
Ultra Deep Sequencing of a Baculovirus Population Reveals Widespread Genomic Variations


Abstract
:
1. Introduction

2. Materials and Methods
2.1. Virus Amplification and DNA Extraction
2.2. Sequencing, Consensus Genome Assembly and Annotation
2.3. Mutation Detection and Analyses
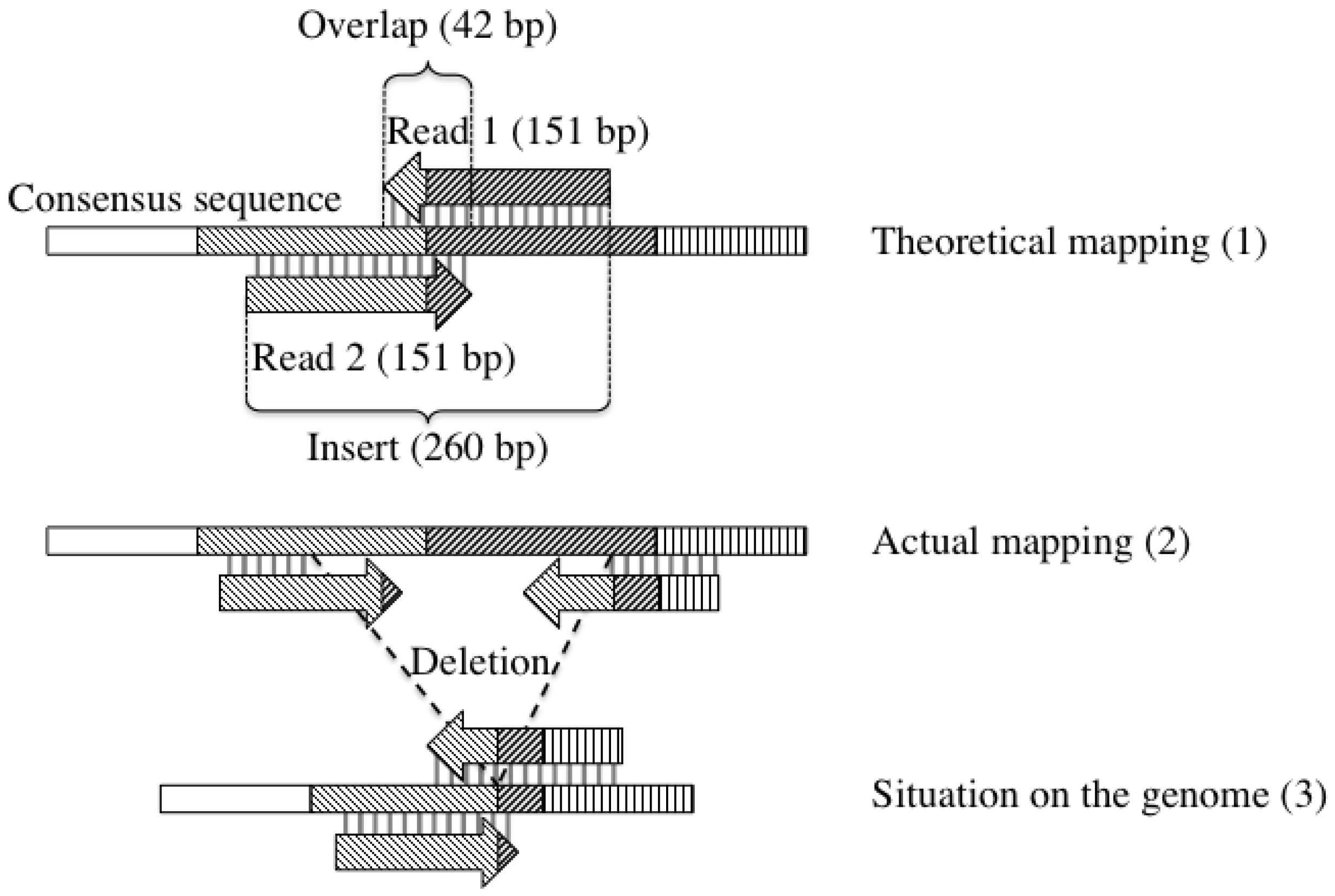
2.4. Detection of Large Deletions
3. Results and Discussion
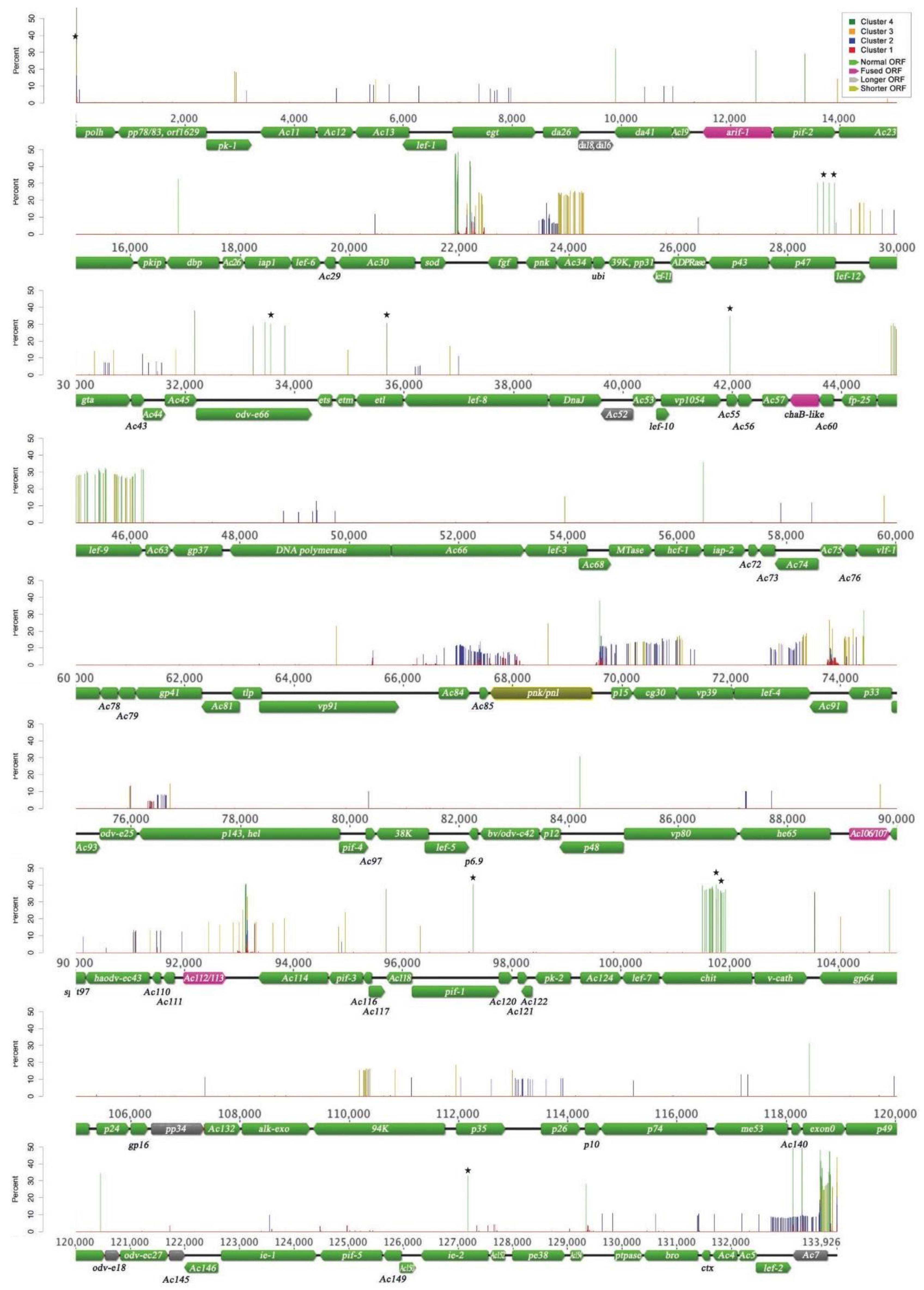
3.1. AcMNPV-WP10 Genome Sequence and Annotation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
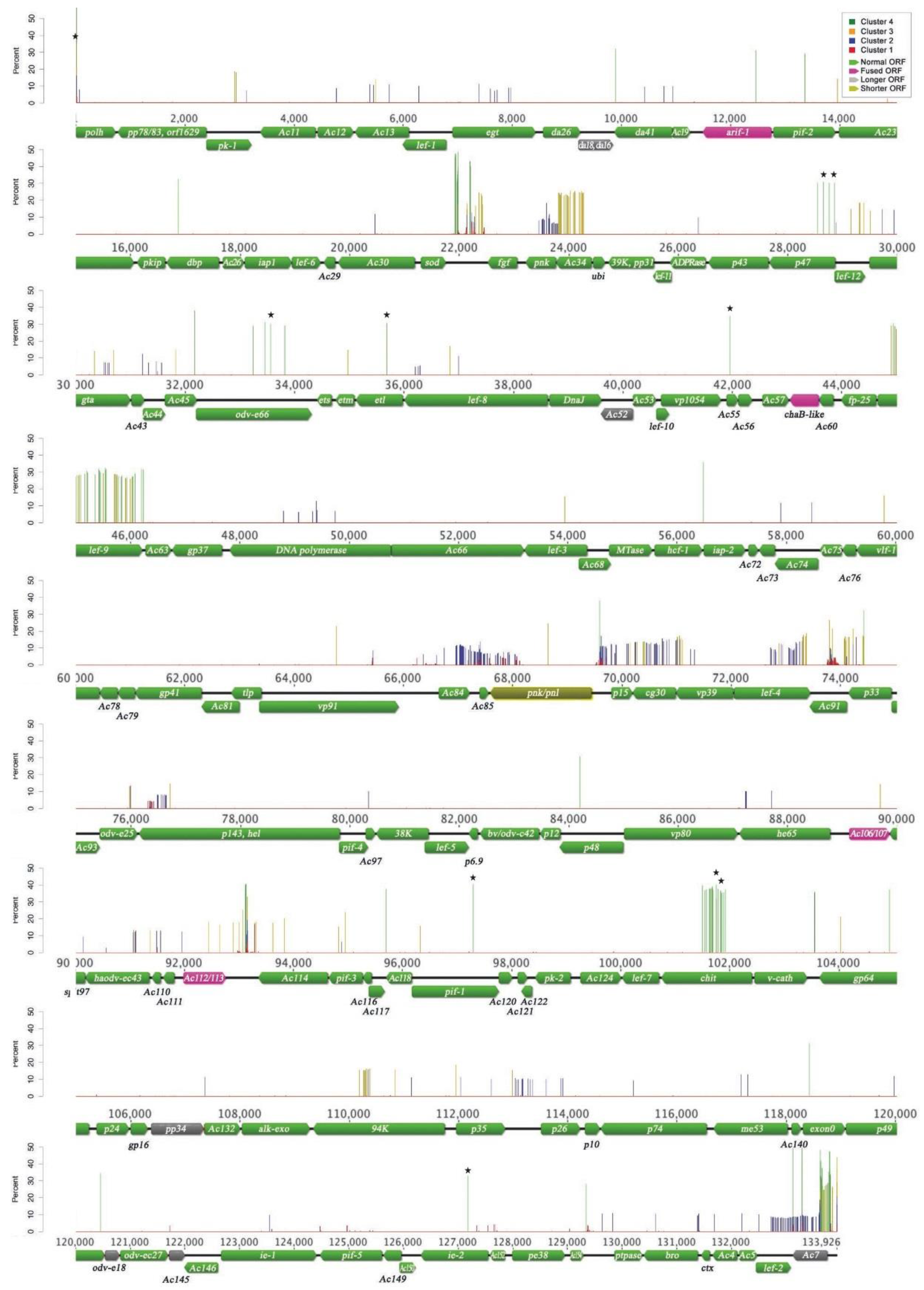
{kind=link}
| WP10/C6 sequence a | Position on WP10 b | Position on C6 c | Type | WP10 gene d | C6 gene |
|---|---|---|---|---|---|
| -/A | 9707 | 14,226 | Indel | Ac17 | Ac17 |
| G/A | 11,651 | 16,171 | SNP | Ac20/21 | Ac20 |
| CG/- | 11,685 | 16,205 | Indel | Ac20/21 | Ac20 |
| AC/- | 11,690 | 16,208 | Indel | Ac20/21 | Ac20 |
| G/C | 11,692 | 16,208 | SNP | Ac20/21 | Ac20 |
| -/G | 11,698 | 16,214 | Indel | Ac20/21 | Ac20 |
| -/C | 11,788 | 16,305 | Indel | Ac20/21 | Ac21 |
| -/G | 38,855 | 44,372 | Indel | Ac52 | Ac52 |
| G/T | 43,197 | 47,714 | SNP | Ac58/59 | Ac58 |
| A/- | 43,398 | 47,914 | Indel | Ac58/59 | Ac59 |
| -/CGACGGTCGAGGG | 67,379 | 71,893 | Indel | Non-coding e | Non-coding e |
| -/TATAATTTTT | 69,604 | 74,134 | Indel | Non-codinge | Ac86 |
| A/- | 89,218 | 93,749 | Indel | Ac106/107 | Ac106 |
| A/- | 89,288 | 93,818 | Indel | Ac106/107 | Ac106 |
| C/- | 89,326 | 93,865 | Indel | Ac106/107 | Ac106 |
| CA/- | 89,414 | 93,953 | Indel | Ac106/107 | Ac106 |
| C/A | 89,417 | 93,954 | SNP | Ac106/107 | Ac106 |
| G/- | 89,447 | 93,983 | Indel | Ac106/107 | Ac106 |
| CG/- | 89,497 | 94,033 | Indel | Ac106/107 | Ac106 |
| A/G | 89,573 | 94,107 | SNP | Ac106/107 | Ac107 |
| ATTTGG/- | 89,576 | 94,110 | Indel | Ac106/107 | Ac107 |
| A/- | 89,587 | 94,114 | Indel | Ac106/107 | Ac107 |
| -/A | 92,249 | 96,777 | Indel | Ac112/113 | Ac112 |
| G/A | 92,440 | 96,968 | SNP | Ac112/113 | Ac113 |
| T/C | 92,635 | 97,163 | SNP | Ac112/113 | Ac113 |
| C/T | 92,885 | 97,413 | SNP | Ac112/113 | Ac113 |
| T/C | 92,998 | 97,526 | SNP | Ac112/113 | Ac113 |
| G/A | 93,065 | 97,593 | SNP | Ac112/113 | Ac113 |
| G/- | 107,127 | 111,645 | Indel | Ac131 | Ac131 |
| -/T | 120,584 | 125,113 | Indel | Ac143 | Ac143 |
| -/T | 120,586 | 125,116 | Indel | Ac143 | Ac143 |
| -/A | 121,748 | 126,790 | Indel | Ac145 | Ac145 |
