
The Role of the Equine Herpesvirus Type 1 (EHV-1) US3-Encoded Protein Kinase in Actin Reorganization and Nuclear Egress


Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses and Cells
2.2. Plasmids
2.3. Mutagenesis
2.4. Western Blotting
2.5. Virus Growth Assay
2.6. Labeling of the Actin Cytoskeleton
2.7. Adhesion Assay
2.8. Electron Microscopy
2.9. Statistical Analysis
3. Results
3.1. Generation of Mutant, Recombinant, and Revertant Viruses
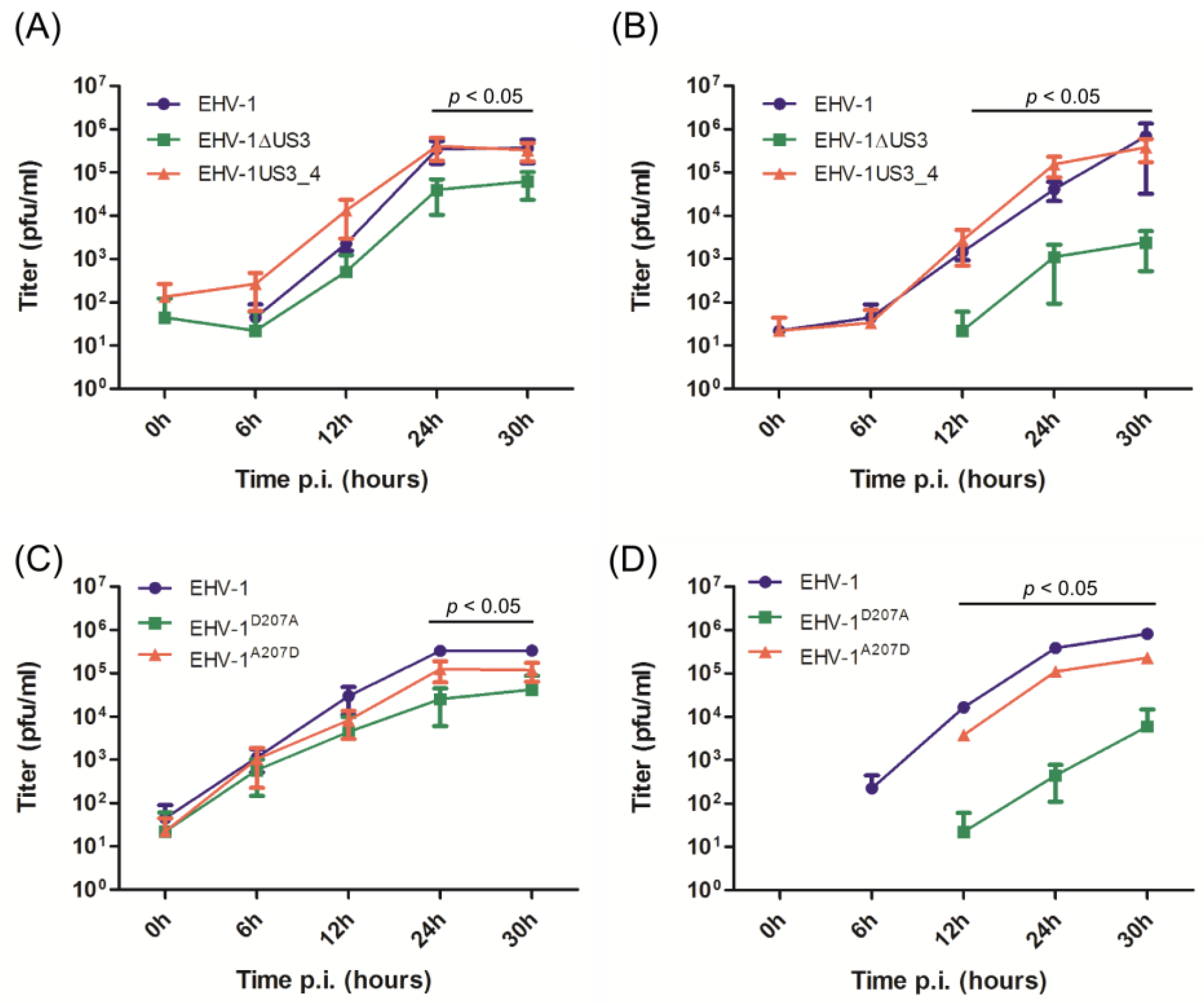
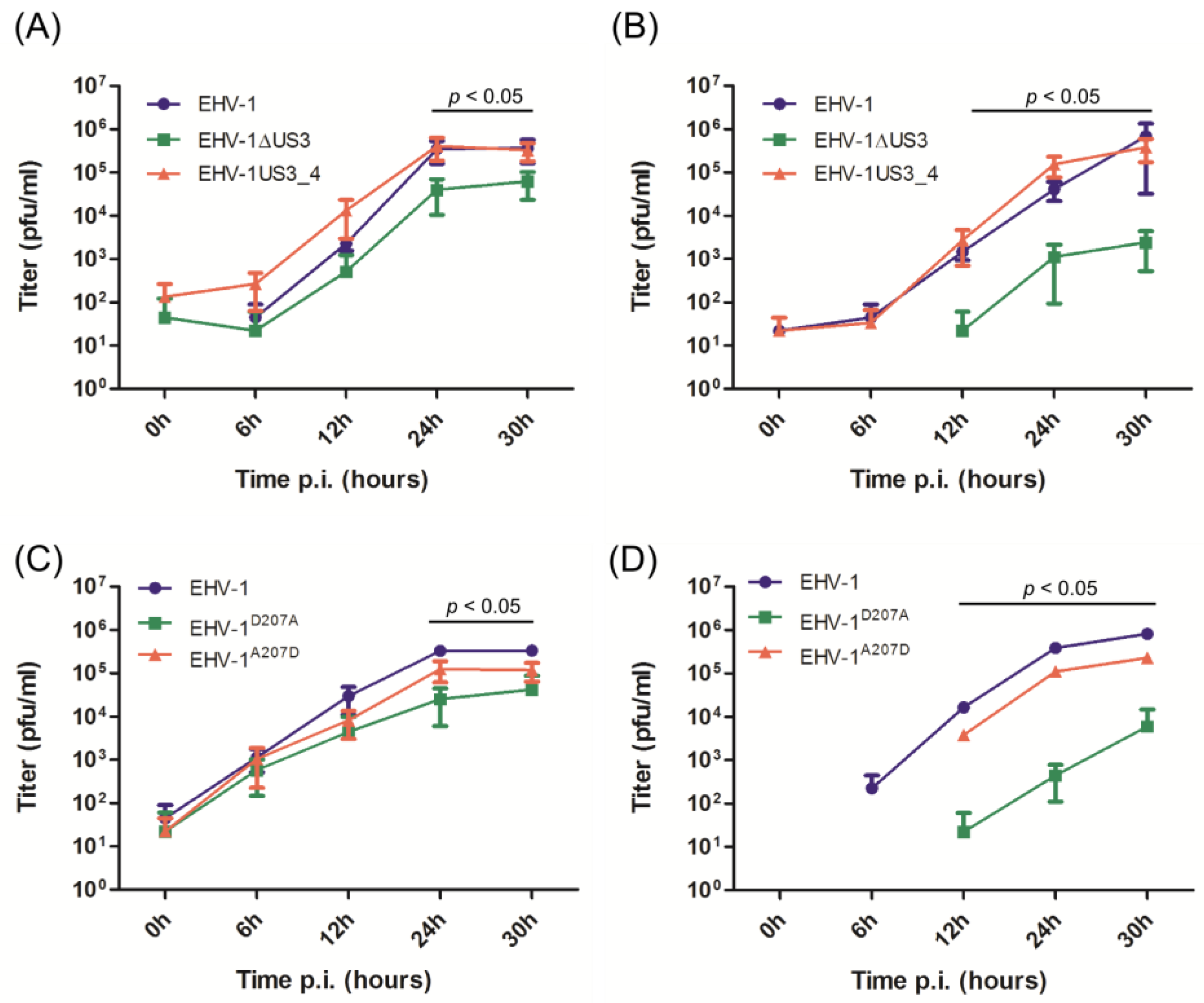
3.2. Virus Growth Deficit of the EHV-1 US3_1 Mutants Is Rescued by US3_4
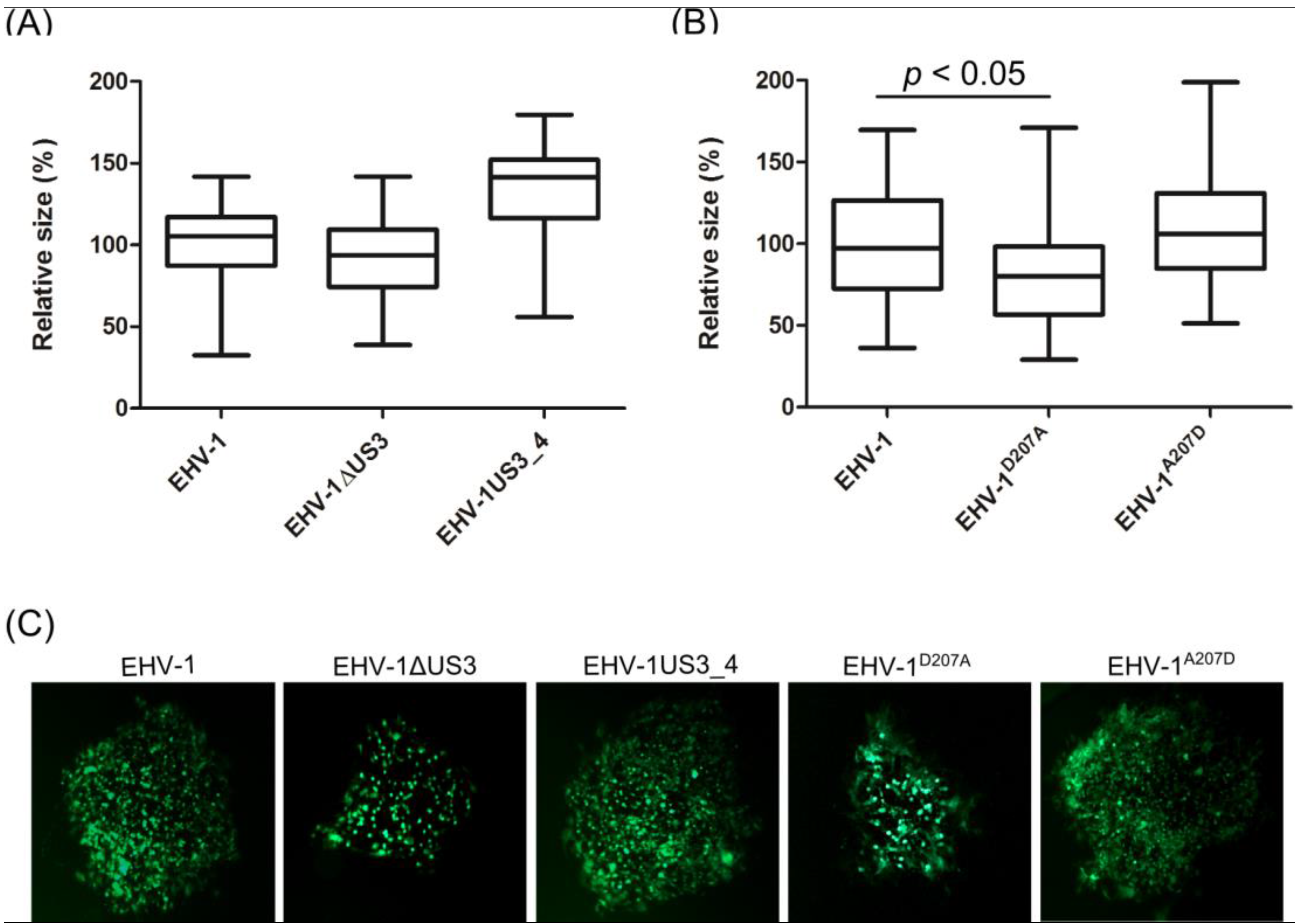
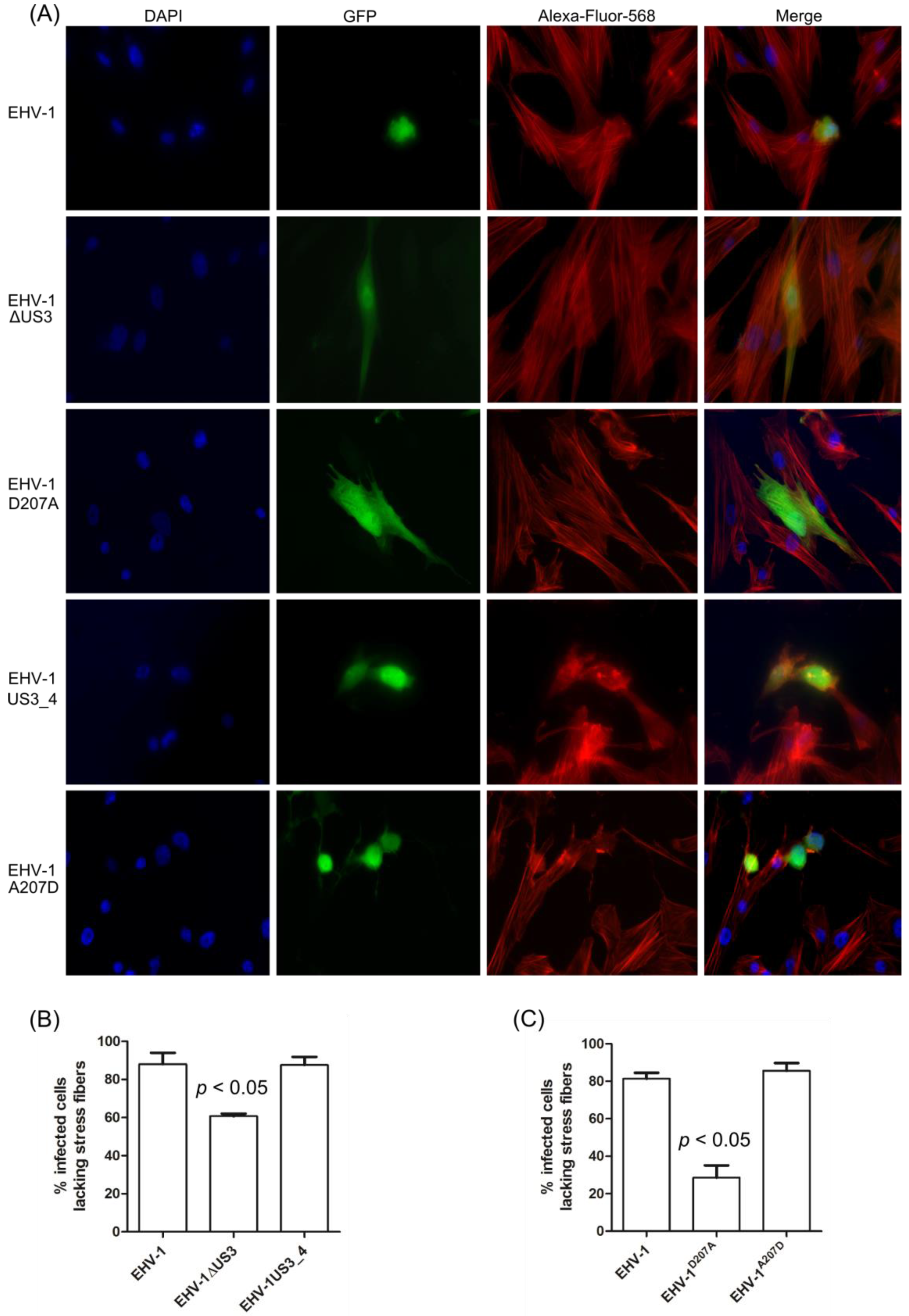
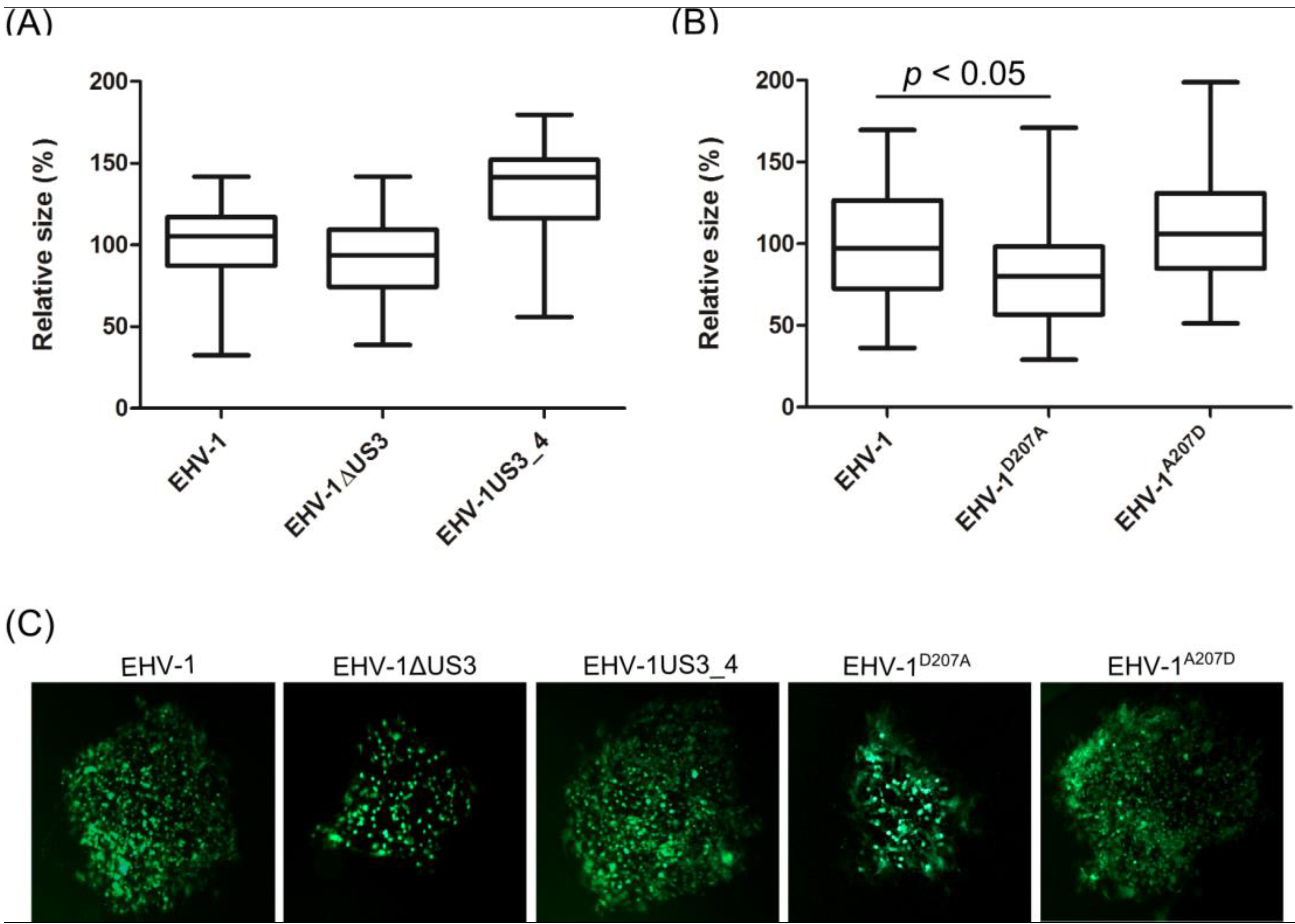
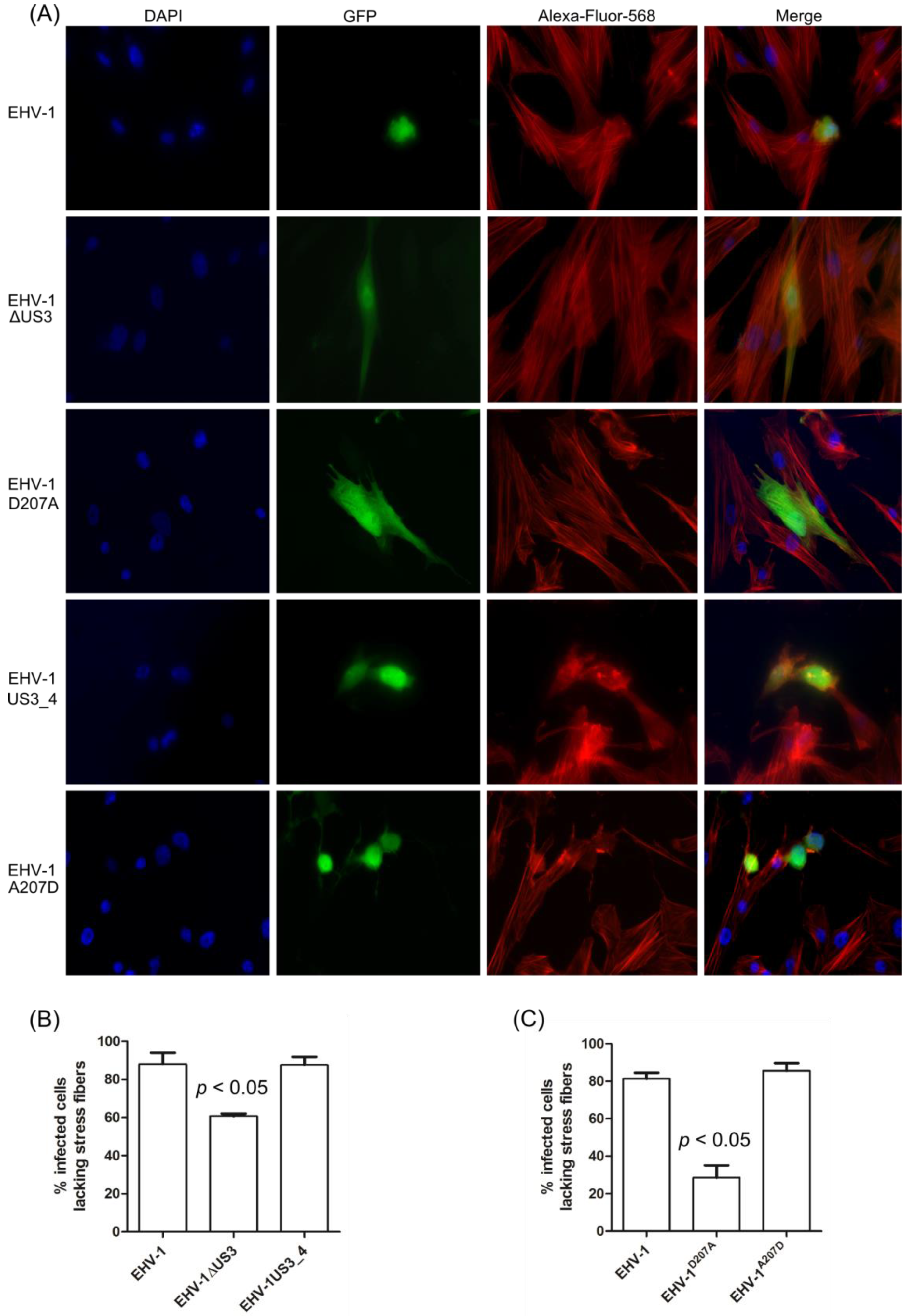
3.3. Morphological Changes and Actin Stress Fiber Disassembly
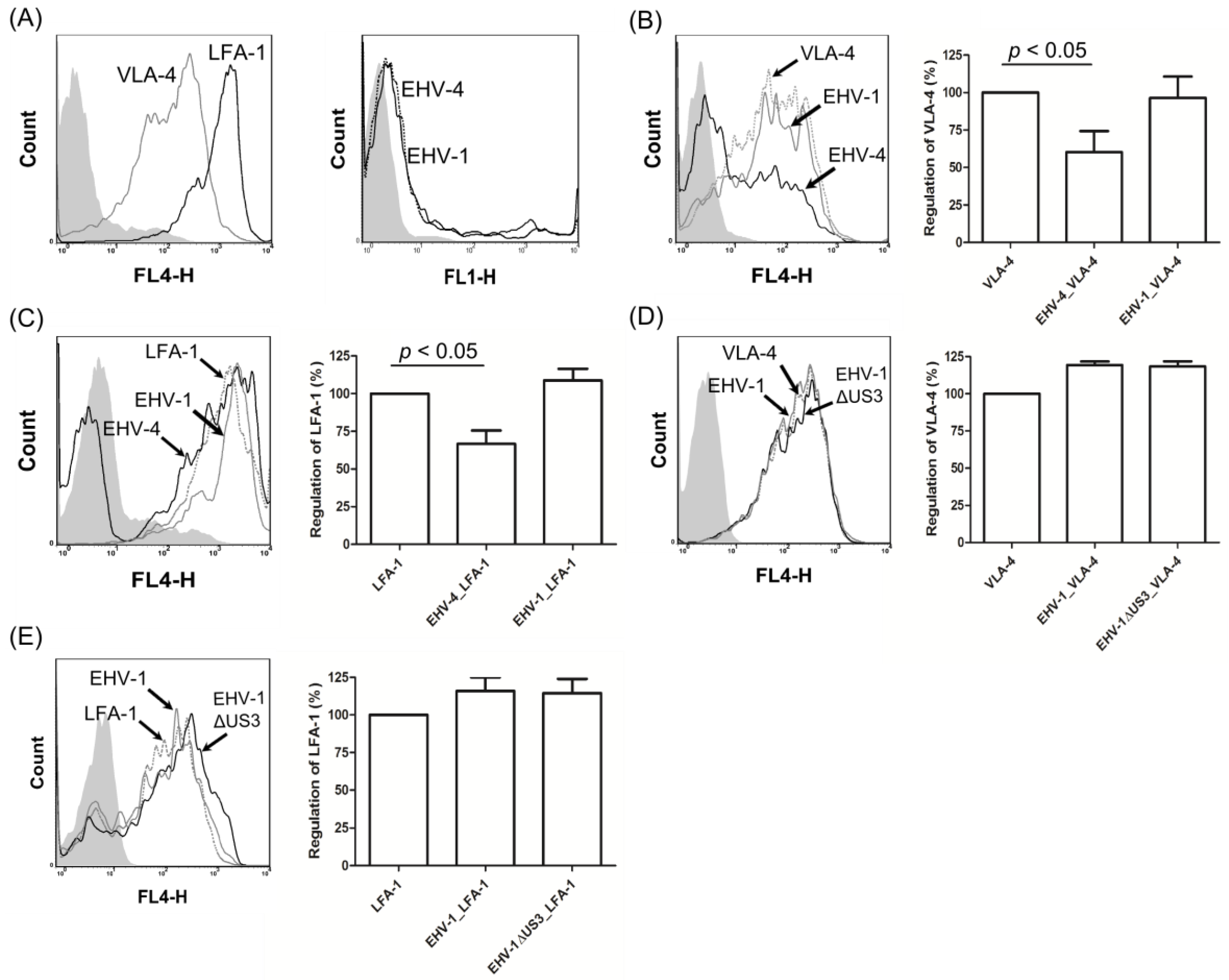
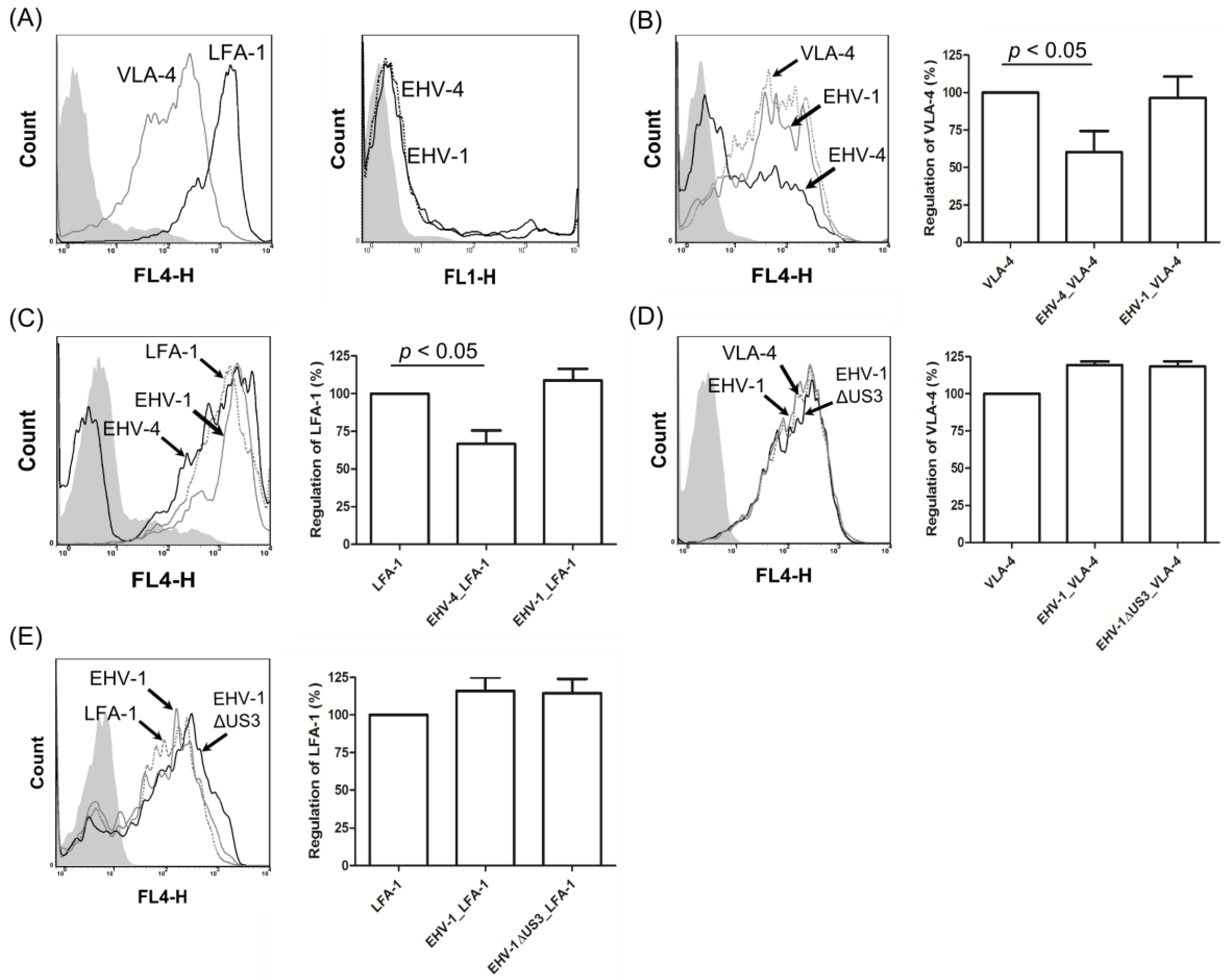
3.4. EHV-1 US3 Does Not Affect the Expression of Adhesion Molecules on the Surface of PBMC
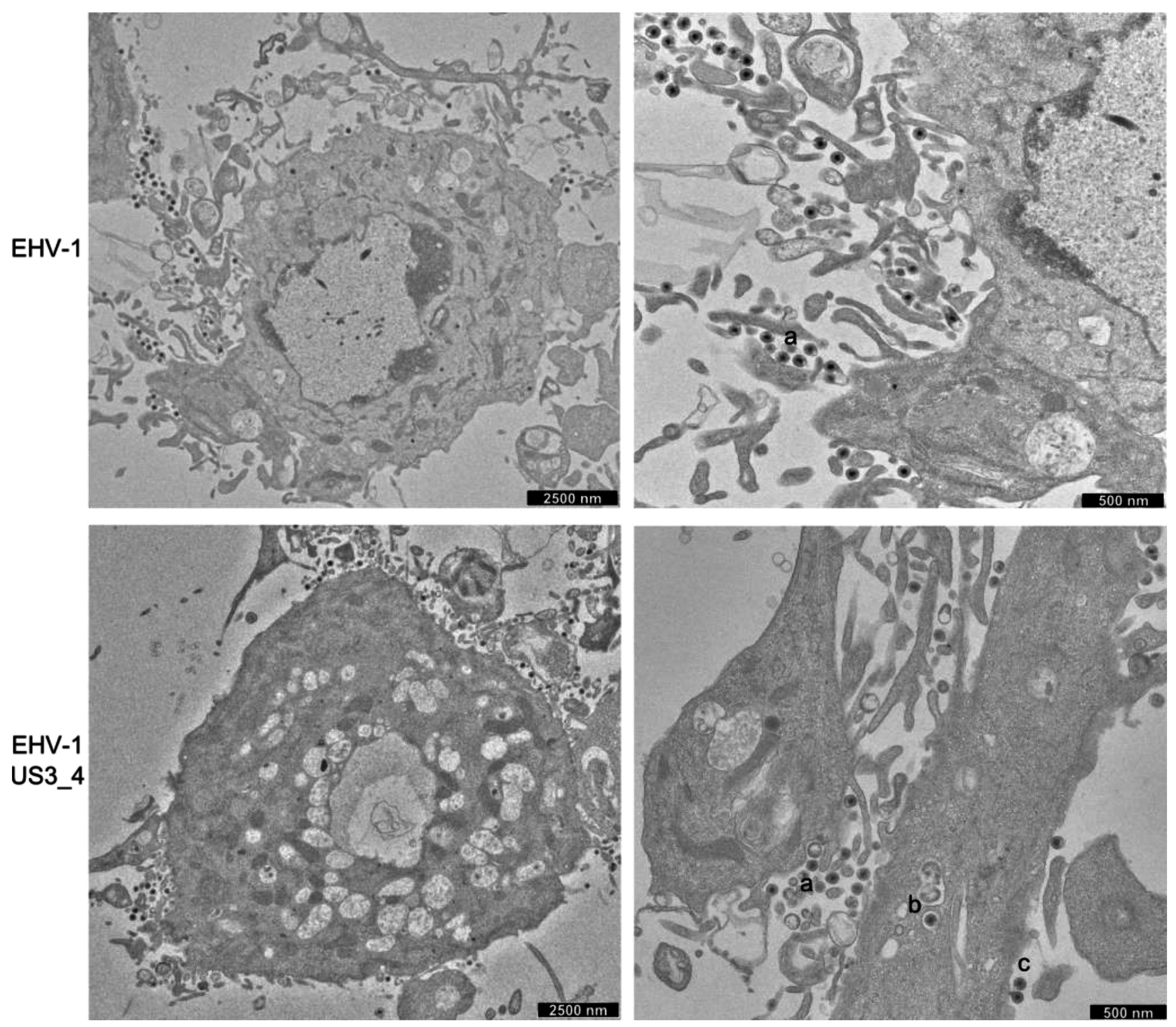
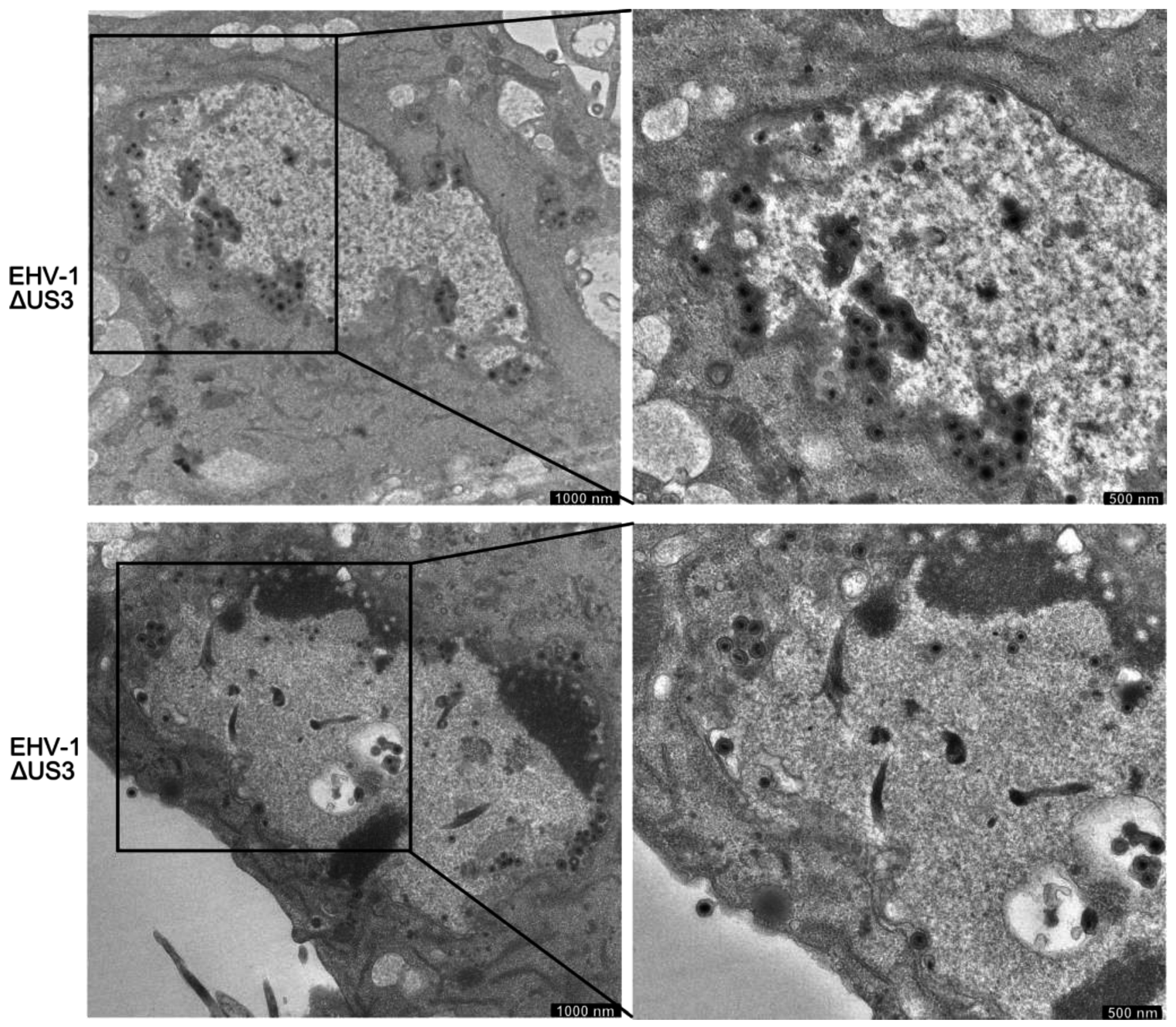
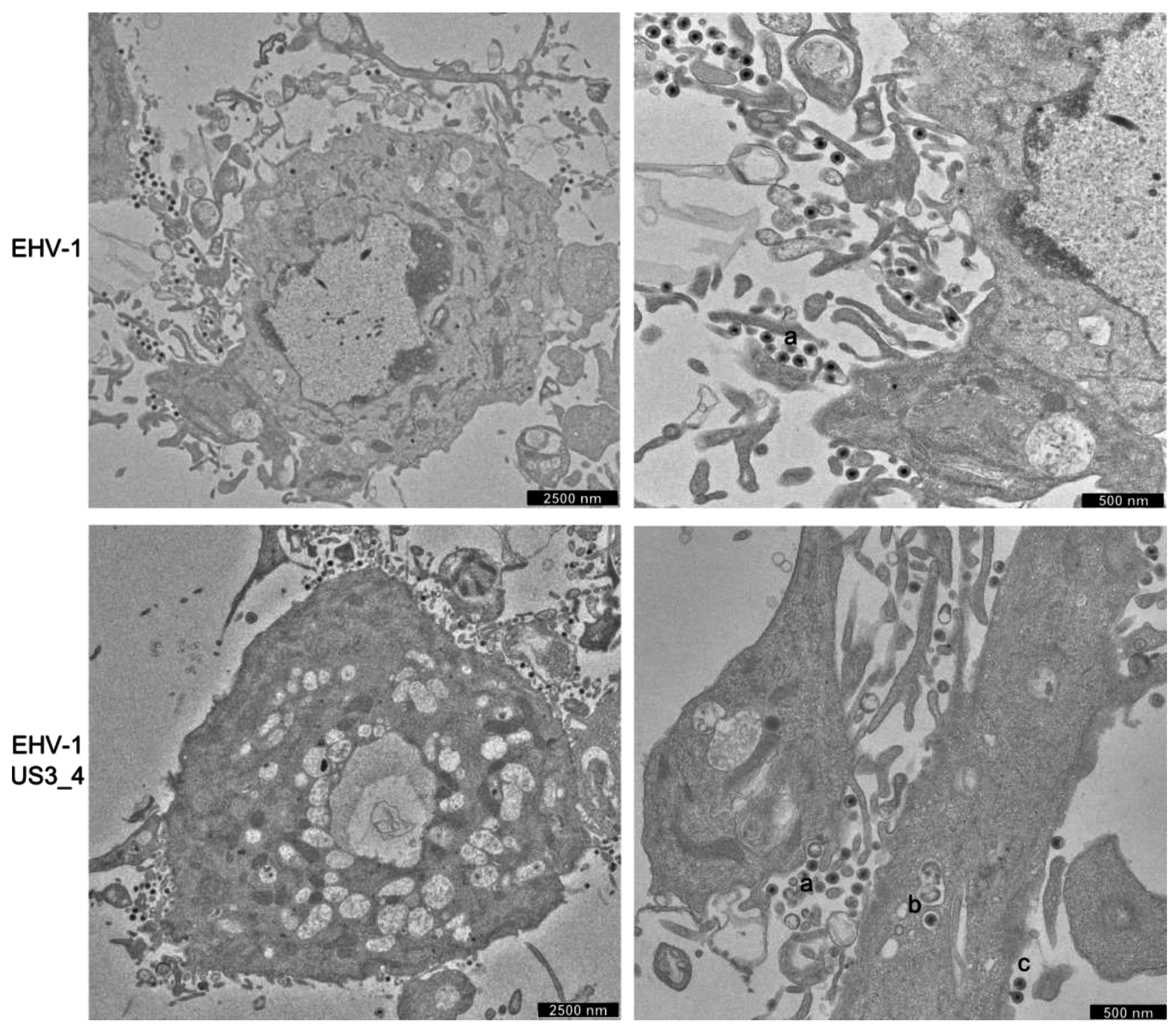
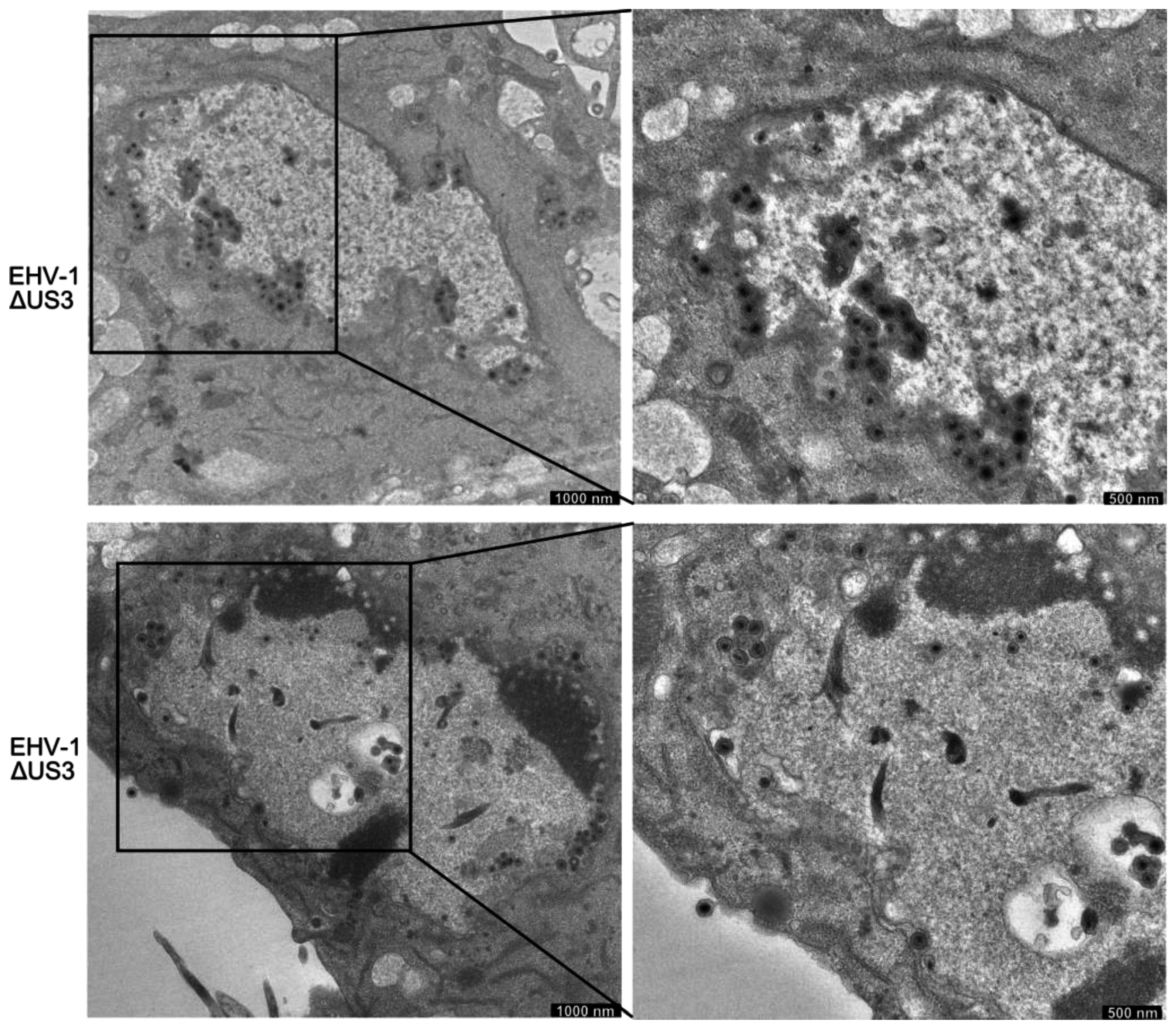
3.5. Accumulation of Enveloped Virions within Perinuclear Vesicles in the Absence of pUS3
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Calton, C.M.; Randall, J.A.; Adkins, M.W.; Banfield, B.W. The pseudorabies virus serine/threonine kinase Us3 contains mitochondrial, nuclear and membrane localization signals. Virus Genes 2004, 29, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Del Medico Zajac, M.P.; Ladelfa, M.F.; Kotsias, F.; Muylkens, B.; Thiry, J.; Thiry, E.; Romera, S.A. Biology of bovine herpesvirus 5. Vet. J. 2010, 184, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Colle, C.F., 3rd; Flowers, C.C.; O’Callaghan, D.J. Open reading frames encoding a protein kinase, homolog of glycoprotein gX of pseudorabies virus, and a novel glycoprotein map within the unique short segment of equine herpesvirus type 1. Virology 1992, 188, 545–557. [Google Scholar] [CrossRef]
