B.K.; Kalams, S.A.; Walker, B.D.; Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 1998, 391, 397–401. [Google Scholar] [PubMed]
- Noviello, C.M.; Benichou, S.; Guatelli, J.C. Cooperative binding of the class I major histocompatibility complex cytoplasmic domain and human immunodeficiency virus type 1 Nef to the endosomal AP-1 complex via its mu subunit. J. Virol. 2008, 82, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Singh, R.; Homann, S.; Yang, H.; Guatelli, J.; Xiong, Y. Structural basis of evasion of cellular adaptive immunity by HIV-1 Nef. Nat. Struct. Mol. Biol. 2012, 19, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Wonderlich, E.R.; Williams, M.; Collins, K.L. The tyrosine binding pocket in the adaptor protein 1 (AP-1) mu1 subunit is necessary for Nef to recruit AP-1 to the major histocompatibility complex class I cytoplasmic tail. J. Biol. Chem. 2008, 283, 3011–3022. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.R.; Roeth, J.F.; Williams, M.; Filzen, T.M.; Fleis, R.I.; Collins, K.L. HIV-1 Nef disrupts antigen presentation early in the secretory pathway. J. Biol. Chem. 2005, 280, 12840–12848. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.A.; Filzen, T.; Carter, C.C.; Schaefer, M.; Collins, K.L. HIV-1 Nef disrupts intracellular trafficking of major histocompatibility complex class I, CD4, CD8, and CD28 by distinct pathways that share common elements. J. Virol. 2011, 85, 6867–6881. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.H.; Thomas, L.; Ruby, C.E.; Atkins, K.M.; Morris, N.P.; Knight, Z.A.; Scholz, I.; Barklis, E.; Weinberg, A.D.; Shokat, K.M.; et al. HIV-1 Nef assembles a Src family kinase-ZAP-70/Syk-PI3K cascade to downregulate cell-surface MHC-I. Cell Host Microbe 2007, 1, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Atkins, K.M.; Thomas, L.; Youker, R.T.; Harriff, M.J.; Pissani, F.; You, H.; Thomas, G. HIV-1 Nef binds PACS-2 to assemble a multikinase cascade that triggers major histocompatibility complex class I (MHC-I) down-regulation: Analysis using short interfering RNA and knock-out mice. J. Biol. Chem. 2008, 283, 11772–11784. [Google Scholar] [CrossRef] [PubMed]
- Blagoveshchenskaya, A.D.; Thomas, L.; Feliciangeli, S.F.; Hung, C.H.; Thomas, G. HIV-1 Nef downregulates MHC-I by a PACS-1- and PI3K-regulated ARF6 endocytic pathway. Cell 2002, 111, 853–866. [Google Scholar] [CrossRef]
- Dikeakos, J.D.; Atkins, K.M.; Thomas, L.; Emert-Sedlak, L.; Byeon, I.J.; Jung, J.; Ahn, J.; Wortman, M.D.; Kukull, B.; Saito, M.; et al. Small molecule inhibition of HIV-1-induced MHC-I down-regulation identifies a temporally regulated switch in Nef action. Mol. Biol. Cell 2010, 21, 3279–3292. [Google Scholar] [CrossRef] [PubMed]
- Wonderlich, E.R.; Leonard, J.A.; Kulpa, D.A.; Leopold, K.E.; Norman, J.M.; Collins, K.L. ADP ribosylation factor 1 activity is required to recruit AP-1 to the major histocompatibility complex class I (MHC-I) cytoplasmic tail and disrupt MHC-I trafficking in HIV-1-infected primary T cells. J. Virol. 2011, 85, 12216–12226. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Rosales, T.; Rose, J.J.; Chowdhury, B.; Knutson, J.R.; Venkatesan, S. HIV-1 Nef binds a subpopulation of MHC-I throughout its trafficking itinerary and down-regulates MHC-I by perturbing both anterograde and retrograde trafficking. J. Biol. Chem. 2010, 285, 30884–308905. [Google Scholar] [CrossRef] [PubMed]
- Dikeakos, J.D.; Thomas, L.; Kwon, G.; Elferich, J.; Shinde, U.; Thomas, G. An interdomain binding site on HIV-1 Nef interacts with PACS-1 and PACS-2 on endosomes to down-regulate MHC-I. Mol. Biol. Cell 2012, 23, 2184–2197. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, S.; Erdtmann, L.; Benichou, S.; Berlioz-Torrent, C.; Liu, L.; Benarous, R.; Heard, J.M.; Schwartz, O. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 1998, 8, 483–495. [Google Scholar] [CrossRef]
