
Mate-Pair Sequencing as a Powerful Clinical Tool for the Characterization of Cancers with a DNA Viral Etiology

{kind=link}
Abstract
:1. Introduction
2. HPV and Cervical Cancer
3. HPV and Other Anogenital Cancers
4. HPV and OPSCC
5. Recent Studies Detecting HPV Integrations by Next Generation Sequencing
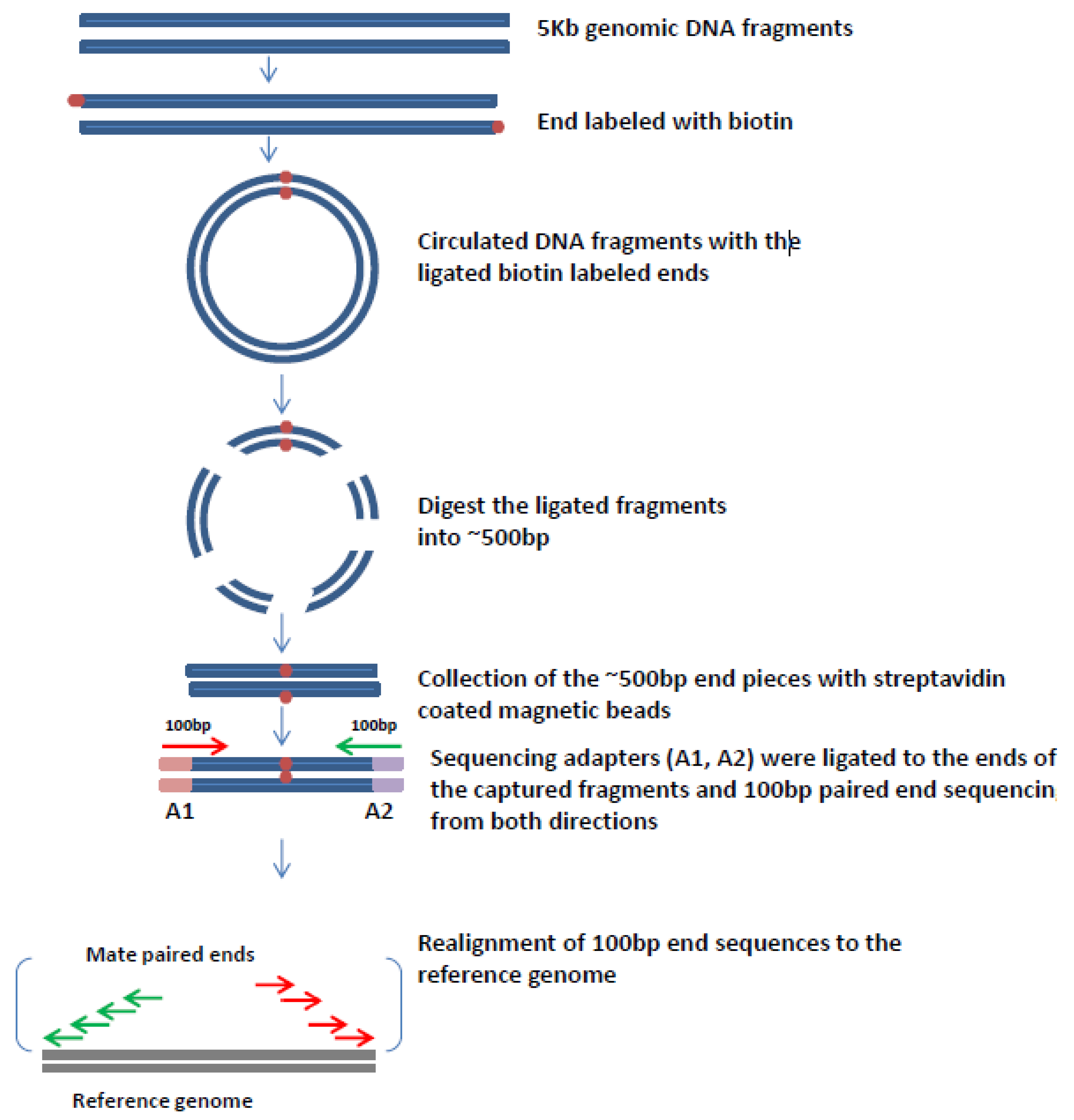
6. Mate Pair Sequencing
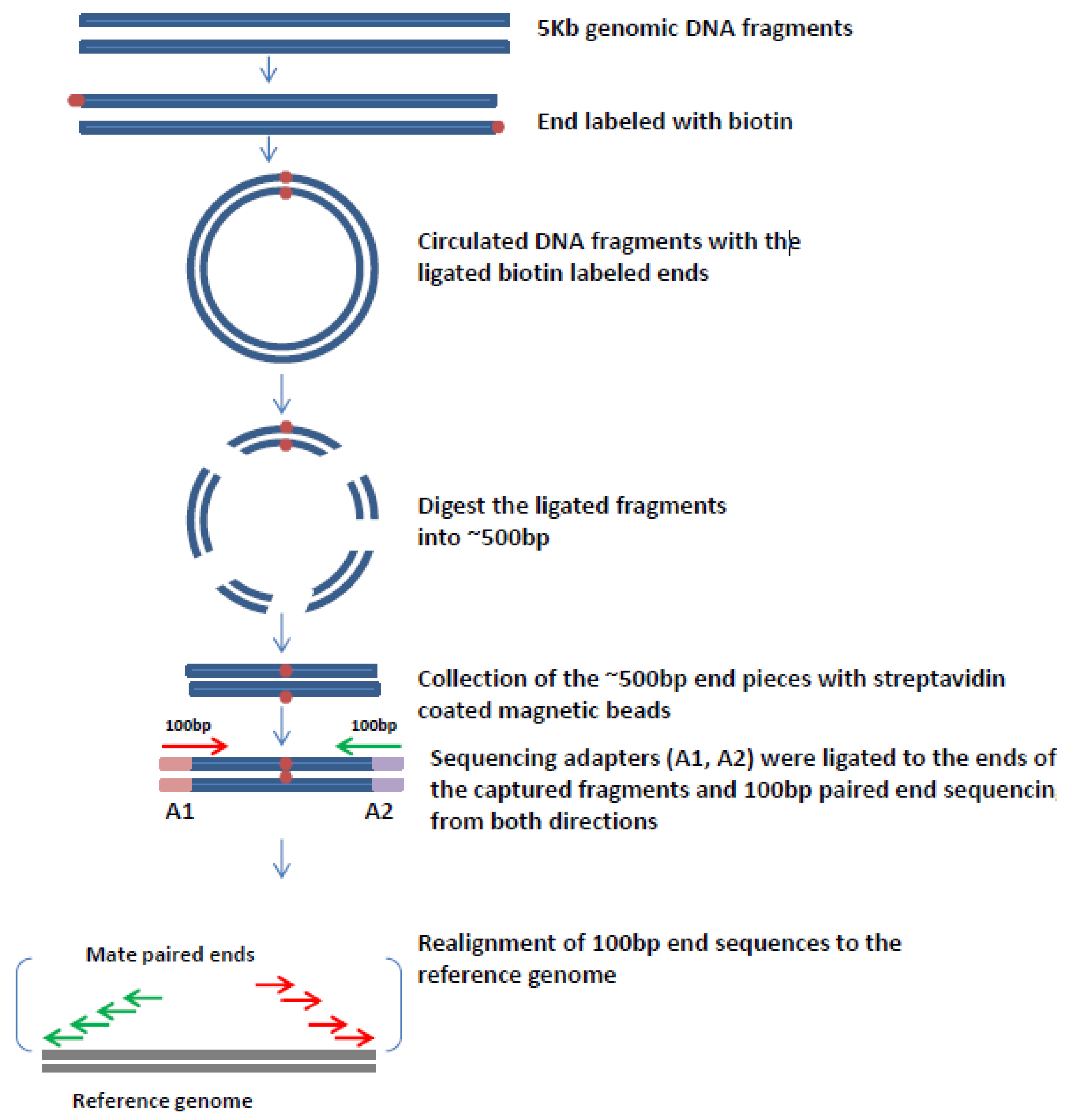
7. Mate-Pair Sequencing to Characterize OPSCCs
8. Other Information Provided by Mate-Pair Sequencing
9. Digital PCR to Monitor the Clinical Course of Cancer
10. Mate-Pair Sequencing As a Powerful Clinical Tool for Cancers with a Viral Etiology
Conflicts of Interest
References
- Zur Hausen, H. Viruses in human cancers. Science 1991, 254, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Zettl, A.; Lee, S.S.; Rüdiger, T.; Starostik, P.; Marino, M.; Kirchner, T.; Ott, M.; Müller-Hermelink, H.K.; Ott, G. Epstein-Barr virus-associated B-cell lymphoproliferative disorders in angloimmunoblastic T-cell lymphoma and peripheral T-cell lymphoma, unspecified. Am. J. Clin. Pathol. 2002, 117, 368–379. [Google Scholar] [PubMed]
- Carbone, A.; Gloghini, A.; Dotti, G. EBV-associated lymphoproliferative disorders: Classification and treatment. Oncologist 2008, 13, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Flavell, K.J.; Murray, P.G. Hodgkin’s disease and Epstein-Barr virus. Mol. Pathol. 2000, 53, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.A.; Wu, J.M.; Tunkel, D.E.; Ishman, S.L. Nasopharyngeal carcinoma: The role of the Epstein-Barr virus. Medscape J. Med. 2008, 10, 165. [Google Scholar] [CrossRef] [PubMed]
- Moscicki, A.B.; Schiffman, M.; Burchell, A.; Albero, G.; Giuliano, A.R.; Goodman, M.T.; Kjaer, S.K.; Palefsky, J. Updating the natural history of human papillomavirus and anogenital cancers. Vaccine 2012, 20, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Lavanchy, D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 2004, 11, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.M.; Pecoraro, G.; Defendi, V. Genetic analysis of in vitro progression of human papillomavirus-transfected human cervical cells. Cancer Res. 1993, 53, 1167–1171. [Google Scholar] [PubMed]
- Montgomery, K.D.; Tedford, K.L.; McDougall, J.K. Genetic instability of chromosome 3 in HPV-immortalized and tumorigenic human keratinocytes. Genes Chromosom. Cancer 1995, 14, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Uejima, H.; Mitsuya, K.; Kugoh, H.; Horikawa, I.; Oshimura, M. Normal human chromosome 2 induces cellular senescence in the human cervical carcinoma cell line SiHa. Genes Chromosom. Cancer 1995, 14, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.X.; Manos, M.M.; Muñoz, N.; Sherman, M.; Jansen, A.M.; Peto, J.; Schiffman, M.H.; Moreno, V.; Kurman, R.; Shah, K.V. Prevalence of human papillomavirus in cervical cancer: A worldwide perspective. International biological study on cervical cancer (IBSCC) Study Group. J. National Cancer Inst. 1995, 87, 796–802. [Google Scholar] [CrossRef]