- Telford, E.A.; Watson, M.S.; Perry, J.; Cullinane, A.A.; Davison, A.J. The DNA sequence of equine herpesvirus-4. J. Gen. Virol. 1998, 79, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.; Van den Broeke, C.; Favoreel, H.W. Viral serine/threonine protein kinases. J. Virol. 2011, 85, 1158–1173. [Google Scholar] [CrossRef] [PubMed]
- Deruelle, M.J.; Favoreel, H.W. Keep it in the subfamily: The conserved alphaherpesvirus US3 protein kinase. J. Gen. Virol. 2011, 92, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Yamamoto, M.; Ohno, T.; Kodaira, H.; Nishiyama, Y.; Kawaguchi, Y. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J. Virol. 2005, 79, 9325–9331. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Huber, M.T. Directed egress of animal viruses promotes cell-to-cell spread. J. Virol. 2002, 76, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zaichick, S.V.; Bohannon, K.P.; Smith, G.A. Alphaherpesviruses and the cytoskeleton in neuronal infections. Viruses 2011, 3, 941–981. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Enquist, L.W. Break ins and break outs: Viral interactions with the cytoskeleton of Mammalian cells. Annu. Rev. Cell. Dev. Biol. 2002, 18, 135–161. [Google Scholar] [CrossRef] [PubMed]
- Van den Broeke, C.; Radu, M.; Deruelle, M.; Nauwynck, H.; Hofmann, C.; Jaffer, Z.M.; Chernoff, J.; Favoreel, H.W. Alphaherpesvirus US3-mediated reorganization of the actin cytoskeleton is mediated by group A p21-activated kinases. Proc. Natl. Acad. Sci. USA 2009, 106, 8707–8712. [Google Scholar] [CrossRef] [PubMed]
