
The Influenza Virus H5N1 Infection Can Induce ROS Production for Viral Replication and Host Cell Death in A549 Cells Modulated by Human Cu/Zn Superoxide Dismutase (SOD1) Overexpression


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cells and Viruses
2.3. Antibodies
2.4. Plasmid Construction
2.5. Biochemical Assays
2.6. ROS Detection
2.7. Apoptosis Detection
2.8. Real-Time Quantitative RT-PCR (qRT-PCR) Assay
2.9. Western Blot
2.10. Confocal Microscopy
2.11. ATP Detection
2.12. Determination of the Mitochondrial Membrane Potential (MMP)
2.13. Statistical Analysis
3. Results
3.1. H5N1 Infection Increased Cellular ROS Level in A549 Cells
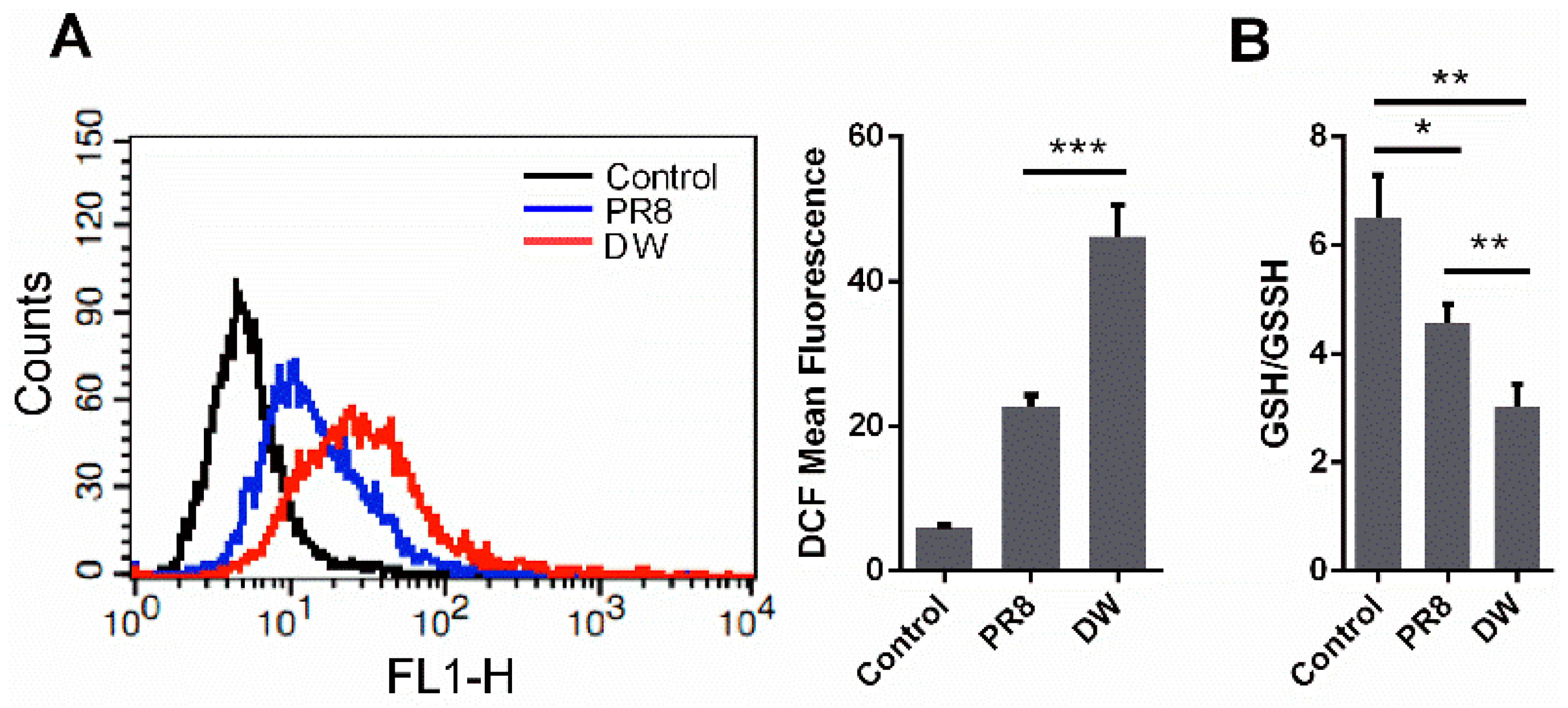
3.2. H5N1 Infection Modified the Expression of Oxidant and Antioxidant Enzymes
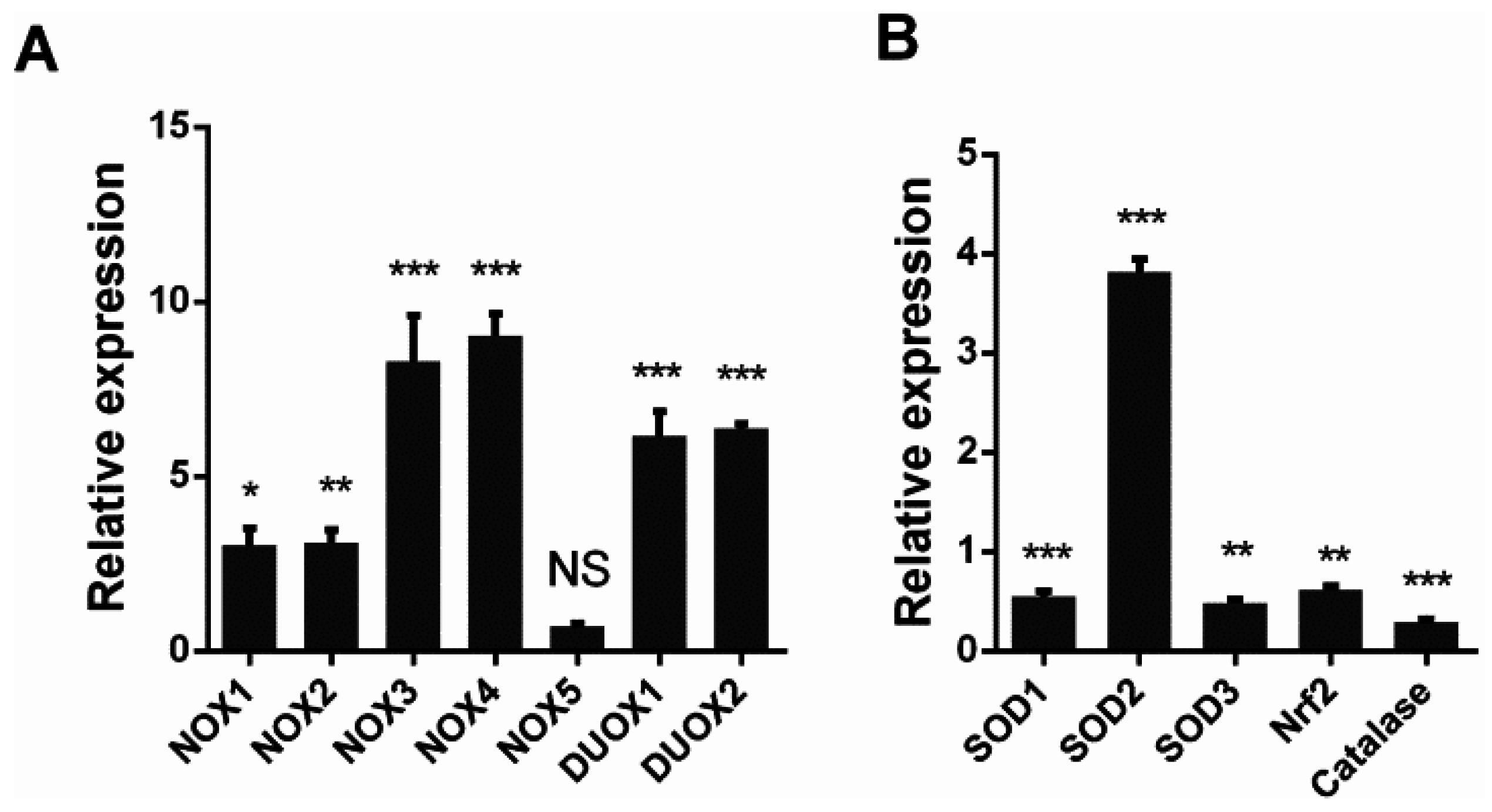
3.3. SOD1 Disrupted H5N1 Replication in A549 Cells
3.4. SOD1 Overexpression Reduced H5N1-Induced ROS Production and Attenuated Cytokine Response
3.5. SOD1 Overexpression Inhibited NF-κB and p38 Pathway and Disrupted the Nuclear Export of Viral NP
3.6. SOD1 Overexpression Rescued H5N1-Induced Apoptosis
3.7. SOD1 Prevented Mitochondrial Dysfunction Caused by H5N1
4. Discussion
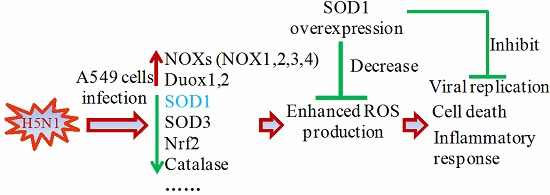
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.; Chau, T.N.; Hoang, D.M.; Chau, N.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.A.; Plowden, J.K.; Garcia-Sastre, A.; Katz, J.M.; Tumpey, T.M. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008, 4, e1000115. [Google Scholar] [CrossRef] [PubMed]
- De Mochel, N.S.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2010, 52, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Polyak, S.J.; Bano, N.; Qiu, W.C.; Carithers, R.L.; Shuhart, M.; Gretch, D.R.; Das, A. Hepatitis C virus induces oxidative stress, DNA damage and modulates the DNA repair enzyme NEIL1. J. Gastroenterol. Hepatol. 2010, 25, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Dosal, R.; Horan, K.A.; Rahbek, S.H.; Ichijo, H.; Chen, Z.J.; Mieyal, J.J.; Hartmann, R.; Paludan, S.R. HSV infection induces production of ROS, which potentiate signaling from pattern recognition receptors: Role for S-glutathionylation of TRAF3 and 6. PLoS Pathog. 2011, 7, e1002250. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Noguchi, Y.; Ijiri, S.; Setoguchi, K.; Suga, M.; Zheng, Y.M.; Dietzschold, B.; Maeda, H. Pathogenesis of influenza virus-induced pneumonia: Involvement of both nitric oxide and oxygen radicals. Proc. Natl. Acad. Sci. USA 1996, 93, 2448–2453. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Forman, H.J. Redox signaling and the MAP kinase pathways. BioFactors 2003, 17, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-κB activation by reactive oxygen species: Fifteen years later. Biochem. Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Asehnoune, K.; Strassheim, D.; Mitra, S.; Kim, J.Y.; Abraham, E. Involvement of reactive oxygen species in toll-like receptor 4-dependent activation of NF-κB. J. Immunol. 2004, 172, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Brydon, E.W.; Morris, S.J.; Sweet, C. Role of apoptosis and cytokines in influenza virus morbidity. FEMS Microbiol. Rev. 2005, 29, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Akaike, T.; Hamamoto, T.; Suzuki, F.; Hirano, T.; Maeda, H. Oxygen radicals in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science 1989, 244, 974–976. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll-Like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating nadph oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, D.B.; Choi, A.M. Influenza virus induces expression of antioxidant genes in human epithelial cells. Free Radic. Biol. Med. 1994, 16, 821–824. [Google Scholar] [CrossRef]
- Knobil, K.; Choi, A.M.; Weigand, G.W.; Jacoby, D.B. Role of oxidants in influenza virus-induced gene expression. Am. J. Physiol. 1998, 274, L134–L142. [Google Scholar] [PubMed]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Selemidis, S.; Sobey, C.G.; Wingler, K.; Schmidt, H.H.; Drummond, G.R. NADPH oxidases in the vasculature: molecular features, roles in disease and pharmacological inhibition. Pharmacol. Ther. 2008, 120, 254–291. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Stambas, J.; Bozinovski, S.; Broughton, B.R.; Drummond, G.R.; Selemidis, S. Inhibition of NOX 2 oxidase activity ameliorates influenza a virus-induced lung inflammation. PLoS Pathog. 2011, 7, e1001271. [Google Scholar] [CrossRef] [PubMed]
- Amatore, D.; Sgarbanti, R.; Aquilano, K.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.R.; Palamara, A.T. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell Microbial. 2015, 17, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [PubMed]