| ATCTG/- | 133,286 | 3919 | Indel | Ac7 | Ac7 |
| TATTT/- | 133,602 | 4229 | Indel | Ac7 | Ac7 |
| AACAACGCTGCAT/- | 133,610 | 4232 | Indel | Ac7 | Ac7 |
| ACATTA/- | 133,625 | 4234 | Indel | Ac7 | Ac7 |
| ATTTCGGCTT/- | 133,808 | 4411 | Indel | Non-coding e | Non-coding e |
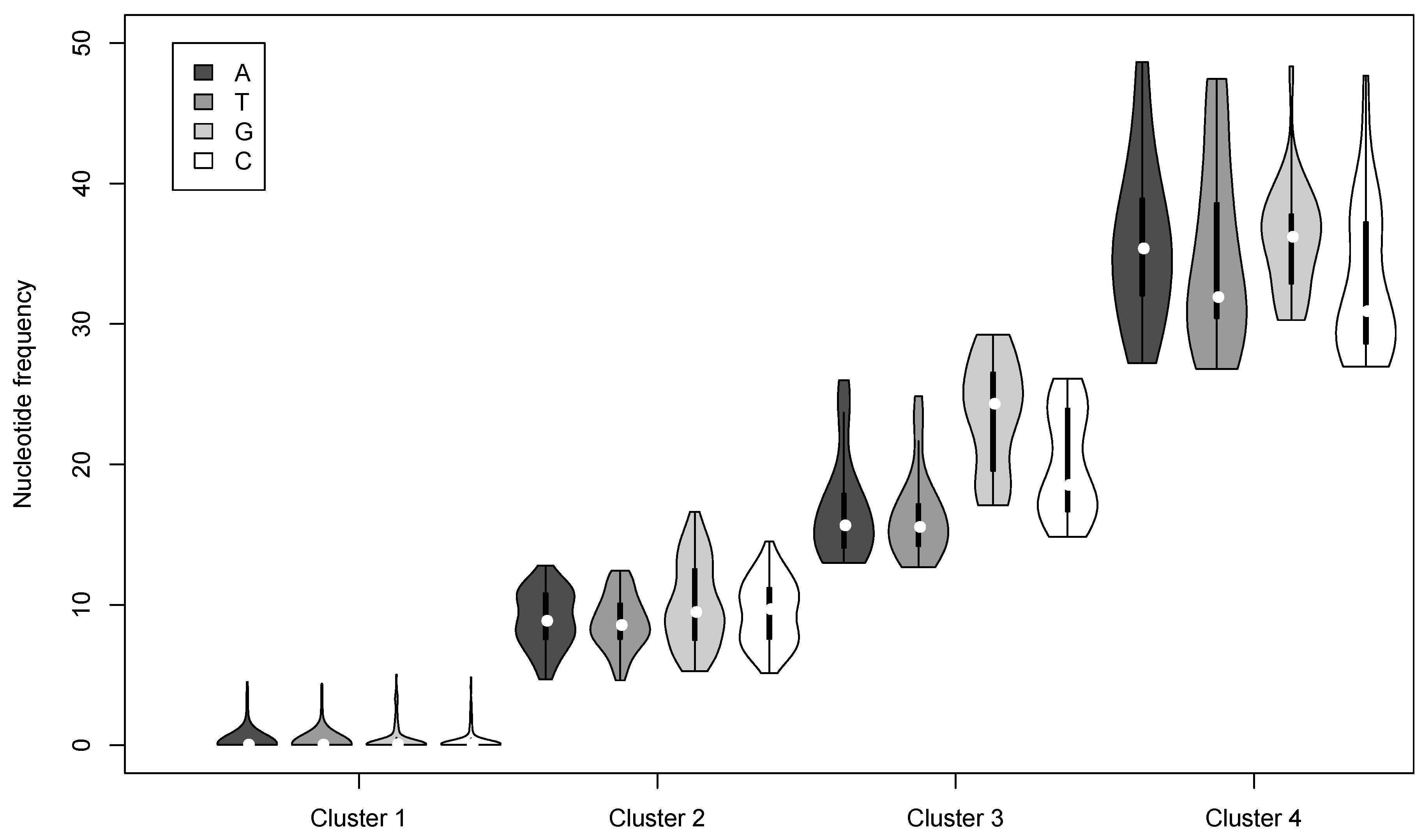
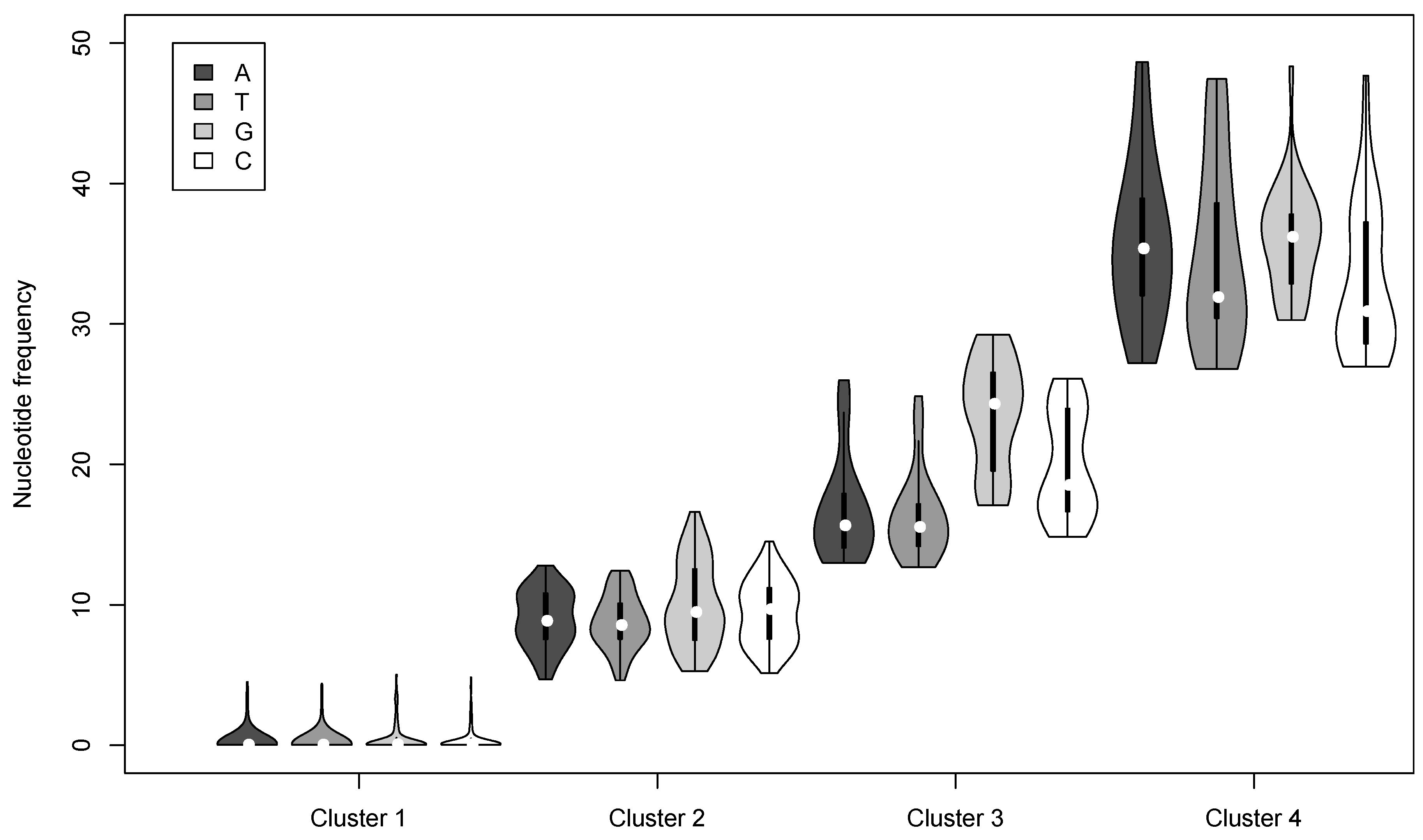
3.2. Nucleotide Variation in the AcMNPV-WP10 Genome Population
| Cluster | Nucleotide | Number of loci | Mean Frequency a | # per genome b | # per OB c |
|---|---|---|---|---|---|
| 1 | A | 903 | 0.23 | 1.39 | 137.71 |
| T | 969 | 0.20 | 1.44 | ||
| G | 328 | 0.44 | 0.87 | ||
| C | 361 | 0.36 | 0.89 | ||
| 2 | A | 98 | 8.97 | 7.07 | 933.75 |
| T | 104 | 8.77 | 7.41 | ||
| G | 112 | 10.02 | 9.79 | ||
| C | 86 | 9.43 | 6.86 | ||
| 3 | A | 39 | 16.75 | 5.94 | 842.56 |
| T | 41 | 16.31 | 6.35 | ||
| G | 46 | 23.59 | 9.72 | ||
| C | 38 | 19.92 | 6.07 | ||
| 4 | A | 25 | 35.82 | 6.03 | 901.33 |
| T | 25 | 34.86 | 5.10 | ||
| G | 30 | 35.99 | 7.95 | ||
| C | 38 | 32.91 | 10.96 |
| Nt a | Gene function b | Position c | Perc. d | Cluster | Gene e | Codon Change | AA change | AA class change f |
|---|---|---|---|---|---|---|---|---|
| C | Accessory | 101,605 | 37.55 | 4 | Ac126/chitinase | TCA->GCA | S->A | NP->No polarity |
| C | Accessory | 101,638 | 38.05 | 4 | Ac126/chitinase | AAA->GAA | K->E | BP->AP |
| T | Accessory | 101,647 | 38.08 | 4 | Ac126/chitinase | CCC->ACC | P->T | No polarity->NP |
| G | Accessory | 101,716 | 40.18 | 4 | Ac126/chitinase | GGT->CGT | G->R | No polarity->BP |
| G | Accessory | 101,793 | 36.25 | 4 | Ac126/chitinase | AGA->AGC | R->S | BP->NP |
| A | Accessory | 101,851 | 35.62 | 4 | Ac126/chitinase | GGC->TGC | G->C | No polarity->NP |
| C | Accessory | 101,884 | 37.25 | 4 | Ac126/chitinase | TCA->GCA | S->A | NP->No polarity |
| T | Host interaction | 110,347 | 15.71 | 3 | Ac134/p94 | CGC->AGC | R->S | BP->NP |
| G | BV specific | 14,832 | 19.28 | 3 | Ac23/f-protein | ACA->GCA | T->A | NP->No polarity |
| T | BV specific | 14,833 | 18.27 | 3 | Ac23/f-protein | ACA->ATA | T->I | NP->No polarity |
| A | BV specific | 103,990 | 21.18 | 3 | Ac128/gp64 | TCG->TTG | S->L | NP->No polarity |
| A | ODV specific | 2 | 36.71 | 4 | Ac8/polyhedrin | ATG->AAG | M->K | No polarity->BP |
| A | ODV specific | 4 | 38.94 | 4 | Ac8/polyhedrin | CCG->ACG | P->T | No polarity->NP |
| T | ODV specific | 4 | 32.48 | 4 | Ac8/polyhedrin | CCG->TCG | P->S | No polarity->NP |