- Van den Broeke, C.; Radu, M.; Nauwynck, H.J.; Chernoff, J.; Favoreel, H.W. Role of group A p21-activated kinases in the anti-apoptotic activity of the pseudorabies virus US3 protein kinase. Virus Res. 2011, 155, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.P.; Bryans, J.T. Molecular epizootiology, pathogenesis, and prophylaxis of equine herpesvirus-1 infections. Prog. Vet. Microbiol. Immunol. 1986, 2, 78–144. [Google Scholar] [PubMed]
- Patel, J.R.; Heldens, J. Equine herpesviruses 1 (EHV-1) and 4 (EHV-4)--epidemiology, disease and immunoprophylaxis: A brief review. Vet. J. 2005, 170, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Azab, W.; Osterrieder, N. Equine herpesviruses type 1 (EHV-1) and 4 (EHV-4)-Masters of co-evolution and a constant threat to equids and beyond. Vet. Microbiol. 2013, 167, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Spiesschaert, B.; Goldenbogen, B.; Taferner, S.; Schade, M.; Mahmoud, M.; Klipp, E.; Osterrieder, N.; Azab, W. Role of gB and pUS3 in EHV-1 transfer between PBMC and endothelial cells: A dynamic in vitro model. J. Virol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C. Herpesvirus assembly and egress. J. Virol. 2002, 76, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Effect of the pseudorabies virus US3 protein on nuclear membrane localization of the UL34 protein and virus egress from the nucleus. J. Gen. Virol. 2001, 82, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- Ryckman, B.J.; Roller, R.J. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 2004, 78, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Tischer, B.K.; Trapp, S.; Osterrieder, N. The protein encoded by the US3 orthologue of Marek’s disease virus is required for efficient de-envelopment of perinuclear virions and involved in actin stress fiber breakdown. J. Virol. 2005, 79, 3987–3997. [Google Scholar] [CrossRef] [PubMed]