- Suliman, H.B.; Ryan, L.K.; Bishop, L.; Folz, R.J. Prevention of influenza-induced lung injury in mice overexpressing extracellular superoxide dismutase. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L69–L78. [Google Scholar] [PubMed]
- Rivas-Estilla, A.M.; Bryan-Marrugo, O.L.; Trujillo-Murillo, K.; Perez-Ibave, D.; Charles-Nino, C.; Pedroza-Roldan, C.; Rios-Ibarra, C.; Ramirez-Valles, E.; Ortiz-Lopez, R.; Islas-Carbajal, M.C.; et al. Cu/Zn superoxide dismutase (SOD1) induction is implicated in the antioxidative and antiviral activity of acetylsalicylic acid in HCV-expressing cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1264–G1273. [Google Scholar] [CrossRef] [PubMed]
- Dimayuga, F.O.; Wang, C.; Clark, J.M.; Dimayuga, E.R.; Dimayuga, V.M.; Bruce-Keller, A.J. SOD1 overexpression alters ROS production and reduces neurotoxic inflammatory signaling in microglial cells. J. Neuroimmunol. 2007, 182, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Pyo, C.W.; Shin, N.; Jung, K.I.; Choi, J.H.; Choi, S.Y. Alteration of Copper-Zinc superoxide dismutase 1 expression by influenza A virus is correlated with virus replication. Biochem. Biophys. Res. Commun. 2014, 450, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Pleschka, S.; Wolff, T.; Ehrhardt, C.; Hobom, G.; Planz, O.; Rapp, U.R.; Ludwig, S. Influenza virus propagation is impaired by inhibition of the RAF/MEK/ERK signalling cascade. Nat. Cell Biol. 2001, 3, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, L.; De Chiara, G.; Sgarbanti, R.; Amatore, D.; Aquilano, K.; Marcocci, M.E.; Serafino, A.; Torcia, M.; Cozzolino, F.; Ciriolo, M.R.; et al. Bcl-2 expression and P38MAPK activity in cells infected with influenza A virus: Impact on virally induced apoptosis and viral replication. J. Biol. Chem. 2009, 284, 16004–16015. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.; Pyo, C.W.; Jung, K.I.; Choi, S.Y. Influenza a virus PB1-F2 is involved in regulation of cellular redox state in alveolar epithelial cells. Biochem. Biophys. Res. Commun. 2015, 459, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Ristow, M. Mitochondria and metabolic homeostasis. Antioxid. Redox Signal. 2013, 19, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Dada, L.A.; Sznajder, J.I. Mitochondrial Ca2+ and ROS take center stage to orchestrate TNF-α-mediated inflammatory responses. J. Clin. Investig. 