- Pollicino, T.; Saitta, C.; Raimondo, G. Hepatocellular carcinoma: The point of view of the hepatitis B virus. Carcinogenesis 2011, 32, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Intl. J. Cancer 2008, 127, 2893–2917. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Muñoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Zur Hausen, H. Papillomaviruses in human cancers. Proc. Assoc. Am. Physicians 1999, 111, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Lacey, C.J.; Lowndes, C.M.; Shah, K.V. Chapter 4: Burden and management of non-cancerous HPV-related conditions: HPV-6/11 disease. Vaccine 2006, 24 (Suppl. 3), 35–41. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Bray, F. Chapter 2: The burden of HPV-related cancers. Vaccine 2006, 24 (Suppl. 3), 11–25. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.C.; Gravitt, P.E.; Cheng, S.; Wheeler, C.M. Generation of entire human papillomavirus genomes by long PCR: Frequency of errors produced during amplification. Genome Res. 1995, 5, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Allen-Hoffmann, B.L.; Lambert, P.F. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 1995, 69, 2989–2997. [Google Scholar] [PubMed]
- Cullen, A.P.; Reid, R.; Campion, M.; Lörincz, A.T. Analysis of the physical state of different human papillomavirus DNAs in intraepithelial and invasive cervical neoplasm. J. Virol. 1991, 65, 606–612. [Google Scholar] [PubMed]
- Pirami, L.; Giachè, V.; Becciolini, A. Analysis of HPV16, 18, 31, and 35 DNA in pre-invasive and invasive lesions of the uterine cervix. J. Clin. Pathol. 1997, 50, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Hudelist, G.; Manavi, M.; Pischinger, K.I.; Watkins-Riedel, T.; Singer, C.F.; Kubista, E.; Czerwenka, K.F. Physical state and expression of HPV DNA in benign and dysplastic cervical tissue: Different levels of viral integration are correlated with lesion grade. Gynecol. Oncol. 2004, 92, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Arias-Pulido, H.; Peyton, C.L.; Joste, N.E.; Vargas, H.; Wheeler, C.M. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J. Clin. Microbiol. 2006, 44, 1755–1762. [Google Scholar] [CrossRef] [PubMed]
- Vinokurova, S.; Wentzensen, N.; Kraus, I.; Klaes, R.; Driesch, C.; Melsheimer, P.; Kisseljov, F.; Dürst, M.; Schneider, A.; von Knebel Doeberitz, M. Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res. 2008, 68, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Badaracco, G.; Venuti, A.; Sedati, A.; Marcante, M.L. HPV16 and HPV18 in genital tumors: Significantly different levels of viral integration and correlation to tumor invasiveness. J. Med. Virol. 2002, 67, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.; Ono, T.; Ishimoto, A.; Dowhanick, J.J.; Frizzell, M.A.; Howley, P.M.; Sakai, H. Mechanisms of human papillomavirus E2-mediated repression of viral oncogene expression and cervical cancer cell growth inhibition. J. Virol. 