- Mou, F.; Wills, E.; Baines, J.D. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J. Virol. 2009, 83, 5181–5191. [Google Scholar] [CrossRef] [PubMed]
- Goodman, L.B.; Loregian, A.; Perkins, G.A.; Nugent, J.; Buckles, E.L.; Mercorelli, B.; Kydd, J.H.; Palu, G.; Smith, K.C.; Osterrieder, N.; Davis-Poynter, N. A point mutation in a herpesvirus polymerase determines neuropathogenicity. PLoS Pathog. 2007, 3, e160. [Google Scholar] [CrossRef] [PubMed]
- Azab, W.; Kato, K.; Arii, J.; Tsujimura, K.; Yamane, D.; Tohya, Y.; Matsumura, T.; Akashi, H. Cloning of the genome of equine herpesvirus 4 strain TH20p as an infectious bacterial artificial chromosome. Arch. Virol. 2009, 154, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Wang, J.; Shen, J.; Liu, M.; Jin, X.; Tang, G.; Chu, P.K. Intracellular pathways and nuclear localization signal peptide-mediated gene transfection by cationic polymeric nanovectors. Biomaterials 2012, 33, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Birnboim, H.C.; Doly, J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979, 7, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [PubMed]
- Azab, W.; Osterrieder, N. Glycoproteins D of equine herpesvirus type 1 (EHV-1) and EHV-4 determine cellular tropism independently of integrins. J. Virol. 2012, 86, 2031–2044. [Google Scholar] [CrossRef] [PubMed]
- Colle, C.F.; O’Callaghan, D.J. Localization of the Us protein kinase of equine herpesvirus type 1 is affected by the cytoplasmic structures formed by the noval IR6 protein. Virology 1996, 220, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Wellington, J.E.; Gooley, A.A.; Love, D.N.; Whalley, J.M. N-terminal sequence analysis of equine herpesvirus 1 glycoproteins D and B and evidence for internal cleavage of the gene 71 product. J. Gen. Virol. 