| A | ODV specific | 5 | 12.99 | 3 | Ac8/polyhedrin | CCG->CAG | P->Q | No polarity->NP |
| A | ODV specific | 13,917 | 14.02 | 3 | Ac22/pif-2 | GGG->AGG | G->R | No polarity->BP |
| A | ODV specific | 33,571 | 30.25 | 4 | Ac46/odv-e66 | GCC->ACC | A->T | No polarity->NP |
| G | ODV specific | 64,772 | 22.98 | 3 | Ac83/vp91 | ACC->GCC | T->A | NP->No polarity |
| A | ODV specific | 70,144 | 13.09 | 3 | Ac88/cg30 | CAA->AAA | Q->K | NP->BP |
| A | ODV specific | 70,234 | 13.58 | 3 | Ac88/cg30 | GCC->TCC | A->S | No polarity->NP |
| A | ODV specific | 70,268 | 13.58 | 3 | Ac88/cg30 | ACA->ATA | T->I | NP->No polarity |
| A | ODV specific | 70,381 | 13.68 | 3 | Ac88/cg30 | GAC->TAC | D->Y | AP->NP |
| T | ODV specific | 70,399 | 13.78 | 3 | Ac88/cg30 | GCG->ACG | A->T | No polarity->NP |
| G | ODV specific | 94,932 | 23.84 | 3 | Ac115/pif-3 | GGT->CGT | G->R | No polarity->BP |
| C | ODV specific | 97,271 | 40.46 | 4 | Ac119/pif-1 | TCG->CCG | S->P | NP->No polarity |
| T | PA | 71,103 | 14.86 | 3 | Ac89/vp39 | CGC->AGC | R->S | BP->NP |
| C | PA | 74,220 | 21.51 | 3 | Ac92/p33 | AAA->GAA | K->E | BP->AP |
| C | PA | 74,391 | 17.50 | 3 | Ac92/p33 | AAA->GAA | K->E | BP->AP |
| A | PA | 74,421 | 32.40 | 4 | Ac92/p33 | CAC->TAC | H->Y | BP->NP |
| G | PA | 84,226 | 30.75 | 4 | Ac103/p45 | ACT->CCT | T->P | NP->No polarity |
| T | PA | 91,064 | 13.25 | 3 | Ac109/odv-ec43 | GAG->AAG | E->K | AP->BP |
| A | Replication | 29,534 | 13.78 | 3 | Ac42/gta | GCG->ACG | A->T | No polarity->NP |
| A | Transcription | 28,878 | 30.16 | 4 | Ac41/lef-12 | GAC->AAC | D->N | AP->NP |
| G | Transcription | 29,337 | 18.47 | 3 | Ac41/lef-12 | ACA->GCA | T->A | NP->No polarity |
| T | Transcription | 29,346 | 18.55 | 3 | Ac41/lef-12 | CCA->TCA | P->S | No polarity->NP |
| A | Transcription | 36,848 | 17.11 | 3 | Ac50/lef-8 | CAC->TAC | H->Y | BP->NP |
| G | Transcription | 45,913 | 26.33 | 3 | Ac62/lef-9 | AAA->GAA | K->E | BP->AP |
| C | Transcription | 68,643 | 24.53 | 3 | Ac86/pnk/pnl | ACA->GCA | T->A | NP->No polarity |
| T | Transcription | 73,303 | 16.73 | 3 | Ac90/lef-4 | TCG->TTG | S->L | NP->No polarity |
| T | Transcription | 73,356 | 16.95 | 3 | Ac90/lef-4 | CCG->TCG | P->S | No polarity->NP |
| G | Unknown | 23,830 | 24.44 | 3 | Ac34 | AAT->CAT | N->H | NP->BP |
| G | Unknown | 23,932 | 22.47 | 3 | Ac34 | TAT->CAT | Y->H | NP->BP |