2011, 121, 1683–1685. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Role of mitochondria as the gardens of cell death. Cancer Chemother. Pharmacol. 2006, 57, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Peterhans, E.; Grob, M.; Burge, T.; Zanoni, R. Virus-induced formation of reactive oxygen intermediates in phagocytic cells. Free Radic. Res. Commun. 1987, 3, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Masella, R.; di Benedetto, R.; Vari, R.; Filesi, C.; Giovannini, C. Novel mechanisms of natural antioxidant compounds in biological systems: involvement of Glutathione and Glutathione-related enzymes. J. Nutr. Biochem. 2005, 16, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Reshi, M.L.; Su, Y.C.; Hong, J.R. RNA Viruses: ROS-mediated cell death. Int. J. Cell Biol. 2014, 2014, 467452. [Google Scholar] [CrossRef] [PubMed]
- Ciriolo, M.R.; Palamara, A.T.; Incerpi, S.; Lafavia, E.; Bue, M.C.; de Vito, P.; Garaci, E.; Rotilio, G. Loss of GSH, oxidative stress, and decrease of intracellular pH as sequential steps in viral infection. J. Biol. Chem. 1997, 272, 2700–2708. [Google Scholar] [CrossRef] [PubMed]
- Garaci, E.; Palamara, A.T.; di Francesco, P.; Favalli, C.; Ciriolo, M.R.; Rotilio, G. Glutathione inhibits replication and expression of viral proteins in cultured cells infected with Sendai virus. Biochem. Biophys. Res. Commun. 1992, 188, 1090–1096. [Google Scholar] [CrossRef]
- Nucci, C.; Palamara, A.T.; Ciriolo, M.R.; Nencioni, L.; Savini, P.; D’Agostini, C.; Rotilio, G.; Cerulli, L.; Garaci, E. Imbalance in corneal redox state during Herpes Simplex Virus 1-induced keratitis in rabbits. Effectiveness of exogenous glutathione supply. Exp. Eye Res. 2000, 70, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Nencioni, L.; Iuvara, A.; Aquilano, K.; Ciriolo, M.R.; Cozzolino, F.; Rotilio, G.; Garaci, E.; Palamara, A.T. Influenza A virus replication is dependent on an antioxidant pathway that involves GSH and Bcl-2. FASEB J. 2003, 17, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Braakman, I.; Helenius, J.; Helenius, A. Role of ATP and disulphide bonds during protein folding in the endoplasmic reticulum. Nature 1992, 356, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Sgarbanti, R.; Nencioni, L.; Amatore, D.; Coluccio, P.; Fraternale, A.; Sale, P.; Mammola, C.L.; Carpino, G.; Gaudio, E.; Magnani, M.; et al. Redox regulation of the influenza hemagglutinin maturation process: A new cell-mediated strategy for anti-influenza therapy. Antioxid. Redox Signal. 