- Cohen, G.B.; Gandhi, R.T.; Davis, D.M.; Mandelboim, O.; Chen, B.K.; Strominger, J.L.; Baltimore, D. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 1999, 10, 661–671. [Google Scholar] [CrossRef]
- Stumptner-Cuvelette, P.; Morchoisne, S.; Dugast, M.; Le Gall, S.; Raposo, G.; Schwartz, O.; Benaroch, P. HIV-1 Nef impairs MHC class II antigen presentation and surface expression. Proc. Natl. Acad. Sci. USA 2001, 98, 12144–12149. [Google Scholar] [CrossRef] [PubMed]
- Schindler, M.; Wurfl, S.; Benaroch, P.; Greenough, T.C.; Daniels, R.; Easterbrook, P.; Brenner, M.; Munch, J.; Kirchhoff, F. Down-modulation of mature major histocompatibility complex class II and up-regulation of invariant chain cell surface expression are well-conserved functions of human and simian immunodeficiency virus nef alleles. J. Virol. 2003, 77, 10548–10556. [Google Scholar] [CrossRef] [PubMed]
- Ghiglione, Y.; Rodriguez, A.M.; De Candia, C.; Carobene, M.; Benaroch, P.; Schindler, M.; Salomon, H.; Turk, G. HIV-mediated up-regulation of invariant chain (CD74) correlates with generalized immune activation in HIV+ subjects. Virus Res. 2012, 163, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.; Verghese, D.A.; Das, S.R.; Jameel, S.; George, A.; Bal, V.; Mayor, S.; Rath, S. HIV-1 Nef promotes endocytosis of cell surface MHC class II molecules via a constitutive pathway. J. Immunol. 2009, 183, 2415–2424. [Google Scholar] [CrossRef] [PubMed]
- Kadri, N.; Wagner, A.K.; Ganesan, S.; Karre, K.; Wickstrom, S.; Johansson, M.H.; Hoglund, P. Dynamic Regulation of NK Cell Responsiveness. Curr. Top. Microbiol. Immunol. 2015, 395, 95–114. [Google Scholar]
- Alter, G.; Altfeld, M. NK cells in HIV-1 infection: Evidence for their role in the control of HIV-1 infection. J. Intern. Med. 2009, 265, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Luteijn, R.; Sciaranghella, G.; van Lunzen, J.; Nolting, A.; Dugast, A.S.; Ghebremichael, M.S.; Altfeld, M.; Alter, G. Early viral replication in lymph nodes provides HIV with a means by which to escape NK-cell-mediated control. Eur. J. Immunol. 2011, 41, 2729–2740. [Google Scholar] [CrossRef] [PubMed]
- Cerboni, C.; Neri, F.; Casartelli, N.; Zingoni, A.; Cosman, D.; Rossi, P.; Santoni, A.; Doria, M. Human immunodeficiency virus 1 Nef protein downmodulates the ligands of the activating receptor NKG2D and inhibits natural killer cell-mediated cytotoxicity. J. Gen. Virol. 2007, 88, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Fausther-Bovendo, H.; Sol-Foulon, N.; Candotti, D.; Agut, H.; Schwartz, O.; Debre, P.; Vieillard, V. HIV escape from natural killer cytotoxicity: Nef inhibits NKp44L expression on CD4+ T cells. AIDS 2009, 23, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Falco, M.; Marcenaro, E.; Romeo, E.; Bellora, F.; Marras, D.; Vely, F.; Ferracci, G.; Moretta, L.; Moretta, A.; Bottino, C. Homophilic interaction of NTBA, a member of the CD2 molecular family: Induction of cytotoxicity and cytokine release in human NK cells. Eur. J. Immunol. 2004, 34, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.H.; Sowrirajan, B.; Davis, Z.B.; Ward, J.P.; Campbell, E.M.; Planelles, V.; Barker, E. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe 2010, 8, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, P.W.; DePaula-Silva, A.B.; Szaniawski, M.; Barker, E.; Bosque, A.; Planelles, V. HIV-1 Vpu utilizes both cullin-RING ligase (CRL) dependent and independent mechanisms to downmodulate host proteins. Retrovirology 2015, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Bolduan, S.; Hubel, P.; Reif, T.; Lodermeyer, V.; Hohne, K.; Fritz, J.V.; Sauter, D.; Kirchhoff, F.; Fackler, O.T.; Schindler, M.; et al. HIV-1 Vpu affects the anterograde transport and the glycosylation pattern of NTB-A. Virology 2013, 440, 190–203. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shibuya, A.; Campbell, D.; Hannum, C.; Yssel, H.; Franz-Bacon, K.; McClanahan, T.; Kitamura, T.; Nicholl, J.; Sutherland, G.R.; Lanier, L.L.; Phillips, J.H. DNAM-1, a novel adhesion molecule involved in the cytolytic function of T lymphocytes. Immunity 1996, 4, 573–581. [Google Scholar] [CrossRef]