2000, 74, 3752–3760. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Dyson, N.; Howley, P.M.; Münger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Münger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.; Lambert, P.F. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: Implications for cervical carcinogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 1654–1658. [Google Scholar] [CrossRef] [PubMed]
- Peitsaro, P.; Johansson, B.; Syrjänen, S. Integrated human papillomavirus type 16 is frequently found in cervical cancer precursors as demonstrated by a novel quantitative real-time PCR technique. J. Clin. Microbiol. 2002, 40, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.; Herrington, C.S.; Graham, A.K.; Evans, M.F.; McGee, J.O. In situ human papillomavirus (HPV) genotyping of cervical intraepithelial neoplasia in South African and British patients: Evidence for putative HPV integration in vivo. J. Clin. Pathol. 1991, 44, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Klaes, R.; Woerner, S.M.; Ridder, R.; Wentzensen, N.; Duerst, M.; Schneider, A.; Lotz, B.; Melsheimer, P.; von Knebel Doeberitz, M. Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res. 1999, 59, 6132–6136. [Google Scholar] [PubMed]
- Cartwright, N.H.; Cassia, L.J.; Easton, A.J.; Morris, A.G. Detection of human papillomavirus in vulval carcinoma using semi-nested PCR and restriction enzyme typing: A rapid and sensitive technique. Clin. Mol. Pathol. 1996, 49, M236–M239. [Google Scholar] [CrossRef] [PubMed]
- Luft, F.; Klaes, R.; Nees, M.; Dürst, M.; Heilmann, V.; Melsheimer, P.; von Knebel Doeberitz, M. Detection of integrated papillomavirus sequences by ligation-mediated PCR (DIPS-PCR) and molecular characterization in cervical cancer cells. Intl. J. Cancer 2001, 92, 9–17. [Google Scholar] [CrossRef]
- Thorland, E.C.; Myers, S.L.; Gostout, B.S.; Smith, D.I. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene 2003, 22, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Ferber, M.J.; Thorland, E.C.; Brink, A.A.; Rapp, A.K.; Phillips, L.A.; McGovern, R.; Gostout, B.S.; Cheung, T.H.; Chung, T.K.; Fu, W.Y.; et al. Preferential integration of human papillomavirus type 18 near the c-myc locus in cervical carcinoma. Oncogene 2003, 22, 7233–7242. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Shen, K.; McBride, A.A. Papillomavirus genomes associate with BRD4 to replicate at fragile sites in the host genome. PLoS Pathog. 2014, 10, e1004117. [Google Scholar] [CrossRef] [PubMed]
- Anorlu, R.I. What is the significance of the HPV epidemic? Can. J. Urol. 2008, 15, 3860–3865. [Google Scholar] [PubMed]