2011, 15, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Liu, T.; Castro, S.M.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am. J. Respir. Cell Mol. Biol. 2009, 41, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Minc, E.; de Coppet, P.; Masson, P.; Thiery, L.; Dutertre, S.; Amor-Gueret, M.; Jaulin, C. The human Copper-Zinc superoxide dismutase gene (SOD1) proximal promoter is regulated by SP1, EGR-1, and WT1 via non-canonical binding sites. J. Biol. Chem. 1999, 274, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Afonso, V.; Santos, G.; Collin, P.; Khatib, A.M.; Mitrovic, D.R.; Lomri, N.; Leitman, D.C.; Lomri, A. Tumor necrosis factor-α down-regulates human Cu/Zn superoxide dismutase 1 promoter via JNK/AP-1 signaling pathway. Free Radic. Biol. Med. 2006, 41, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Wurzer, W.J.; Ehrhardt, C.; Pleschka, S.; Berberich-Siebelt, F.; Wolff, T.; Walczak, H.; Planz, O.; Ludwig, S. NF-κB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and FAS/FASl is crucial for efficient influenza virus propagation. J. Biol. Chem. 2004, 279, 30931–30937. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326 Pt 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sanghavi, D.M.; Thelen, M.; Thornberry, N.A.; Casciola-Rosen, L.; Rosen, A. Caspase-mediated proteolysis during apoptosis: Insights from apoptotic neutrophils. FEBS Lett. 1998, 422, 179–184. [Google Scholar] [CrossRef]
- Slee, E.A.; Adrain, C.; Martin, S.J. Serial killers: Ordering caspase activation events in apoptosis. Cell Death Differ. 1999, 6, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Wurzer, W.J.; Planz, O.; Ehrhardt, C.; Giner, M.; Silberzahn, T.; Pleschka, S.; Ludwig, S. Caspase 3 activation is essential for efficient influenza virus propagation. EMBO J. 2003, 22, 2717–2728. [Google Scholar] [CrossRef] [PubMed]