| T | Unknown | 23,938 | 22.88 | 3 | Ac34 | GAG->AAG | E->K | AP->BP |
| C | Unknown | 24,013 | 22.45 | 3 | Ac34 | CGC->GGC | R->G | BP->No polarity |
| G | Unknown | 24,025 | 22.20 | 3 | Ac34 | AAT->CAT | N->H | NP->BP |
| G | Unknown | 24,112 | 24.62 | 3 | Ac34 | TAT->CAT | Y->H | NP->BP |
| G | Unknown | 24,205 | 24.73 | 3 | Ac34 | GGG->CGG | G->R | No polarity->BP |
| G | Unknown | 24,226 | 24.57 | 3 | Ac34 | GGG->CGG | G->R | No polarity->BP |
| G | Unknown | 24,263 | 24.26 | 3 | Ac34 | GAT->GCT | D->A | AP->No polarity |
| C | Unknown | 24,267 | 24.15 | 3 | Ac34 | AAT->AAG | N->K | NP->BP |
| T | Unknown | 24,277 | 24.28 | 3 | Ac34 | CAG->AAG | Q->K | NP->BP |
| C | Unknown | 41,973 | 34.83 | 4 | Ac55 | TTG->TCG | L->S | No polarity->NP |
| A | Unknown | 73,784 | 25.98 | 3 | Ac91 | CCA->TCA | P->S | No polarity->NP |
| T | Unknown | 74,061 | 15.53 | 3 | Ac91 | TTA->TAA | L-> * | No polarity->None |
| A | Unknown | 89,718 | 14.28 | 3 | Ac106/107 | CCA->ACA | P->T | No polarity->NP |
| G | Unknown | 93,822 | 20.30 | 3 | Ac114 | AAT->CAT | N->H | NP->BP |
| C | Unknown | 133,289 | 37.26 | 4 | Ac7/orf603 | CTG->CGG | L->R | No Polarity->BP |
| T | Unknown | 133,648 | 37.68 | 4 | Ac7/orf603 | CCA->ACA | P->T | No polarity->NP |
| G | Unknown | 133,708 | 26.94 | 3 | Ac7/orf603 | AAC->CAC | N->H | NP->BP |
| C | Unknown | 133,738 | 27.70 | 4 | Ac7/orf603 | AAG->GAG | K->E | BP->AP |
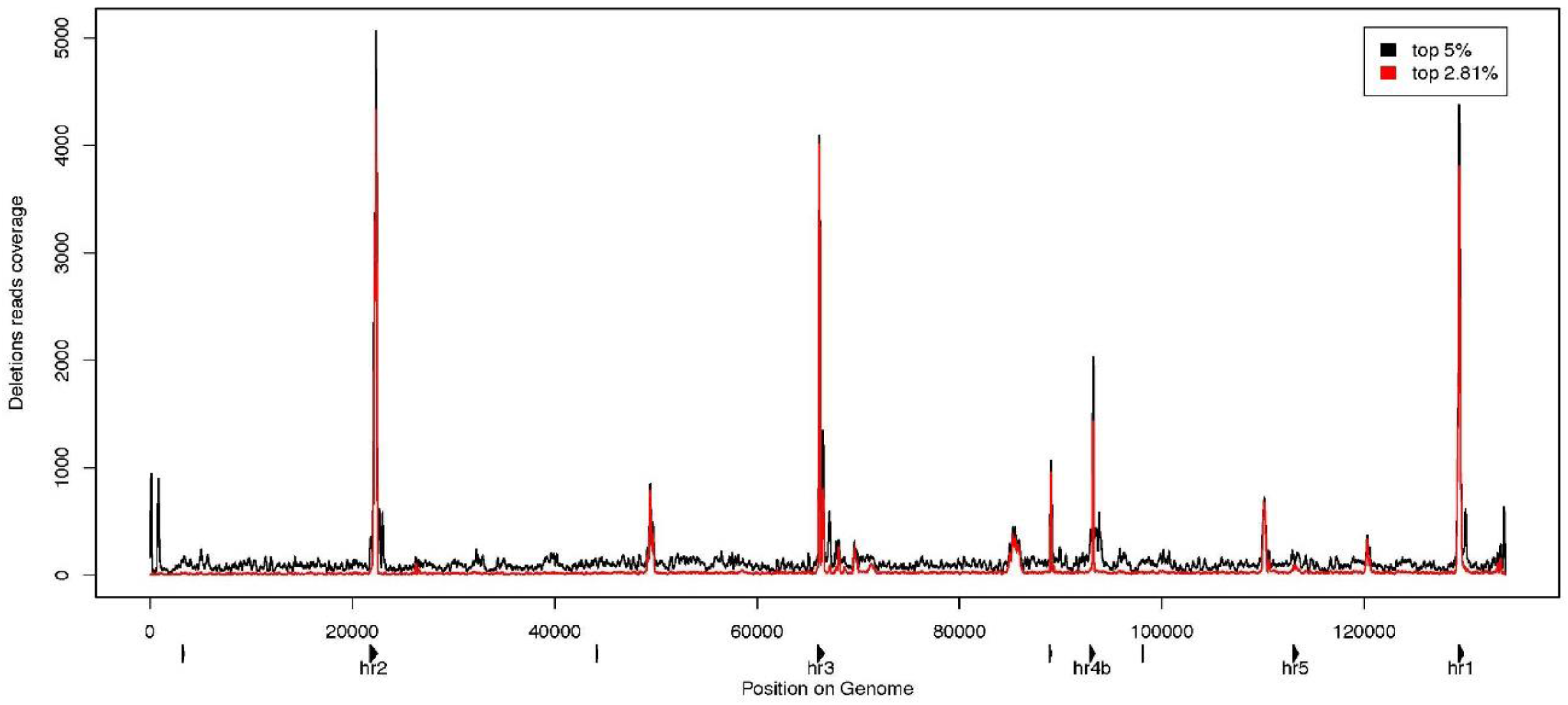
3.3. Characterization of Large Deletions
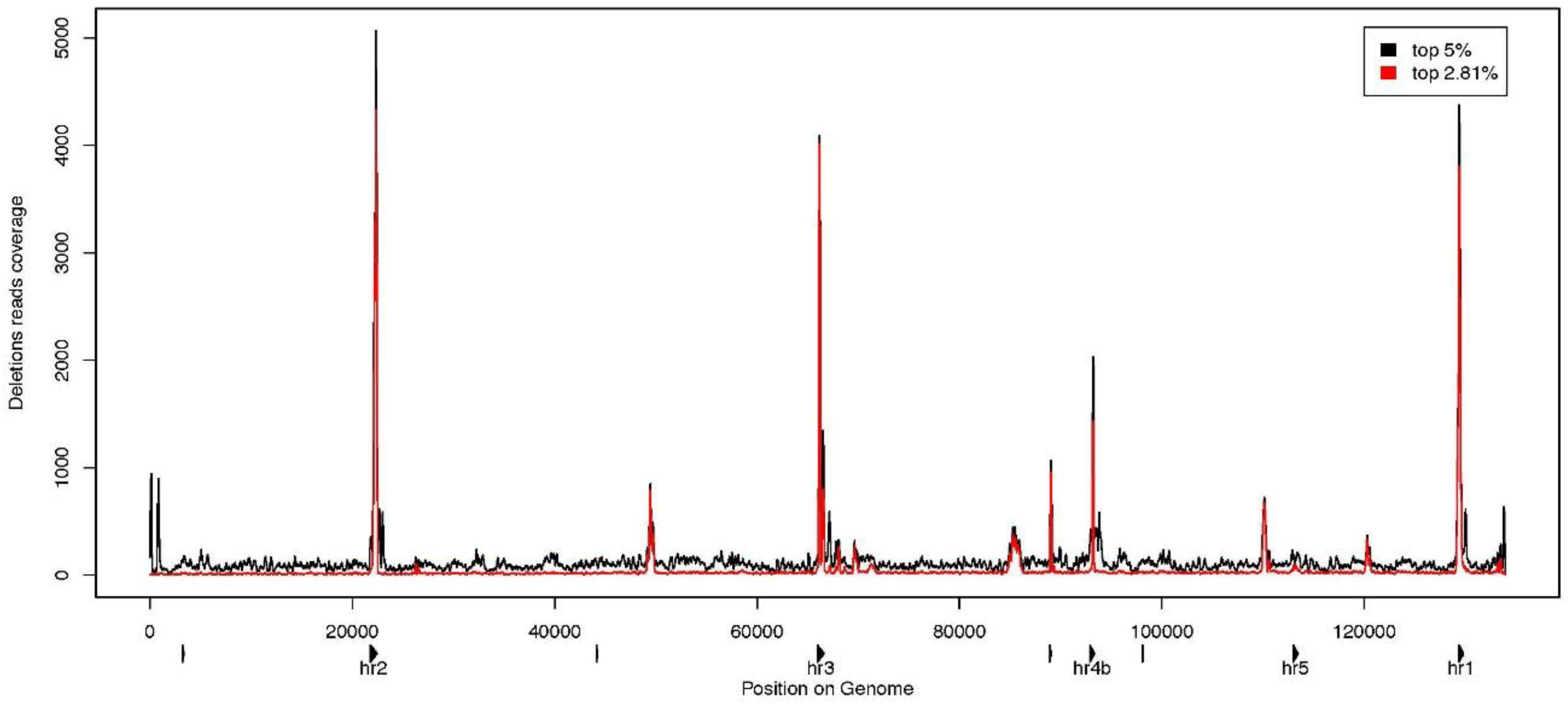
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Data Deposition
Conflicts of Interest
References and Notes
- Darwin, C. On the Origins of Species by Means of Natural Selection; Murray: London, UK, 1859. [Google Scholar]
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press: Cambridge, UK, 1985. [Google Scholar]
- Masel, J. Genetic drift. Curr. Biol. 2011, 21, R837–R838. [Google Scholar] [CrossRef] [PubMed]
- Gordo, I.; Charlesworth, B. The degeneration of asexual haploid populations and the speed of Muller's ratchet. Genetics 2000, 154, 1379–1387. [Google Scholar] [PubMed]
- Barraclough, T.G.; Fontaneto, D.; Ricci, C.; Herniou, E.A. Evidence for inefficient selection against deleterious mutations in cytochrome oxidase I of asexual bdelloid rotifers. Mol. Biol. Evol. 2007, 24, 1952–1962. [Google Scholar] [CrossRef] [PubMed]
