
Intracellular Trafficking of Baculovirus Particles: A Quantitative Study of the HearNPV/HzAM1 Cell and AcMNPV/Sf9 Cell Systems

Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses, Cell Lines, and Medium
2.2. Infection Set Up
2.3. Cell Fractionation into Cytoplasmic and Nuclear Fractions
2.4. Western Blotting
2.5. Real-Time Quantitative PCR
2.6. Pulse Field Gel Electrophoresis (PFGE)
2.7. Electron Microscopy
3. Results
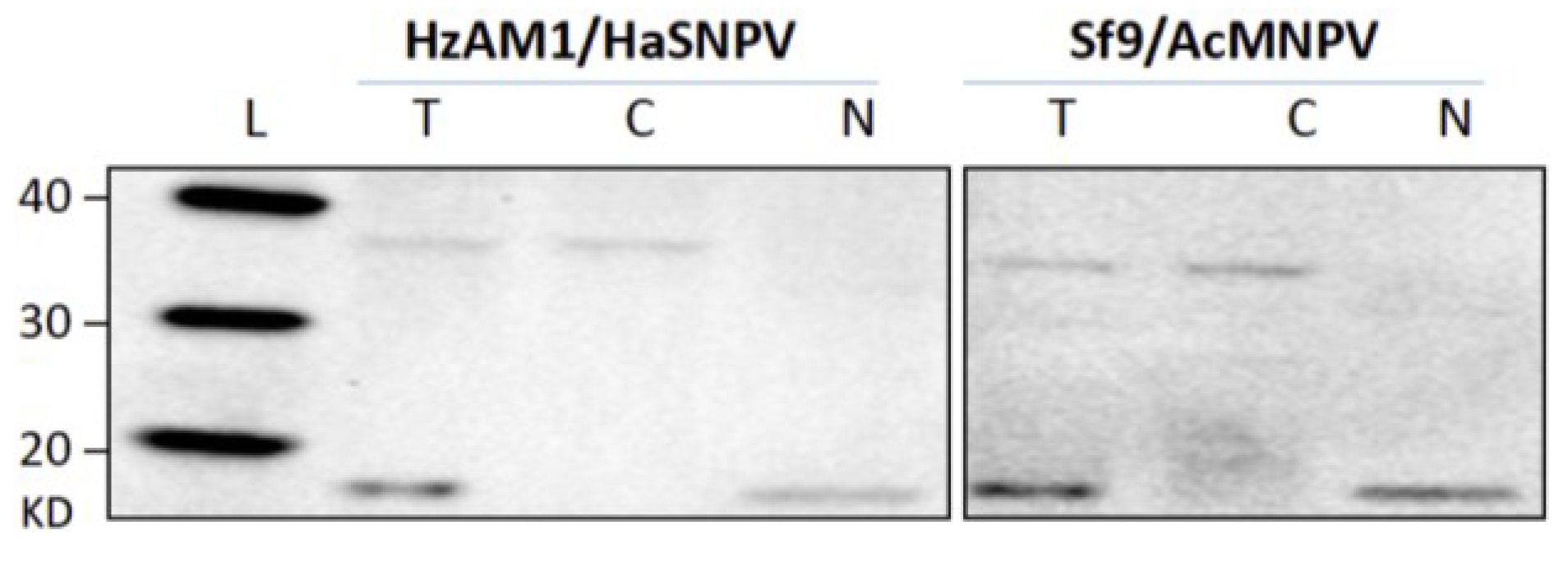
3.1. Cell Fractionation
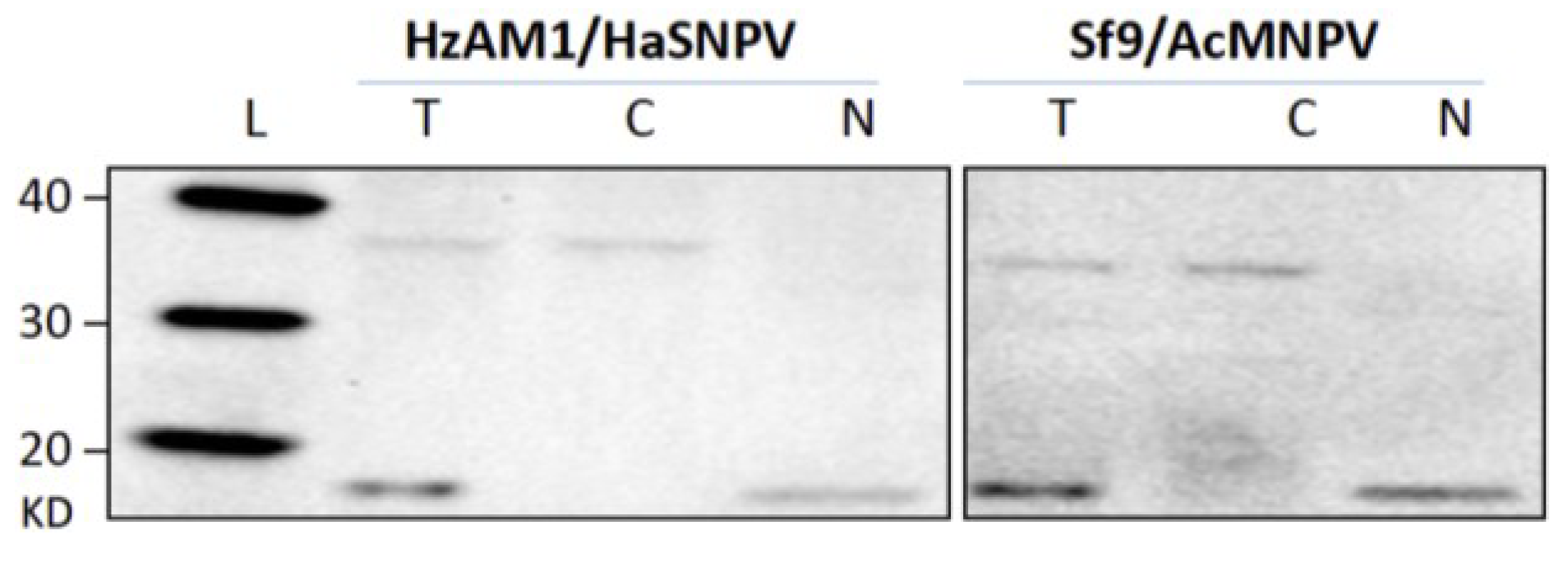
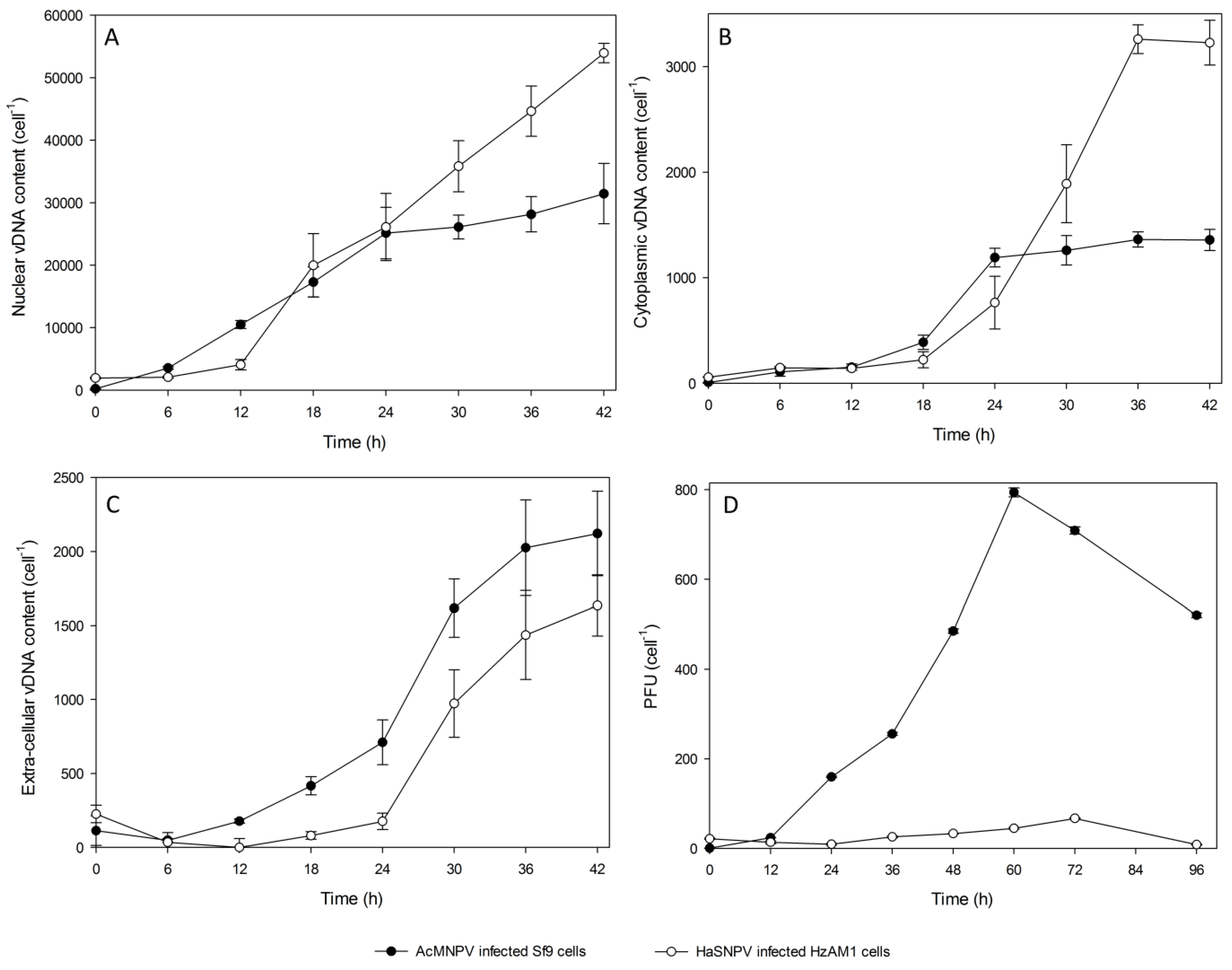
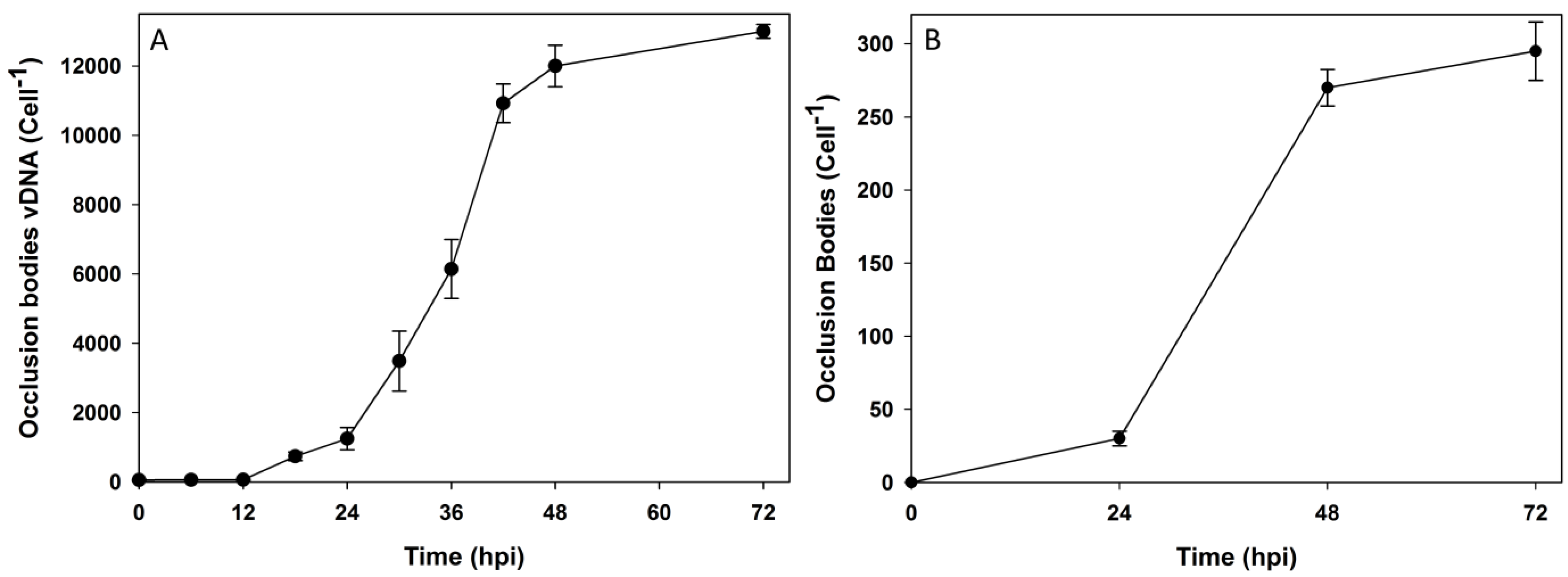
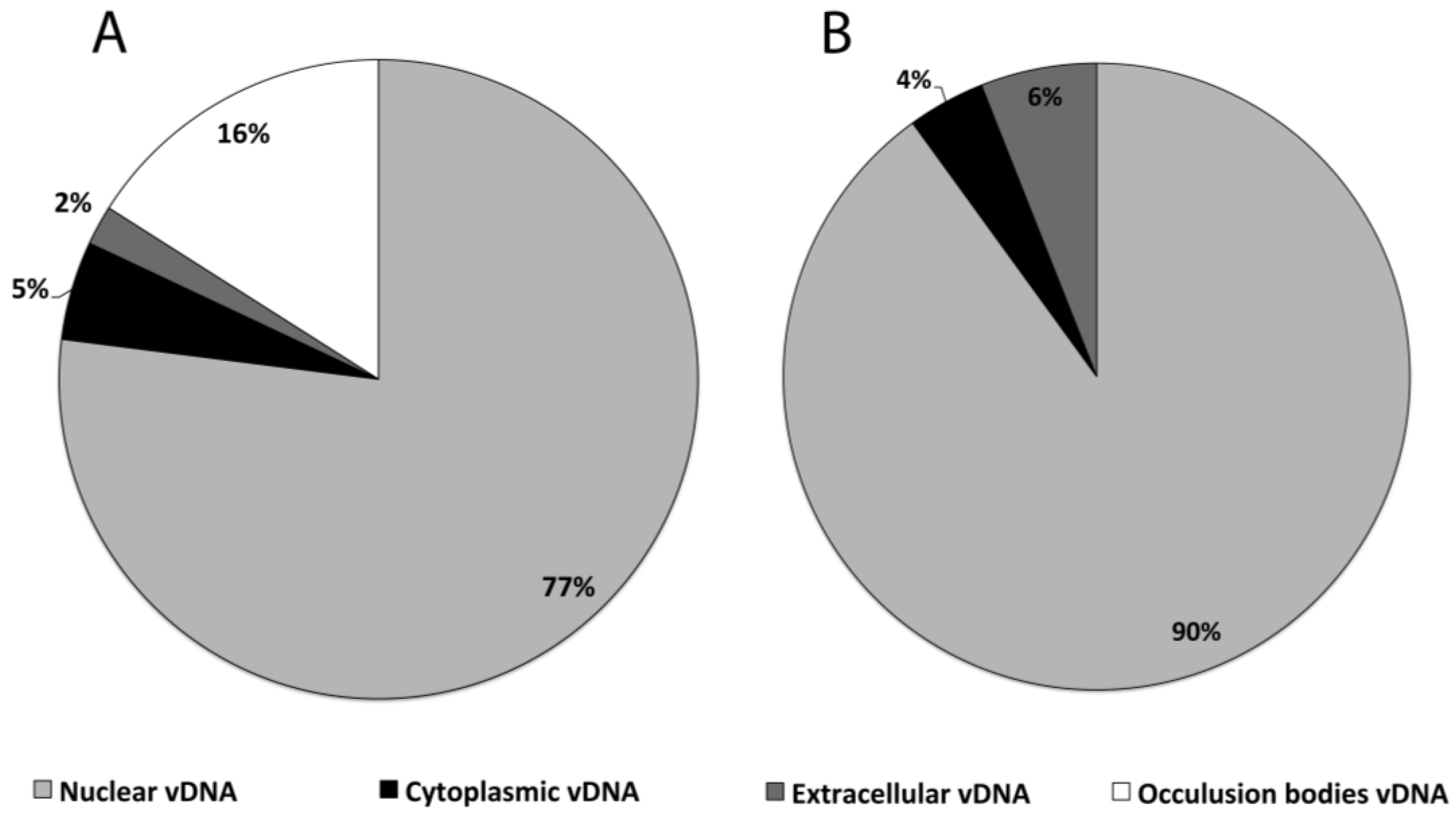
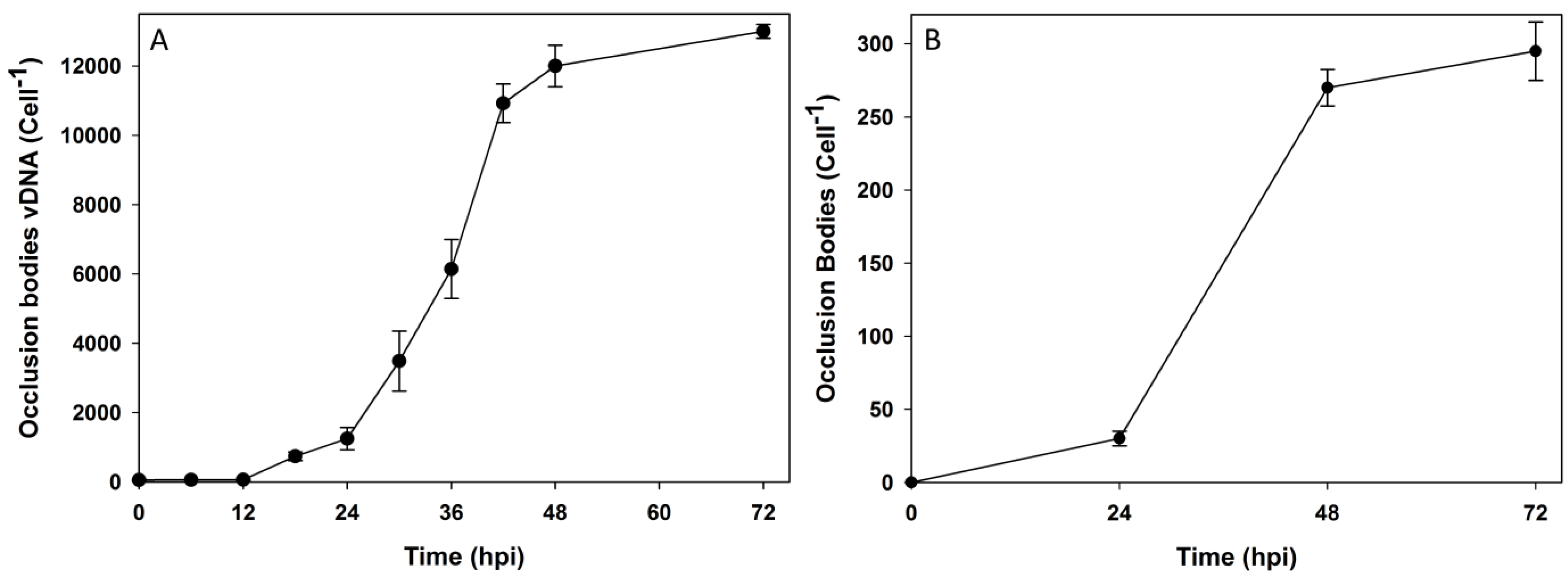
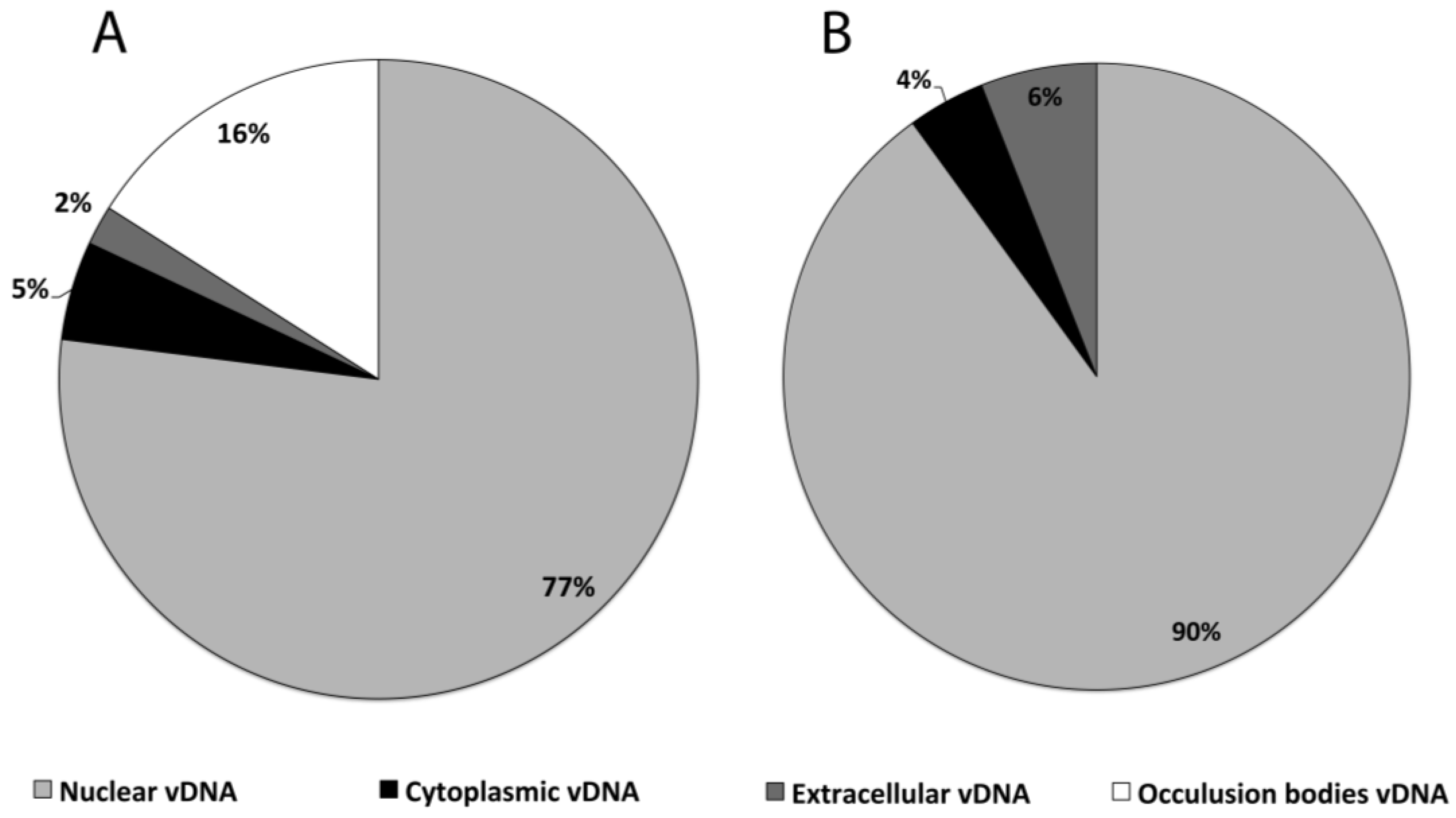
3.2. Subcellular Populations of vDNA in Host Cells
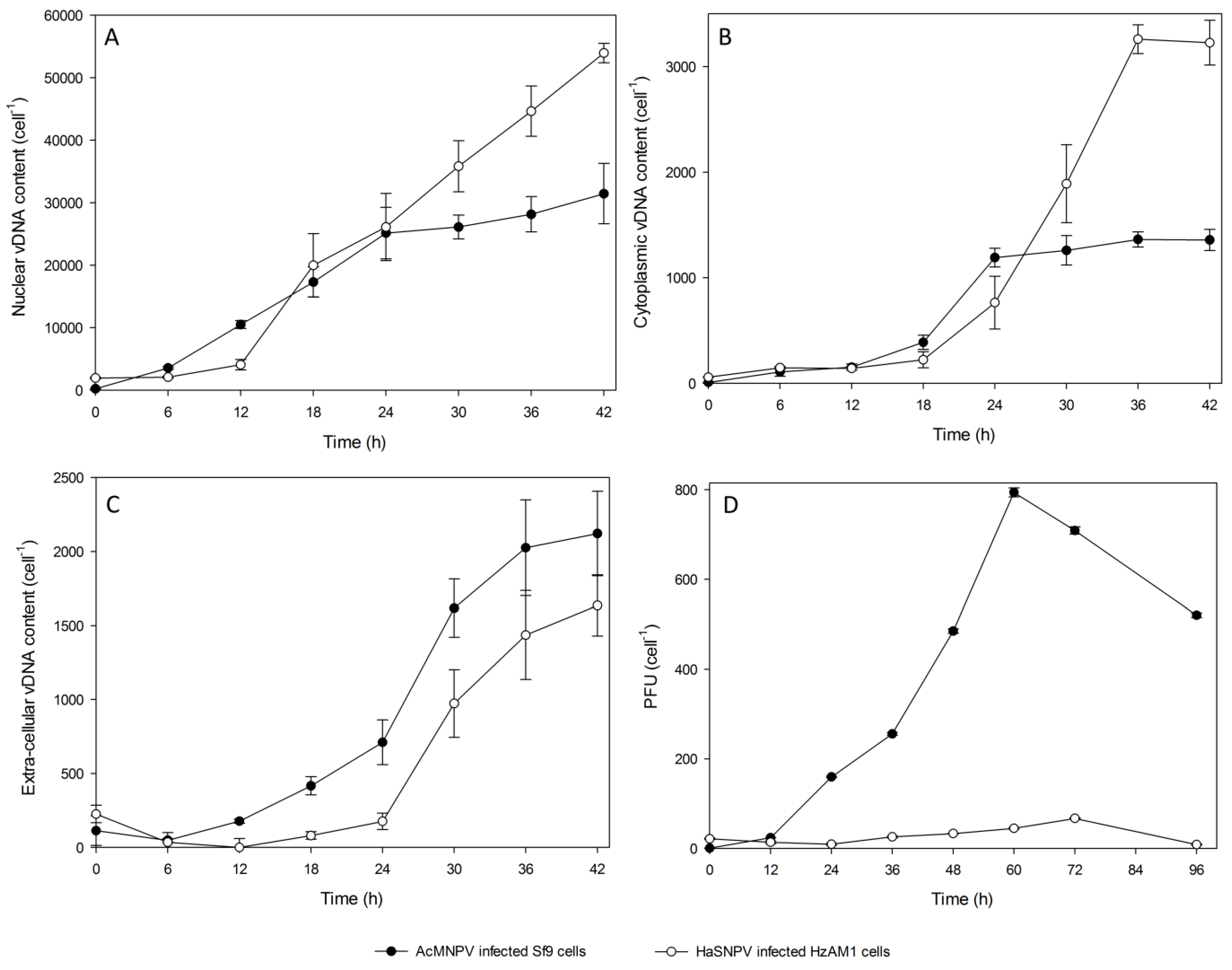

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (hpi) | Nuclear vDNA/cell | Cytoplasmic vDNA/Cell | Extracellular vDNA/cell | OB vDNA/cell | Total progeny vDNA | Nuclear % of total | Cytoplasmic % of total | Extracellular % of total | OB % of total |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 2 ± 2 | 60 ± 13 | 279 ± 52 | 0 | 341 | 1 | 17 | 82 | 0 |