- Geiler, J.; Michaelis, M.; Naczk, P.; Leutz, A.; Langer, K.; Doerr, H.W.; Cinatl, J., Jr. N-acetyl-l-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549 cells infected with highly pathogenic H5N1 influenza a virus. Biochem. Pharmacol. 2010, 79, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N.; Susin, S.A.; Marchetti, P.; Hirsch, T.; Gomez-Monterrey, I.; Castedo, M.; Kroemer, G. Mitochondrial control of nuclear apoptosis. J. Exp. Med. 1996, 183, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome C by the mitochondrial channel VDAC. Nature 1999, 399, 483–487. [Google Scholar] [PubMed]
- Zamarin, D.; Garcia-Sastre, A.; Xiao, X.; Wang, R.; Palese, P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005, 1, e4. [Google Scholar] [CrossRef] [PubMed]
- Ricci, J.E.; Gottlieb, R.A.; Green, D.R. Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J. Cell Biol. 2003, 160, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Schlegel, J. Mitochondrial calcium release induced by prooxidants. Toxicol. Lett. 1993, 67, 119–127. [Google Scholar] [CrossRef]
- Ueda, M.; Daidoji, T.; Du, A.; Yang, C.S.; Ibrahim, M.S.; Ikuta, K.; Nakaya, T. Highly pathogenic H5N1 avian influenza virus induces extracellular Ca2+ influx, leading to apoptosis in avian cells. J. Virol. 2010, 84, 3068–3078. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Wang, R.; Zou, W.; Sun, X.; Liu, X.; Zhao, L.; Wang, S.; Jin, M. The Influenza Virus H5N1 Infection Can Induce ROS Production for Viral Replication and Host Cell Death in A549 Cells Modulated by Human Cu/Zn Superoxide Dismutase (SOD1) Overexpression. Viruses 2016, 8, 13. https://doi.org/10.3390/v8010013
Lin X, Wang R, Zou W, Sun X, Liu X, Zhao L, Wang S, Jin M. The Influenza Virus H5N1 Infection Can Induce ROS Production for Viral Replication and Host Cell Death in A549 Cells Modulated by Human Cu/Zn Superoxide Dismutase (SOD1) Overexpression. Viruses. 2016; 8(1):13. https://doi.org/10.3390/v8010013
Chicago/Turabian StyleLin, Xian, Ruifang Wang, Wei Zou, Xin Sun, Xiaokun Liu, Lianzhong Zhao, Shengyu Wang, and Meilin Jin. 2016. "The Influenza Virus H5N1 Infection Can Induce ROS Production for Viral Replication and Host Cell Death in A549 Cells Modulated by Human Cu/Zn Superoxide Dismutase (SOD1) Overexpression" Viruses 8, no. 1: 13. https://doi.org/10.3390/v8010013
APA StyleLin, X., Wang, R., Zou, W., Sun, X., Liu, X., Zhao, L., Wang, S., & Jin, M. (2016). The Influenza Virus H5N1 Infection Can Induce ROS Production for Viral Replication and Host Cell Death in A549 Cells Modulated by Human Cu/Zn Superoxide Dismutase (SOD1) Overexpression. Viruses, 8(1), 13. https://doi.org/10.3390/v8010013