1996, 77, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Spiesschaert, B.; Stephanowitz, H.; Krause, E.; Osterrieder, N.; Azab, W. Glycoprotein B of equine herpesvirus type 1 has two recognition sites for subtilisin-like proteases that are cleaved by furin. J. Gen. Virol. 2016, 97, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, J.; O’Callaghan, D.J.; Osterrieder, N. Cloning of the genomes of equine herpesvirus type 1 (EHV-1) strains KyA and racL11 as bacterial artificial chromosomes (BAC). J. Vet. Med. B Infect. Dis Vet. Public Health 2002, 49, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Brzozowska, A.; Rychlowski, M.; Lipinska, A.D.; Bienkowska-Szewczyk, K. Point mutations in BHV-1 Us3 gene abolish its ability to induce cytoskeletal changes in various cell types. Vet. Microbiol. 2010, 143, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Ladelfa, M.F.; Kotsias, F.; Del Medico Zajac, M.P.; Van den Broeke, C.; Favoreel, H.; Romera, S.A.; Calamante, G. Effect of the US3 protein of bovine herpesvirus 5 on the actin cytoskeleton and apoptosis. Vet. Microbiol. 2011, 153, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Takashima, Y.; Tamura, H.; Xuan, X.; Otsuka, H. Identification of the US3 gene product of BHV-1 as a protein kinase and characterization of BHV-1 mutants of the US3 gene. Virus Res. 1999, 59, 23–34. [Google Scholar] [CrossRef]
- Jacob, T.; Van den Broeke, C.; Grauwet, K.; Baert, K.; Claessen, C.; De Pelsmaeker, S.; Van Waesberghe, C.; Favoreel, H.W. Pseudorabies virus US3 leads to filamentous actin disassembly and contributes to viral genome delivery to the nucleus. Vet. Microbiol. 