- Johnson, L.G.; Madeleine, M.M.; Newcomer, L.M.; Schwartz, S.M.; Daling, J.R. Anal cancer incidence and survival: The surveillance, epidemiology, and end results experience, 1973–2000. Cancer 2004, 101, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Brewster, D.H.; Bhatti, L.A. Increasing incidence of squamous cell carcinoma of the anus in Scotland, 1975–2002. Br. J. Cancer 2006, 95, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Edgren, G.; Sparén, P. Risk of anogenital cancer after diagnosis of cervical intraepithelial neoplasia: A prospective population-based study. Lancet Oncol. 2007, 8, 311–316. [Google Scholar] [CrossRef]
- Rabkin, C.S.; Biggar, R.J.; Melbye, M.; Curtis, R.E. Second primary cancers following anal and cervical carcinoma: Evidence of shared etiologic factors. Am. J. Epidemiol. 1992, 136, 54–58. [Google Scholar] [PubMed]
- Wentzensen, N.; Ridder, R.; Klaes, R.; Vinokurova, S.; Schaefer, U.; Doeberitz, M.V. Characterization of viral-cellular fusion transcripts in a large series of HPV16 and 18 positive anogenital lesions. Oncogene 2002, 21, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Ziegert, C.; Wentzensen, N.; Vinokurova, S.; Kisseljov, F.; Einenkel, J.; Hoeckel, M.; von Knebel Doeberitz, M. A comprehensive analysis of HPV integration loci in anogenital lesions combining transcript and genome-based amplification techniques. Oncogene 2003, 22, 3977–3984. [Google Scholar] [CrossRef] [PubMed]
- Licitra, L.; Bernier, J.; Grandi, C.; Merlano, M.; Bruzzi, P.; Lefebvre, J.L. Cancer of the oropharynx. Crit. Rev. Oncol. Hematol. 2002, 41, 107–122. [Google Scholar] [CrossRef]
- Fakhry, C.; Gillison, M.L.; D’Souza, G. Tobacco use and oral HPV-16 infection. JAMA 2014, 312, 1465–1467. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.A. Human Papillomavirus: Confronting the Epidemic—A Urologist’s Perspective. Rev. Urol. 2005, 7, 135–144. [Google Scholar] [PubMed]
- Gao, G.; Chernock, R.D.; Gay, H.A.; Thorstad, W.L.; Zhang, T.R.; Wang, H.; Ma, X.J.; Luo, Y.; Lewis, J.S.; Wang, X. A novel RT-PCR method for quantification of human papillomavirus transcripts in archived tissues and its application in oropharyngeal cancer prognosis. Intl. J. Cancer 2013, 132, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Mirghani, H.; Amen, F.; Blanchard, P.; Moreau, F.; Guigay, J.; Hartl, D.M.; Lacau St Guily, J. Treatment de-escalation in HPV-positive oropharyngeal carcinoma: Ongoing trials, critical issues and perspectives. Intl. J. Cancer 2015, 136, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, M.X.; Su, N.; Wang, L.C.; Wu, X.; Bui, S.; Nielsen, A.; Vo, H.T.; Nguyen, N.; Luo, Y.; et al. RNAscope for in situ detection of transcriptionally active human papillomavirus in head and neck squamous cell carcinoma. J. Vis. Exp. 2014, 85. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, W.J.; Chen, R.W.; Leivo, I.; Mäkitie, A.; Bäck, L.; Kontio, R.; Suuronen, R.; Lindqvist, C.; Auvinen, E.; Molijn, A.; et al. Prevalence and physical status of human papillomavirus in squamous cell carcinomas of the head and neck. Intl. J. Cancer 2003, 107, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Hasegawa, M.; Kiyuna, A.; Matayoshi, S.; Uehara, T.; Agena, S.; Yamashita, Y.; Ogawa, K.; Maeda, H.; Suzuki, M. Viral load, physical