| 6 | 134 ± 14 | 87 ± 14 | 137 ± 53 | 2 ± 1 | 360 | 37 | 24 | 38 | 1 |
| 12 | 6667 ± 432 | 94 ± 10 | 129 ± 72 | 62 ± 18 | 6952 | 96 | 1 | 2 | 1 |
| 18 | 19,967 ± 5069 | 223 ± 76 | 282 ± 127 | 738 ± 125 | 21,210 | 94 | 2 | 1 | 3 |
| 24 | 26,279 ± 6082 | 764 ± 250 | 400 ± 28 | 1247 ± 320 | 28,690 | 92 | 3 | 1 | 4 |
| 30 | 35,837 ± 4594 | 1891 ± 369 | 1128 ± 218 | 3485 ± 866 | 42,341 | 85 | 4 | 3 | 8 |
| Time (hpi) | Nuclear vDNA/cell | Cytoplasmic vDNA/cell | Extracellular vDNA/cell | Total progeny vDNA | Nuclear % of total | Cytoplasmic % of total | Extracellular % of total |
|---|---|---|---|---|---|---|---|
| 0 | 10 ± 6 | 17 ± 0 | 29 ± 10 | 56 | 18 | 30 | 52 |
| 6 | 186 ± 100 | 21 ± 4 | 2 ± 1 | 209 | 89 | 10 | 1 |
| 12 | 4606 ± 634 | 153 ± 30 | 168 ± 14 | 4927 | 94 | 3 | 3 |
| 18 | 17,306 ± 2403 | 389 ± 67 | 417 ± 61 | 18,112 | 96 | 2 | 2 |
| 24 | 25,146 ± 4113 | 1191 ± 78 | 711 ± 151 | 27,048 | 93 | 4 | 3 |