2015, 177, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Finnen, R.L.; Roy, B.B.; Zhang, H.; Banfield, B.W. Analysis of filamentous process induction and nuclear localization properties of the HSV-2 serine/threonine kinase Us3. Virology 2010, 397, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Purves, F.C.; Spector, D.; Roizman, B. The herpes simplex virus 1 protein kinase encoded by the US3 gene mediates posttranslational modification of the phosphoprotein encoded by the UL34 gene. J. Virol. 1991, 65, 5757–5764. [Google Scholar] [PubMed]
- Reynolds, A.E.; Ryckman, B.J.; Baines, J.D.; Zhou, Y.; Liang, L.; Roller, R.J. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 2001, 75, 8803–8817. [Google Scholar] [CrossRef] [PubMed]
- Van Minnebruggen, G.; Favoreel, H.W.; Jacobs, L.; Nauwynck, H.J. Pseudorabies virus US3 protein kinase mediates actin stress fiber breakdown. J. Virol. 2003, 77, 9074–9080. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; van Bokhoven, A.; Jansen, C.F.; Bussemakers, M.J.; Schalken, J.A. Coordinate recruitment of E-cadherin and ALCAM to cell-cell contacts by alpha-catenin. Biochem. Biophys. Res. Commun. 2000, 267, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Oppenheimer-Marks, N.; Davis, L.S.; Bogue, D.T.; Ramberg, J.; Lipsky, P.E. Differential utilization of ICAM-1 and VCAM-1 during the adhesion and transendothelial migration of human T lymphocytes. J. Immunol. 1991, 147, 2913–2921. [Google Scholar] [PubMed]
- Swart, G.W.; Lunter, P.C.; Kilsdonk, J.W.; Kempen, L.C. Activated leukocyte cell adhesion molecule (ALCAM/CD166): signaling at the divide of melanoma cell clustering and cell migration? Cancer Metastasis Rev. 2005, 24, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Hieshima, K.; Haskell, C.; Baba, M.; Nagira, M.; Nishimura, M.; Kakizaki, M.; Takagi, S.; Nomiyama, H.; Schall, T.J.; Yoshie, O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 1997, 91, 521–530. [Google Scholar] [CrossRef]