status, and E6/E7 mRNA expression of human papillomavirus in head and neck squamous cell carcinoma. Head Neck 2013, 35, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Mooren, J.J.; Kremer, B.; Claessen, S.M.; Voogd, A.C.; Bot, F.J.; Peter Klussmann, J.; Huebbers, C.U.; Hopman, A.H.; Ramaekers, F.C.; Speel, E.J. Chromosome stability in tonsillar squamous cell carcinoma is associated with HPV16 integration and indicates a favorable prognosis. Intl. J. Cancer 2013, 132, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Ukpo, O.C.; Moore, E.J.; Smith, D.I. Human papillomavirus and oropharyngeal cancer. N. Engl. J. Med. 2007, 357, 1156–1157. [Google Scholar] [PubMed]
- Pett, M.; Coleman, N. Integration of high-risk human papillomavirus: A key event in cervical carcinogenesis? J. Pathol. 2007, 212, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Ferber, M.J.; Cheung, T.H.; Chung, T.K.; Wong, Y.F.; Smith, D.I. The role of viral integration in the development of cervical cancer. Cancer Genet. Cytogenet. 2005, 158, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, M.; Gabriel, S.; Getz, G. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010, 11, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Stephens, P.J.; Pleasance, E.D.; O’Meara, S.; Li, H.; Santarius, T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Hardy, C.; et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008, 40, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Tucker, T.; Marra, M.; Friedman, J.M. Massively parallel sequencing: The next big thing in genetic medicine. Am. J. Hum. Genet. 2009, 85, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Pareek, C.S.; Smoczynski, R.; Tretyn, A. Sequencing technologies and genome sequencing. J. Appl. Genet. 2011, 52, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Adey, A.; Burton, J.N.; Kitzman, J.O.; Hiatt, J.B.; Lewis, A.P.; Martin, B.K.; Qiu, R.; Lee, C.; Shendure, J. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 2013, 500, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Ohta, M.; Inoue, H.; Cotticelli, M.G.; Kastury, K.; Baffa, R.; Palazzo, J.; Siprashvili, Z.; Mori, M.; McCue, P.; Druck, T.; et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell 1996, 84, 587–597. [Google Scholar] [CrossRef]
- Zimonjic, D.B.; Druck, T.; Ohta, M.; Kastury, K.; Croce, C.M.; Popescu, N.C.; Huebner, K. Positions of chromosome 3p14.2 fragile sites (FRA3B) within the FHIT gene. Cancer Res. 1997, 57, 1166–1170. [Google Scholar] [PubMed]
- Liu, C.X.; Musco, S.; Lisitsina, N.M.; Yaklichkin, S.Y.; Lisitsyn, N.A. Genomic organization of a new candidate tumor suppressor gene, LRP1B. Genomics 2000, 69, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Mazoyer, S.; Gayther, S.A.; Nagai, M.A.; Smith, S.A.; Dunning, A.; van Rensburg, E.J.; Albertsen, H.; White, R.; Ponder, B.A. A gene (DLG2) located at 17q12–q21 encodes a new homologue of the Drosophila tumor suppressor dIg-A. Genomics 1995, 28, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.I.; Zhu, Y.; McAvoy, S.; Kuhn, R. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 2006, 232, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.K.; Van Hummelen, P.; Chan, P.K.; Cheung, T.H.; Yim, S.F.; Yu, M.Y.; Ducar, M.D.; Thorner, A.R.; MacConaill, L.E.; Doran, G.; et al. Genomic aberrations in cervical adenocarcinomas in Hong Kong Chinese women. Intl. J. Cancer 2015, 137, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Tannir, N.M.; Williams, M.D.; Chen, Y.; Yao, H.; Zhang, J.; Thompson, E.J.; TCGA Network; Meric-Bernstam, F.; Medeiros, L.J.; et al. Landscape of DNA virus associations across human malignant cancers: Analysis of 3775 cases using RNA-Seq. J. Virol. 2013, 87, 8916–8926. [Google Scholar] [CrossRef] [PubMed]
- Edgren, H.; Murumagi, A.; Kangaspeska, S.; Nicorici, D.; Hongisto, V.; Kleivi, K.; Rye, I.H.; Nyberg, S.; Wolf, M.; Borresen-Dale, A.L.; et al. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.W.; Alaei-Mahabadi, B.; Samuelsson, T.; Lindh, M.; Larsson, E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.J.; Cheville, J.C.; Zarei, S.; Johnson, S.H.; Sikkink, R.A.; Kosari, F.; Feldman, A.L.; Eckloff, B.W.; Karnes, R.J.; Vasmatzis, G. Mate pair sequencing of whole-genome-amplified DNA following laser capture microdissection of prostate cancer. DNA Res. 2012, 19, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.L.; Dogan, A.; Smith, D.I.; Law, M.E.; Ansell, S.M.; Johnson, S.H.; Porcher, J.C.; Ozsan, N.; Wieben, E.D.; Eckloff, B.W.; et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood 2011, 117, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Vasmatzis, G.; Johnson, S.H.; Knudson, R.A.; Ketterling, R.P.; Braggio, E.; Fonseca, R.; Viswanatha, D.S.; Law, M.E.; Kip, N.S.; Ozsan, N.; et al. Genome-wide analysis reveals recurrent structural abnormalities of TP63 and other p53-related genes in peripheral T-cell lymphomas. Blood 2012, 120, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Mead, D.A.; Lucas, S.; Copeland, A.; Lapidus, A.; Cheng, J.F.; Bruce, D.C.; Goodwin, L.A.; Pitluck, S.; Chertkov, O.; Zhang, X.; et al. Complete genome sequence of Paenibacillus strain Y4.12MC10, a novel paenibacillus lautus strain isolated from Obsidian Hot Spring in Yellowstone National Park. Stand. Genomic Sci. 2012, 6, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Chander, Y.; Koelbl, J.; Puckett, J.; Moser, M.J.; Klingele, A.J.; Liles, M.R.; Carrias, A.; Mead, D.A.; Schoenfeld, T.W. A novel thermostable polymerase for RNA and DNA loop-mediated isothermal amplification (LAMP). Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Johnson, S.H.; Kasperbauer, J.L.; Eckloff, B.W.; Tombers, N.M.; Vasmatzis, G.; Smith, D.I. Mate pair sequencing of oropharyngeal squamous cell carcinomas reveals that HPV integration occurs much less frequently than in cervical cancer. J. Clin. Virol. 2014, 59, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Lasho, T.; Johnson, S.H.; Smith, D.I.; Crispino, J.D.; Pardanani, A.; Vasmatzis, G.; Tefferi, A. Identification of submicroscopic genetic changes and precise breakpoint mapping in myelofibrosis using high resolution mate-pair sequencing. Am. J. Hematol. 2013, 88, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromoseomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. 2014, 6. [Google Scholar] [CrossRef]