| 30 | 26,109 ± 1906 | 1259 ± 140 | 1618 ± 197 | 28,986 | 90 | 4 | 6 |
| Cell line/virus | Fraction | Peak vDNA/cell |
|---|---|---|
| HzAM1/HearNPV | Nuclear | 54,000 ± 1,500 |
| HzAM1/HearNPV | Cytoplasm | 3260 ± 140 |
| HzAM1/HearNPV | Extracellular | 1635 ± 206 |
| HzAM1/HearNPV | OB | 11,000 ± 558 |
| HzAM1/HearNPV | Total | 69,895 |
| Sf9/AcMNPV | Nuclear | 31,450 ± 4,835 |
| Sf9/AcMNPV | Cytoplasm | 1360 ± 70 |
| Sf9/AcMNPV | Extracellular | 2120 ± 285 |
| Sf9/AcMNPV | Total | 34,930 |
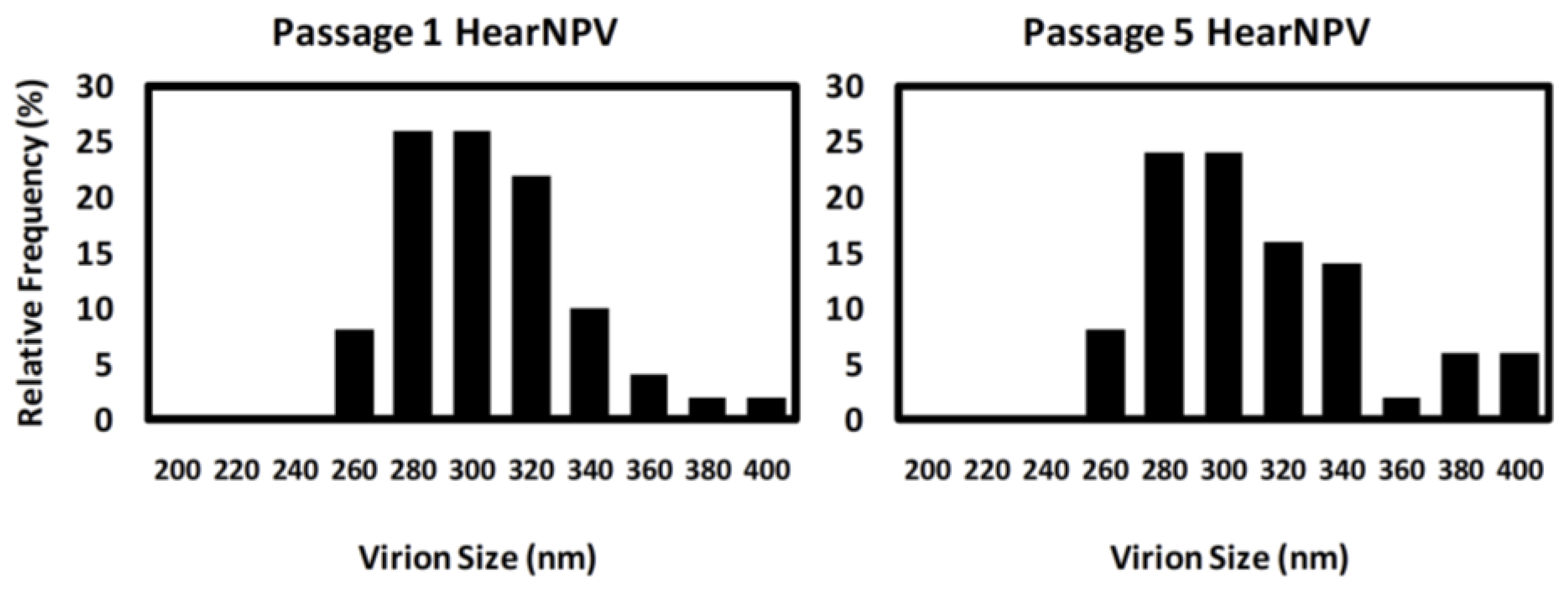
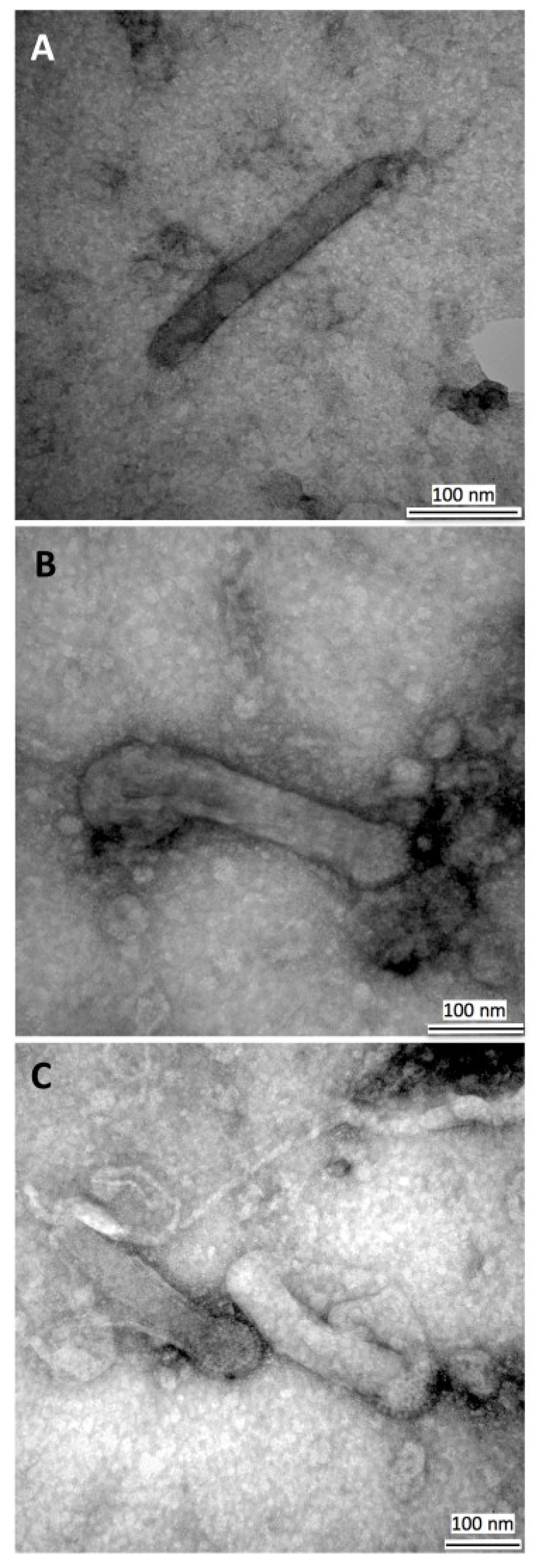
3.3. Transmission Electron Microscopy of HearNPV Virions
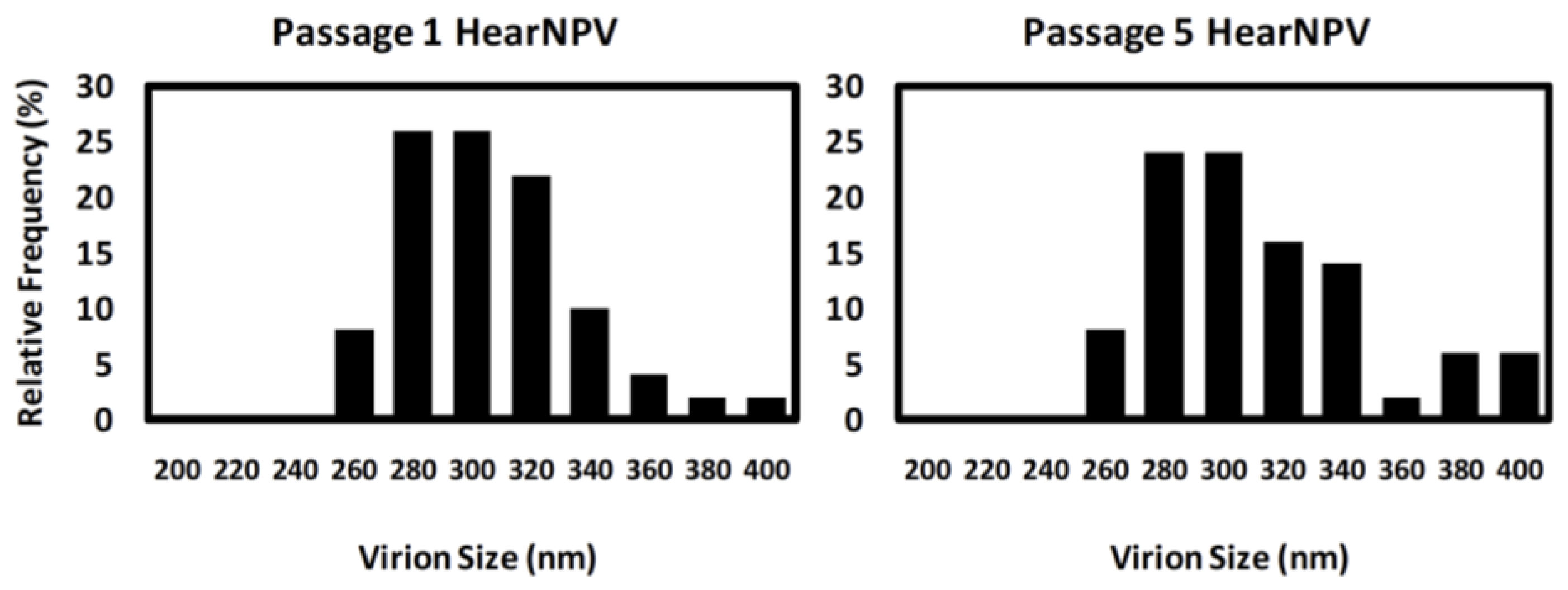
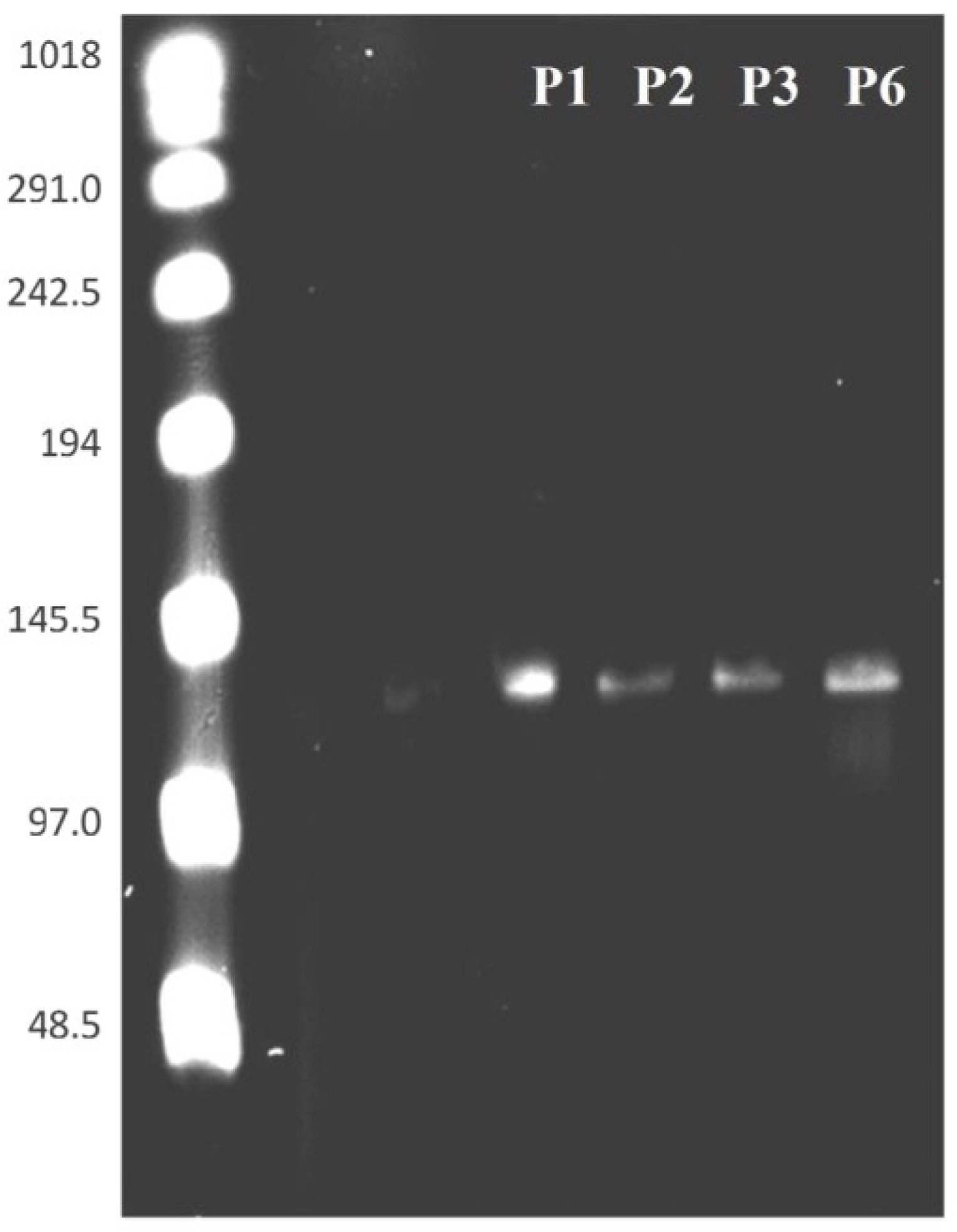
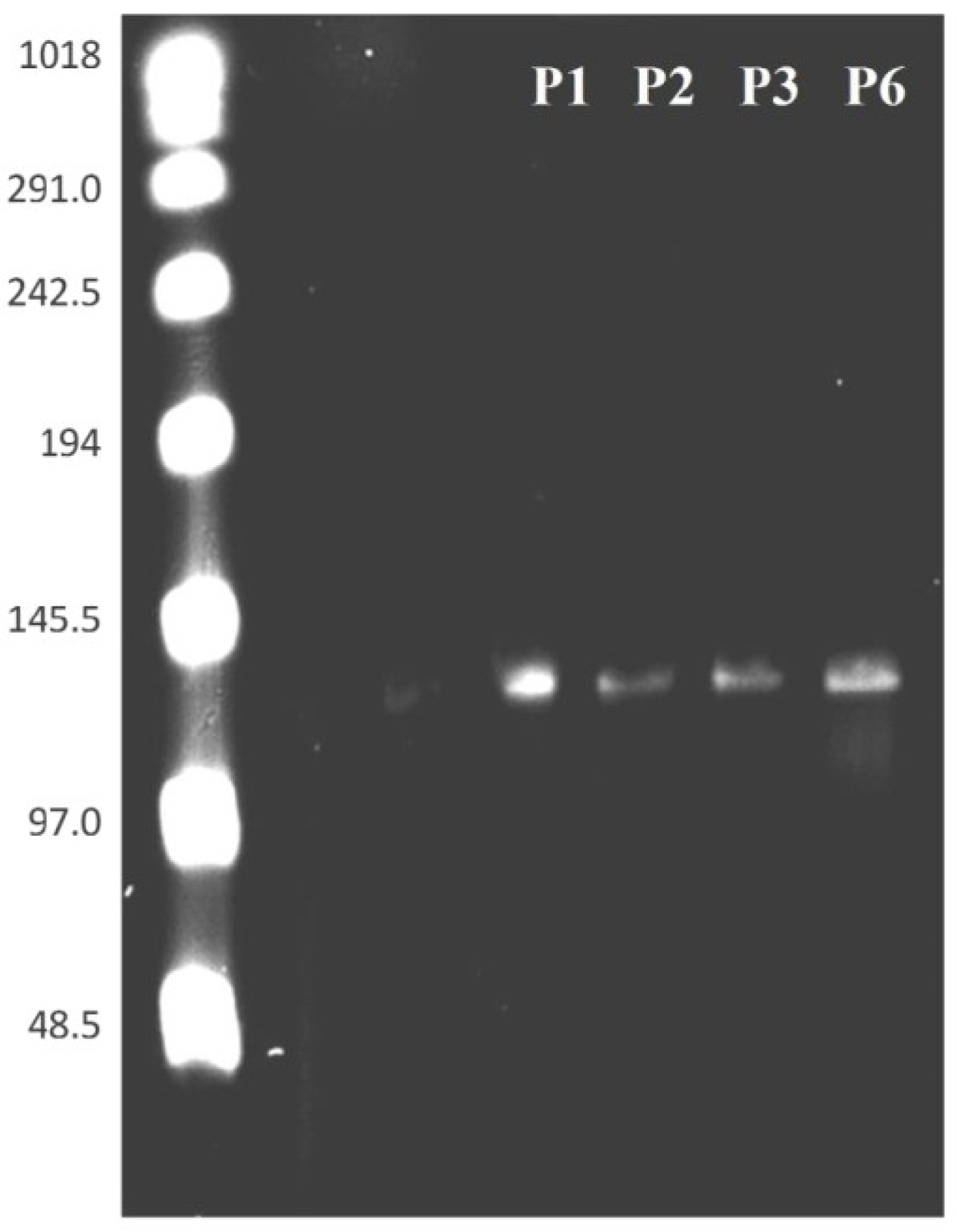
3.4. PFGE of HearNPV at Different Passages
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Szewczyk, B.; Hoyos-Carvajal, L.; Paluszek, M.; Skrzecz, W.; de Souza, M.L. Baculoviruses—Re-emerging biopesticides. Biotechnol. Adv. 2006, 24, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, D.L. Baculovirus-Insect Cell Expression Systems. In Guide to Protein Purification, Second Edition; Burgess, R.R., Deutscher, M.P., Eds.; Academic Press: Waltham, MA, USA, 2009; Volume 463, pp. 191–222. [Google Scholar]
- Grabherr, R.; Ernst, W. Baculovirus for Eukaryotic Protein Display. Curr. Gene Ther. 2010, 10, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Hitchman, R.B.; Possee, R.D.; King, L.A. Baculovirus expression systems for recombinant protein production in insect cells. Recent Pat. Biotechnol. 2009, 3, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Makela, A.R.; Ernst, W.; Grabherr, R.; Oker-Blom, C. Creation of baculovirus display libraries. Cold Spring Harb. Protoc. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.C. Baculoviral vectors for gene delivery: A review. Curr. Gene Ther. 2008, 8, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Dee, K.U.; Shuler, M.L. Mathematical model of the trafficking of acid-dependent enveloped viruses: Application to the binding, uptake, and nuclear accumulation of baculovirus. Biotechnol. Bioeng. 1997, 54, 468–490. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.N.; Groten, C.; Rohrmann, G.F. Identification of the Lymantria dispar nucleopolyhedrovirus envelope fusion protein provides evidence for a phylogenetic division of the Baculoviridae. J. Virol. 2000, 74, 6126–6131. [Google Scholar] [CrossRef] [PubMed]
- Lung, O.; Westenberg, M.; Vlak, J.M.; Zuidema, D.; Blissard, G.W. Pseudotyping Autographa californica multicapsid nucleopolyhedrovirus (AcMNPV): F proteins from group IINPVs are functionally analogous to AcMNPV GP64. J. Virol. 2002, 76, 5729–5736. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Sun, B.; Han, H.; Ouyang, J.F.; Yao, H.C.; Yang, Y.B.; Liu, K.Y.; Li, Y. Application of baculovirus as a delivery vehicle for study of transcription and translation mechanism of parvovirus in non-permissive mammalian cells. J. Virol. Methods 2012, 183, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kost, T.A. Baculovirus Technology: Protein Expression, Drug Discovery, Vaccines, What Next? Vitro Cell. Dev. Biol. Anim. 2012, 48, 10. [Google Scholar] [CrossRef]