- van Kooyk, Y.; Figdor, C.G. Avidity regulation of integrins: the driving force in leukocyte adhesion. Curr. Opin. Cell. Biol. 2000, 12, 542–547. [Google Scholar] [CrossRef]
- Elangbam, C.S.; Qualls, C.W., Jr.; Dahlgren, R.R. Cell adhesion molecules--update. Vet. Pathol. 1997, 34, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.S.; Bivins-Smith, E.R.; Tilley, A.M.; Bentz, G.L.; Chan, G.; Minard, J.; Yurochko, A.D. Roles of phosphatidylinositol 3-kinase and NF-kappaB in human cytomegalovirus-mediated monocyte diapedesis and adhesion: Strategy for viral persistence. J. Virol. 2007, 81, 7683–7694. [Google Scholar] [CrossRef] [PubMed]
- Nelissen, J.M.; Peters, I.M.; de Grooth, B.G.; van Kooyk, Y.; Figdor, C.G. Dynamic regulation of activated leukocyte cell adhesion molecule-mediated homotypic cell adhesion through the actin cytoskeleton. Mol. Biol. Cell. 2000, 11, 2057–2068. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Primer | Sequence |
|---|---|---|
| P1 | US3 1 Hind For | attaagcttatggaaaataaacaatgcga |
| P2 | US3 1 EcoR Rev | attgaattcctaaagttgtgcaaacattg |
| P3 | US3 4 Hind For | attaagcttatggaaaataaacaatacga |
| P4 | US3 4 EcoR Rev | attgaattcctacagattcataaacattg |
| P5 | US3 4 kan ins For | gattctagaggggctgcggtaccttcacgaggatgacgacgataagtaggg |
| P6 | US3 4 kan ins Rev | ccctctagaatctgtcgttctataatcaacaaccaattaaccaattctgattag |
| P7 | US3 1 del For | aagccctatagctttataggcacacgcccacggcatcggagatgactaacctgtttctggaggatgacgacgataagtaggg |
| P8 | US3 1 del Rev | gtcgcccacgctgtctcctcccagaaacaggttagtcatctccgatgccgtgggcgtgtgcaaccaattaaccaattctgattag |
| P9 | US3 4 ins into 1 For | gccgtgcgccaagccctatagctttataggcacacgcccacggcatcggaatggaaaataaacaatacga |
| P10 | US3 4 ins into 1 Rev | tttatacaccgtcgcccacgctgtctcctcccagaaacaggttagtcatcctacagattcataaacattg |
| P11 | US3D207A For | tacctgcacgcacagaggatcatccacagagcggtcaagactgaaaatattttcataaacaggatgacgacgataagtaggg |