- Matusali, G.; Potesta, M.; Santoni, A.; Cerboni, C.; Doria, M. The human immunodeficiency virus type 1 Nef and Vpu proteins downregulate the natural killer cell-activating ligand PVR. J. Virol. 2012, 86, 4496–4504. [Google Scholar] [CrossRef] [PubMed]
- Bolduan, S.; Reif, T.; Schindler, M.; Schubert, U. HIV-1 Vpu mediated downregulation of CD155 requires alanine residues 10, 14 and 18 of the transmembrane domain. Virology 2014, 464–465, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Tupin, E.; Kinjo, Y.; Kronenberg, M. The unique role of natural killer T cells in the response to microorganisms. Nat. Rev. Microbiol. 2007, 5, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Moll, M.; Andersson, S.K.; Smed-Sorensen, A.; Sandberg, J.K. Inhibition of lipid antigen presentation in dendritic cells by HIV-1 Vpu interference with CD1d recycling from endosomal compartments. Blood 2010, 116, 1876–1884. [Google Scholar] [CrossRef] [PubMed]
- Bachle, S.M.; Sauter, D.; Sibitz, S.; Sandberg, J.K.; Kirchhoff, F.; Moll, M. Involvement of a C-terminal motif in the interference of primate lentiviral Vpu proteins with CD1d-mediated antigen presentation. Sci. Rep. 2015, 5, 9675. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, J.K.; Andersson, S.K.; Bachle, S.M.; Nixon, D.F.; Moll, M. HIV-1 Vpu interference with innate cell-mediated immune mechanisms. Curr. HIV Res. 2012, 10, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Knox, K.S.; Kohli, L.M.; He, J.J.; Exley, M.A.; Wilson, S.B.; Brutkiewicz, R.R. Impaired cell surface expression of human CD1d by the formation of an HIV-1 Nef/CD1d complex. Virology 2005, 337, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; McCarthy, C.; Drakesmith, H.; Li, D.; Cerundolo, V.; McMichael, A.J.; Screaton, G.R.; Xu, X.N. HIV-1 down-regulates the expression of CD1d via Nef. Eur. J. Immunol. 2006, 36, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Kelly, H.; Mandraju, R.; Coelho-dos-Reis, J.G.; Tsuji, M. Effects of HIV-1-induced CD1c and CD1d modulation and endogenous lipid presentation on CD1c-restricted T-cell activation. BMC Immunol. 2013, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, D.G.; Nordeng, T.W.; Pedersen, K.; Balk, S.P.; Bakke, O. A critical tyrosine residue in the cytoplasmic tail is important for CD1d internalization but not for its basolateral sorting in MDCK cells. J. Immunol. 1999, 162, 1488–1495. [Google Scholar] [PubMed]
- Ly, D.; Moody, D.B. The CD1 size problem: Lipid antigens, ligands, and scaffolds. Cell. Mol. Life Sci. 2014, 71, 3069–3079. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, P.W.; Famiglietti, M.; Sowrirajan, B.; DePaula-Silva, A.B.; Rodesch, C.; Barker, E.; Bosque, A.; Planelles, V. Downmodulation of CCR7 by HIV-1 Vpu results in impaired migration and chemotactic signaling within CD4(+) T cells. Cell Rep. 2014, 7, 2019–2030. [Google Scholar] [CrossRef] [PubMed]
- Vassena, L.; Giuliani, E.; Koppensteiner, H.; Bolduan, S.; Schindler, M.; Doria, M. HIV-1 Nef and Vpu Interfere with L-Selectin (CD62L) Cell Surface Expression To Inhibit Adhesion and Signaling in Infected CD4+ T Lymphocytes. J. Virol. 2015, 89, 5687–5700. [Google Scholar] [CrossRef] [PubMed]