- Birdsell, J.A.; Wills, C. The Evolutionary Origin and Maintenance of Sexual Recombination: A Review of Contemporary Models. In Evolutionary Biology; Springer US: Boston, MA, USA, 2003; pp. 27–138. [Google Scholar]
- Hermisson, J.; Pennings, P.S. Soft sweeps: Molecular population genetics of adaptation from standing genetic variation. Genetics 2005, 169, 2335–2352. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2005, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, S.; Michalakis, Y.; Blanc, S. Virus population bottlenecks during within-host progression and host-to-host transmission. Curr. Opin. Virol. 2012, 2, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Zhang, J.; Zhou, X.; Li, H. Genetic structure and population variability of tomato yellow leaf curl China virus. J. Virol. 2007, 81, 5902–5907. [Google Scholar] [CrossRef] [PubMed]
- Van Loy, T.; Thys, K.; Tritsmans, L.; Stuyver, L.J. Quasispecies analysis of JC virus DNA present in urine of healthy subjects. PLoS ONE 2013, 8, e70950. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.A.; Tyler, S.D.; Carpenter, J.E.; Jackson, W.; Mori, Y.; Arvin, A.M.; Grose, C. The attenuated genotype of varicella-zoster virus includes an ORF0 transitional stop codon mutation. J. Virol. 2012, 86, 10695–10703. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Kundu, S.; Jensen, N.J.; Gray, E.R.; Jones, M.; Steinberg, S.; Gershon, A.; Kinchington, P.R.; Schmid, D.S.; Balloux, F.; Nichols, R.A.; Breuer, J. Deep sequencing of viral genomes provides insight into the evolution and pathogenesis of varicella zoster virus and its vaccine in humans. Mol. Biol. Evol. 2014, 31, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Gibson, L.; Jensen, J.D.; Kowalik, T.F. Human cytomegalovirus intrahost evolution—A new avenue for understanding and controlling herpesvirus infections. Curr. Opin. Virol. 2014, 8, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Sijmons, S.; Van Ranst, M.; Maes, P. Genomic and functional characteristics of human cytomegalovirus revealed by next-generation sequencing. Viruses 2014, 6, 1049–1072. [Google Scholar] [CrossRef] [PubMed]
- Slack, J.; Arif, B.M. The baculoviruses occlusion-derived virus: Virion structure and function. Adv. Virus Res. 2007, 69, 99–165. [Google Scholar] [PubMed]
- Kawamoto, F.; Asayama, T. Studies on the arrangement patterns of nucleocapsids within the envelopes of nuclear-polyhedrosis virus in the fat-body cells of the brown tail moth, Euproctis similis. J. Invertebr. Pathol. 1975, 26, 47–55. [Google Scholar] [CrossRef]