| P12 | US3D207A Rev | gtttatgaaaatattttcagtcttgaccgctctgtggatgatcctctgtgcgtgcaggtacaaccaattaaccaattctgattag |
| P13 | US3A207D For | tacctgcacgcacagaggatcatccacagagacgtcaagactgaaaatattttcataaacaggatgacgacgataagtaggg |
| P14 | US3A207D Rev | gtttatgaaaatattttcagtcttgacgtctctgtggatgatcctctgtgcgtgcaggtacaaccaattaaccaattctgattag |
| P15 | US3 1 Pre For | ccaactccataaatttcagc |
| P16 | US3 1 Post Rev | ttacagttggtggcactgta |
| Virus | % of virus particles | Total Counted (Particles/Cells) | ||
|---|---|---|---|---|
| Nucleus | Extracellular and Cytoplasmic Vesicles | |||
| Perinuclear area | Intranuclear | |||
| EHV-1 | 6.5 | 0 | 93.5 | 246/10 |
| EHV-1ΔUS3 | 79 | 11 | 10 | 203/12 |
| EHV-1US3_4 | 4 | 0 | 96 | 417/12 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Proft, A.; Spiesschaert, B.; Izume, S.; Taferner, S.; Lehmann, M.J.; Azab, W. The Role of the Equine Herpesvirus Type 1 (EHV-1) US3-Encoded Protein Kinase in Actin Reorganization and Nuclear Egress. Viruses 2016, 8, 275. https://doi.org/10.3390/v8100275
Proft A, Spiesschaert B, Izume S, Taferner S, Lehmann MJ, Azab W. The Role of the Equine Herpesvirus Type 1 (EHV-1) US3-Encoded Protein Kinase in Actin Reorganization and Nuclear Egress. Viruses. 2016; 8(10):275. https://doi.org/10.3390/v8100275
Chicago/Turabian StyleProft, Alexandra, Bart Spiesschaert, Satoko Izume, Selina Taferner, Maik J. Lehmann, and Walid Azab. 2016. "The Role of the Equine Herpesvirus Type 1 (EHV-1) US3-Encoded Protein Kinase in Actin Reorganization and Nuclear Egress" Viruses 8, no. 10: 275. https://doi.org/10.3390/v8100275
APA StyleProft, A., Spiesschaert, B., Izume, S., Taferner, S., Lehmann, M. J., & Azab, W. (2016). The Role of the Equine Herpesvirus Type 1 (EHV-1) US3-Encoded Protein Kinase in Actin Reorganization and Nuclear Egress. Viruses, 8(10), 275. https://doi.org/10.3390/v8100275