- Comerford, I.; Harata-Lee, Y.; Bunting, M.D.; Gregor, C.; Kara, E.E.; McColl, S.R. A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev. 2013, 24, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.I.; Kubes, P. L-selectin: An emerging player in chemokine function. Microcirculation 2003, 10, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Varoqui, H.; Zhu, H.; Yao, D.; Ming, H.; Erickson, J.D. Cloning and functional identification of a neuronal glutamine transporter. J. Biol. Chem. 2000, 275, 4049–4054. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Sugden, S.; Cohen, E.A. Attacking the Supply Lines: HIV-1 Restricts Alanine Uptake to Prevent T Cell Activation. Cell Host Microbe 2015, 18, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Tokarev, A.; Guatelli, J. Misdirection of membrane trafficking by HIV-1 Vpu and Nef: Keys to viral virulence and persistence. Cell. Logist. 2011, 1, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, E.N.; Dikeakos, J.D. HIV-1 Nef: a master manipulator of the membrane trafficking machinery mediating immune evasion. Biochim. Biophys. Acta 2015, 1850, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Andrew, A.; Strebel, K. HIV-1 Vpu targets cell surface markers CD4 and BST-2 through distinct mechanisms. Mol. Asp. Med. 2010, 31, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J. The antiviral activities of tetherin. Curr. Top. Microbiol. Immunol. 2013, 371, 67–104. [Google Scholar]
- Smithgall, T.E.; Thomas, G. Small molecule inhibitors of the HIV-1 virulence factor, Nef. Drug Discov. Today Technol. 2013, 10, e523–e529. [Google Scholar] [CrossRef] [PubMed]
- Mi, Z.; Ding, J.; Zhang, Q.; Zhao, J.; Ma, L.; Yu, H.; Liu, Z.; Shan, G.; Li, X.; Zhou, J.; et al. A small molecule compound IMB-LA inhibits HIV-1 infection by preventing viral Vpu from antagonizing the host restriction factor BST-2. Sci. Rep. 2015, 5, 18499. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nef | Molecule Type | Downregulation | Binding | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Observed with | Potential mechanism | Nef domains required | Experimental model (mechanism) | Ref | Nef domains required | Experimental model (binding) | Ref | ||
| CD4 | Viral receptor/ immunoreceptor | Infection CD4+ T cells | Increased endocytosis via clathrin pathway. Lysosomal degradation. | 160ExxxLL; 174DD; 154EE; 57WLE | Many model cell lines and methodologies | [172] | 57WLE; G95, G96, L97, R106, L110 | Direct in vitro binding with NMR | [45,46] |
| SERINC3/5 | Intrinsic restriction factor | Infection CD4+ T cell | Relocalization to Rab7+ endosomes. | D123, 164LL | Trans. Exp. Jurkat | [108,109] | Not reported | Microscopy Trans. Exp. Jurkat | [108] |
| Tetraspanins | Membrane microstructure organizers | Infection CD4+ T cell | Intracellular sequestration in TGN. | Multiple (diffuse) | Trans. Exp. 293T | [121,122] | Not reported | CoIP Trans. Exp. 293T | [122] |
| MHC-I | Immunoreceptor | Infection CD4+ T cell | 1. Increased endocytosis and sequestration. 2. Shunting of de novo MHC-I from TGN to lysosomes for degradation. | 72PxxPxxP; 62EEEE; R17, R19; 13WxxVxxxM; W113, Y120 | Many model cell lines and methodologies | [173] | Interacts in complex with AP-1 through 13WxxVxxxM; 62EEEE | X-ray crystallo-graphy | [127] |
| MHC-II | Immunoreceptor | Infection MDMs | Increased clathrin-independent, Rab-dependent endocytosis and decreased anterograde transport. Lysosomal degradation. | Multiple (diffuse) | Trans. Exp. U937/KO mouse BMDM | [143] | Not reported | Microscopy Trans. Exp. U937 | [143] |
| NKG2D-L | Ligand of NK cell activating receptor | Infection Jurkat | Not reported | Multiple (diffuse) | N/A | [147] | N/A | N/A | N/A |
| PVR | Ligand of NK cell activating receptor | Infection CD4+ T cell | Intracellular sequestration in perinuclear areas without degradation. | 72PxxPxxP; 62EEEE; F191 | Trans. Exp. HeLa | [154,155] | Not reported | Microscopy Trans. Exp. HeLa | [155] |