- Heidary, M.; Auer, M.; Ulz, P.; Heitzer, E.; Petru, E.; Gasch, C.; Riethdorf, S.; Mauermann, O.; Lafer, I.; Pristauz, G.; et al. The dynamic range of circulating tumor DNA in metastatic breast cancer. Breast Cancer Res. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- Hayden, R.T.; Gu, Z.; Ingersoll, J.; Abdul-Ali, D.; Shi, L.; Pounds, S.; Caliendo, A.M. Comparison of droplet digital PCR to real-time PCR for quantitative detection of cytomegalovirus. J. Clin. Microbiol. 2013, 51, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.O.; Xu, C.W.; Shao, Y.; Wang, H.T.; Wu, Y.F.; Song, Y.Y.; Li, X.B.; Zhang, Z.; Wang, W.J.; Li, L.Q.; et al. Comparison of droplet digital PCR and conventional quantitative PCR for measuring EGFR gene mutation. Exp. Ther. Med. 2015, 9, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Schøler, L.V.; Thomsen, R.; Tobiasen, H.; Vang, S.; Nordentoft, I.; Lamy, P.; Kannerup, A.S.; Mortensen, F.V.; Stribolt, K.; et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 2015. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Fernández-Landázuri, S.; Rodríguez, C.; Zárate, R.; Lozano, M.D.; Zubiri, L.; Perez-Gracia, J.L.; Martín-Algarra, S.; González, A. Quantitative cell-free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin. Chem. 2015, 61, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, I.; Lonetti, A.; Venturi, C.; Ferrari, A.; Papayannidis, C.; Ottaviani, E.; Abbenante, M.C.; Paolini, S.; Bresciani, P.; Potenza, L.; et al. Use of a high sensitive nanofluidic array for the detection of rare copies of BCR-ABL1 transcript in patients with Philadelphia-positive acute lymphoblastic leukemia in complete response. Leuk. Res. 2014, 38, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, G.; Smith, D.I. Mate-Pair Sequencing as a Powerful Clinical Tool for the Characterization of Cancers with a DNA Viral Etiology. Viruses 2015, 7, 4507-4528. https://doi.org/10.3390/v7082831
Gao G, Smith DI. Mate-Pair Sequencing as a Powerful Clinical Tool for the Characterization of Cancers with a DNA Viral Etiology. Viruses. 2015; 7(8):4507-4528. https://doi.org/10.3390/v7082831
Chicago/Turabian StyleGao, Ge, and David I. Smith. 2015. "Mate-Pair Sequencing as a Powerful Clinical Tool for the Characterization of Cancers with a DNA Viral Etiology" Viruses 7, no. 8: 4507-4528. https://doi.org/10.3390/v7082831
APA StyleGao, G., & Smith, D. I. (2015). Mate-Pair Sequencing as a Powerful Clinical Tool for the Characterization of Cancers with a DNA Viral Etiology. Viruses, 7(8), 4507-4528. https://doi.org/10.3390/v7082831