- Kondo, A.; Maeda, S. Host range expansion by recombination of the baculoviruses Bombyx mori nuclear polyhedrosis virus and Autographa californica nuclear polyhedrosis virus. J. Virol. 1991, 65, 3625–3632. [Google Scholar] [PubMed]
- Stiles, B.; Himmerich, S. Autographa californica NPV isolates: Restriction endonuclease analysis and comparative biological activity. J. Invertebr. Pathol. 1998, 72, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Kamita, S.G.; Maeda, S.; Hammock, B.D. High-frequency homologous recombination between baculoviruses involves DNA replication. J. Virol. 2003, 77, 13053–13061. [Google Scholar] [CrossRef] [PubMed]
- Cory, J.S.; Green, B.M.; Paul, R.K.; Hunter-Fujita, F. Genotypic and phenotypic diversity of a baculovirus population within an individual insect host. J. Invertebr. Pathol. 2005, 89, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Bull, J.C.; Godfray, H.C.J.; O'Reilly, D.R. A few-polyhedra mutant and wild-type nucleopolyhedrovirus remain as a stable polymorphism during serial coinfection in Trichoplusia ni. Appl. Environ. Microb. 2003, 69, 2052–2057. [Google Scholar] [CrossRef]
- López-Ferber, M.; Simón, O.; Williams, T.; Caballero, P. Defective or effective? Mutualistic interactions between virus genotypes. Proc. R. Soc. B. 2003, 270, 2249–2255. [Google Scholar]
- Ayres, M.D.; Howard, S.C.; Kuzio, J.; López-Ferber, M.; Possee, R.D. The complete DNA sequence of Autographa californica nuclear polyhedrosis virus. Virology 1994, 202, 586–605. [Google Scholar] [CrossRef] [PubMed]
- Herniou, E.A.; Arif, B.M.; Becnel, B.M.; Blissard, G.W.; Bonning, B.C.; Harrison, R.D.; Jehle, J.A.; Theilmann, D.A.; Vlak, J.M. Family Baculoviridae. In Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Lefkowitz, S.M., Carstens, E.B., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Rohrmann, G.F. Baculovirus Molecular Biology, 3rd ed.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2013. [Google Scholar]
- Garavaglia, M.J.; Miele, S.A.B.; Iserte, J.A.; Belaich, M.N.; Ghiringhelli, P.D. The ac53, ac78, ac101, and ac103 genes are newly discovered core genes in the family Baculoviridae. J. Virol. 2012, 86, 12069–12079. [Google Scholar] [CrossRef] [PubMed]