| CD1d | MHC-like immunoreceptor | Infection Jurkat | Increased rate of internalization. Intracellular sequestration in the TGN. | Multiple (diffuse) | Infection Jurkat; Trans. Exp. 293T/HeLa | [160,161] | Not reported | CoIP Trans. Exp. T2 | [160] |
| CD62L | Adhesion molecule/ homing | Infection CD4+ T cell | Intracellular sequestration. Decreased protein levels. | Multiple (diffuse) | Transd. Jurkat; Trans. Exp. 293T | [166] | Not reported | Microscopy Trans. Exp. 293T | [166] |
| Vpu | Molecule Type | Downregulation | Binding | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Observed with | Potential mechanism | Vpu domains required | Experimental model (mechanism) | Ref | Vpu domains required | Experimental model (binding) | Ref | ||
| CD4 | Viral receptor/ immunoreceptor | Infection CD4+ T cells and MDMs | Degradation via ERAD-like process. | TM; L63; V68; S52,56 | Many model cell lines and methodologies | [53,172] | TM; cytosolic elements | CoIP Trans. Exp. HeLa; NMR | [174] |
| BST2 | Intrinsic restriction factor | Infection CD4+ T cells and MDMs | Altered trafficking leading to intracellular sequestration and lysosomal degradation. Displacement from virus assembly sites. | TM; S52,56; 59ExxxLV | Many model cell lines and methodologies | [175] | TM (A10,14,18, W22) | CoIP Infection HeLa; NMR | [64,74] |
| Tetraspanins | Membrane microstructure organizers | Infection CD4+ T cells | Proteasome and lysosome-dependent degradation. | Undefined | Infection of lymphoid cells; Trans. Exp. 293T | [121,122] | Not reported | CoIP Trans. Exp. 293T | [122] |
| NTB-A | Co-stimulatory receptor. NK cell activating | Infection CD4+ T cells | Intracellular sequestration without degradation. Inhibition of NTB-A glycosylation. | TM (A18) | Trans. Exp. 293T/HeLa | [150,152] | TM | CoIP Trans. Exp. HeLa | [150] |
| PVR | Ligand of NK cell activating receptor | Infection CD4+ T cells | Intracellular sequestration without degradation. | TM (A10,14,18) S52,56? | Trans. Exp. HeLa | [154,155] | TM (A10,14,18) | Microscopy Trans. Exp. HeLa | [155] |
| CD1d | MHC-like immunoreceptor | Infection MDDCs | Intracellular sequestration in early endosomes without degradation. | Undefined | Trans. Exp. 293T | [157] | Not reported | CoIP Trans. Exp. 293T | [157] |
| CCR7 | Homing receptor | Infection CD4+ T cells | Impaired recycling, and intracellular sequestration without degradation. | TM (A14,18, W22) | Trans. Exp. HeLa | [165] | Not reported | CoIP Trans. Exp. 293T | [165] |
| CD62L | Adhesion molecule/ homing | Infection CD4+ T cells | Intracellular sequestration without degradation. | Undefined | Trans. Exp. Jurkat/293T | [166] | Not reported | Microscopy Trans. Exp. 293T | [166] |
| SNAT1 | Metabolite transporter | Infection CD4+ T cells | SCFβ-TrCP-mediated ubiquitination for lysosomal degradation. | S52,56; TM (W22) | Stable Exp. Jurkat/HeLa | [123] | Not reported | CoIP Stable. Exp. HeLa | [123] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugden, S.M.; Bego, M.G.; Pham, T.N.Q.; Cohen, É.A. Remodeling of the Host Cell Plasma Membrane by HIV-1 Nef and Vpu: A Strategy to Ensure Viral Fitness and Persistence. Viruses 2016, 8, 67. https://doi.org/10.3390/v8030067
Sugden SM, Bego MG, Pham TNQ, Cohen ÉA. Remodeling of the Host Cell Plasma Membrane by HIV-1 Nef and Vpu: A Strategy to Ensure Viral Fitness and Persistence. Viruses. 2016; 8(3):67. https://doi.org/10.3390/v8030067
Chicago/Turabian StyleSugden, Scott M., Mariana G. Bego, Tram N.Q. Pham, and Éric A. Cohen. 2016. "Remodeling of the Host Cell Plasma Membrane by HIV-1 Nef and Vpu: A Strategy to Ensure Viral Fitness and Persistence" Viruses 8, no. 3: 67. https://doi.org/10.3390/v8030067
APA StyleSugden, S. M., Bego, M. G., Pham, T. N. Q., & Cohen, É. A. (2016). Remodeling of the Host Cell Plasma Membrane by HIV-1 Nef and Vpu: A Strategy to Ensure Viral Fitness and Persistence. Viruses, 8(3), 67. https://doi.org/10.3390/v8030067