- Cory, J.S. Ecological impacts of virus insecticides: Host range and non-target organisms. In Environmental Impacts of Microbial Insecticides; Hokkanen, H.M., Hajek, A.E., Eds.; Kluwer Academic Publishers: Amsterdam, The Netherlands, 2003; pp. 73–92. [Google Scholar]
- Goulson, D. Can Host Susceptibility to Baculovirus Infection be Predicted from Host Taxonomy or Life History? Environ. Entomol. 2003, 32, 61–70. [Google Scholar] [CrossRef]
- Lee, H.H.; Miller, L.K. Isolation of genotypic variants of Autographa californica nuclear polyhedrosis virus. J. Virol. 1978, 27, 754–767. [Google Scholar] [PubMed]
- Knell, J.D.; Summers, M.D. Investigation of genetic heterogeneity in wild isolates of Spodoptera frugiperda nuclear polyhedrosis virus by restriction endonuclease analysis of plaque-purified variants. Virology 1981, 112, 190–197. [Google Scholar] [CrossRef]
- Maeda, S.; Mukohara, Y.; Kondo, A. Characteristically distinct isolates of the nuclear polyhedrosis virus from Spodoptera litura. J. Gen. Virol. 1990, 71, 2631–2639. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, C.; Chateigner, A.; Ernenwein, L.; Barbe, V.; Bézier, A.; Herniou, E.A.; Cordaux, R. Population genomics supports baculoviruses as vectors of horizontal transfer of insect transposons. Nat. Commun. 2014, 5, 3348. [Google Scholar] [CrossRef] [PubMed]
- Crumb, S.E. The Larvae of the Phalaenidae; United States Department of Agriculture: Washington, DC, USA, 1956. [Google Scholar]
- Vail, P.; Sutter, G.; Jay, D.; Gough, D. Reciprocal infectivity of nuclear polyhedrosis viruses of the cabbage looper and alfalfa looper. J. Invertebr. Pathol. 1971, 17, 383–388. [Google Scholar] [CrossRef]
- Li, H.; Bonning, B.C. Evaluation of the insecticidal efficacy of wild-type and recombinant baculoviruses. Methods Mol. Biol. 2007, 388, 379–404. [Google Scholar] [PubMed]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Institut de Recherche sur la Biologie de l'Insecte. Available online: http://irbi.univ-tours.fr/softwares/Blast2Gb.pl (accessed on 2 July 2015).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- MacQueen, J.B. Some Methods for Classification and Analysis of Multivariate Observations. In Fifth Berkeley Symposium on Mathematical Statistics and Probability; Le Cam, L.M., Neyman, J., Eds.; University of California Press: Berkeley, CA, USA, 1966; p. 17. [Google Scholar]
- Swiss Federal Institute of Technology Zurich. Available online: http://stat.ethz.ch/R-manual/R-devel/library/stats/html/kmeans.html (accessed on 2 July 2015).
- Hintze, J.L.; Nelson, R.D. Violin plots: A box plot-density trace synergism. Am. Stat. 1998, 52, 181–184. [Google Scholar]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [PubMed]
- Carstens, E.B.; Wu, Y. No single homologous repeat region is essential for DNA replication of the baculovirus Autographa californica multiple nucleopolyhedrovirus. J. Gen. Virol. 2007, 88, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-R.; Zhong, S.; Fei, Z.; Hashimoto, Y.; Xiang, J.Z.; Zhang, S.; Blissard, G.W. The transcriptome of the baculovirus Autographa californica multiple nucleopolyhedrovirus in Trichoplusia ni cells. J. Virol. 2013, 87, 6391–6405. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Luria, S.E.; Delbrück, M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics 1943, 28, 491–511. [Google Scholar] [PubMed]
- Drake, J.W. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. USA 1991, 88, 7160–7164. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Gojobori, T.; Li, W.-H. Radical amino acid change versus positive selection in the evolution of viral envelope proteins. Gene 2006, 385, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Thézé, J.; Cabodevilla, O.; Palma, L.; Williams, T.; Caballero, P.; Herniou, E.A. Genomic diversity in European Spodoptera exigua multiple nucleopolyhedrovirus isolates. J. Gen. Virol. 2014, 95, 2297–2309. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; van Oers, M.M.; Hu, Z.; van Lent, J.W.M.; Vlak, J.M. Baculovirus per os infectivity factors form a complex on the surface of occlusion-derived virus. J. Virol. 2010, 84, 9497–9504. [Google Scholar] [CrossRef] [PubMed]
- Simón, O.; Williams, T.; Cerutti, M.; Caballero, P.; López-Ferber, M. Expression of a peroral infection factor determines pathogenicity and population structure in an insect virus. PLoS ONE 2013, 8, e78834. [Google Scholar]
- Bull, J.C.; Godfray, H.C.J.; O'Reilly, D.R. Persistence of an occlusion-negative recombinant nucleopolyhedrovirus in Trichoplusia ni indicates high multiplicity of cellular infection. Appl. Environ. Microb. 2001, 67, 5204–5209. [Google Scholar] [CrossRef] [PubMed]
- Oomens, A.G.; Monsma, S.A.; Blissard, G.W. The baculovirus GP64 envelope fusion protein: Synthesis, oligomerization, and processing. Virology 1995, 209, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Monsma, S.A.; Blissard, G.W. Identification of a membrane fusion domain and an oligomerization domain in the baculovirus GP64 envelope fusion protein. J. Virol. 1995, 69, 2583–2595. [Google Scholar] [PubMed]
- Hawtin, R.E.; Zarkowska, T.; Arnold, K.; Thomas, C.J.; Gooday, G.W.; King, L.A.; Kuzio, J.A.; Possee, R.D. Liquefaction of Autographa californica nucleopolyhedrovirus-infected insects is dependent on the integrity of virus-encoded chitinase and cathepsin genes. Virology 1997, 238, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.J.; Brown, H.L.; Hawes, C.R.; Lee, B.Y.; Min, M.K.; King, L.A.; Possee, R.D. Localization of a baculovirus-induced chitinase in the insect cell endoplasmic reticulum. J. Virol. 1998, 72, 10207–10212. [Google Scholar] [PubMed]
- Braconi, C.T.; Ardisson-Araujo, D.M.P.; Leme, A.F.P.; Oliveira, J.V.D.C.; Pauletti, B.A.; Garcia-Maruniak, A.; Ribeiro, B.M.; Maruniak, J.E.; Zanotto, P.M.D.A. Proteomic analyses of baculovirus Anticarsia gemmatalis multiple nucleopolyhedrovirus budded and occluded virus. J. Gen. Virol. 2014, 95, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Cochran, M.A.; Faulkner, P. Location of Homologous DNA Sequences Interspersed at Five Regions in the Baculovirus AcMNPV Genome. J. Virol. 1983, 45, 961–970. [Google Scholar] [PubMed]
- Pearson, M.; Bjornson, R.; Pearson, G.; Rohrmann, G. The Autographa californica baculovirus genome: Evidence for multiple replication origins. Science 1992, 257, 1382–1384. [Google Scholar] [CrossRef] [PubMed]
- Okano, K.; Vanarsdall, A.L.; Rohrmann, G.F. A baculovirus alkaline nuclease knockout construct produces fragmented DNA and aberrant capsids. Virology 2007, 359, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Marriott, A.C.; Dimmock, N.J. Defective interfering viruses and their potential as antiviral agents. Rev. Med. Virol. 2010, 20, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Voncken, J.W.; van Lier, F.L.; Tramper, J.; Vlak, J.M. Detection and analysis of Autographa californica nuclear polyhedrosis virus mutants with defective interfering properties. Virology 1991, 183, 739–746. [Google Scholar] [CrossRef]
- Li, D.; Aaskov, J. Sub-genomic RNA of defective interfering (D.I.) dengue viral particles is replicated in the same manner as full length genomes. Virology 2014, 468–470, 248–255. [Google Scholar]
- Van Valen, L. Molecular evolution as predicted by natural selection. J. Mol. Evol. 1974, 3, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Taddei, F.; Radman, M.; Maynard-Smith, J.; Toupance, B.; Gouyon, P.-H.; Godelle, B. Role of mutator alleles in adaptive evolution. Nature 1997, 387, 700–702. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Menéndez Arias, L.; Holland, J.J. RNA virus fitness. Rev. Med. Virol. 1997, 7, 87–96. [Google Scholar] [CrossRef]
- Lauring, A.S.; Frydman, J.; Andino, R. The role of mutational robustness in RNA virus evolution. Nat. Rev. Microbiol. 2013, 11, 327–336. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chateigner, A.; Bézier, A.; Labrousse, C.; Jiolle, D.; Barbe, V.; Herniou, E.A. Ultra Deep Sequencing of a Baculovirus Population Reveals Widespread Genomic Variations. Viruses 2015, 7, 3625-3646. https://doi.org/10.3390/v7072788
Chateigner A, Bézier A, Labrousse C, Jiolle D, Barbe V, Herniou EA. Ultra Deep Sequencing of a Baculovirus Population Reveals Widespread Genomic Variations. Viruses. 2015; 7(7):3625-3646. https://doi.org/10.3390/v7072788
Chicago/Turabian StyleChateigner, Aurélien, Annie Bézier, Carole Labrousse, Davy Jiolle, Valérie Barbe, and Elisabeth A. Herniou. 2015. "Ultra Deep Sequencing of a Baculovirus Population Reveals Widespread Genomic Variations" Viruses 7, no. 7: 3625-3646. https://doi.org/10.3390/v7072788
APA StyleChateigner, A., Bézier, A., Labrousse, C., Jiolle, D., Barbe, V., & Herniou, E. A. (2015). Ultra Deep Sequencing of a Baculovirus Population Reveals Widespread Genomic Variations. Viruses, 7(7), 3625-3646. https://doi.org/10.3390/v7072788