- Wu, W.B.; Passarelli, A.L. The Autographa californica M Nucleopolyhedrovirus ac79 Gene encodes an early gene product with structural similarities to UvrC and intron-encoded endonucleases that is required for efficient Budded Virus production. J. Virol. 2012, 86, 5614–5625. [Google Scholar] [CrossRef] [PubMed]
- Pedrini, M.R.S.; Reid, S.; Nielsen, L.K.; Chan, L.C.L. Kinetic characterization of the group II Helicoverpa armigera Nucleopolyhedrovirus propagated in suspension cell cultures: Implications for development of a biopesticides production process. Biotechnol. Progr. 2011, 27, 614–624. [Google Scholar] [CrossRef]
- Westenberg, M.; Wang, H.L.; Ijkel, W.F.J.; Goldbach, R.W.; Vlak, J.M.; Zuidema, D. Furin is involved in Baculovirus envelope fusion protein activation. J. Virol. 2002, 76, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Washburn, J.O.; Trudeau, D.; Wong, J.F.; Volkman, L.E. Early pathogenesis of Autographa californica multiple nucleopolyhedrovirus and Helicoverpa zea single nucleopolyhedro virus in Heliothis virescens: a comparison of the “M” and “S” strategies for establishing fatal infection. J. Gen. Virol. 2003, 84, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Lynn, D.E. Effects of temperature on the susceptibility of insect cells to infection by baculoviruses. Methods Cell. Sci. 2001, 23, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Matindoost, L.; Hu, H.; Chan, L.L.; Nielsen, L.; Reid, S. The effect of cell line, phylogenetics and medium on baculovirus budded virus yield and quality. Arch. Virol. 2014, 159, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.H.; Hillman, C.C.; Zhang, C.X.; Cheng, X.W. Reduction of polyhedrin mRNA and protein expression levels in Sf9 and Hi5 cell lines, but not in Sf21 cells, infected with Autographa californica multiple nucleopolyhedrovirus fp25k mutants. J. Gen. Virol. 2013, 94, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Dickison, V.L.; Willis, L.G.; Sokal, N.R.; Theilmann, D.A. Deletion of AcMNPV ac146 eliminates the production of budded virus. Virology 2012, 431, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Olszewski, J.; Miller, L.K. Identification and characterization of a baculovirus structural protein, VP1054, required for nucleocapsid formation. J. Virol. 1997, 71, 5040–5050. [Google Scholar] [PubMed]
- Russell, R.L.Q.; Funk, C.J.; Rohrmann, G.F. Association of a baculovirus-encoded protein with the capsid basal region. Virology 1997, 227, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.B.; Liang, H.Q.; Kan, J.S.; Liu, C.; Yuan, M.J.; Liang, C.; Yang, K.; Pang, Y. Autographa californica Multiple Nucleopolyhedrovirus 38K Is a Novel Nucleocapsid Protein That Interacts with VP1054, VP39, VP80, and Itself. J. Virol. 2008, 82, 12356–12364. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Miller, L.K. Expression and mutational analysis of the Baculovirus very late factor 1 (vlf-1) gene. Virology 1998, 245, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Petricevich, V.L.; Palomares, L.A.; Gonzalez, M.; Ramirez, O.T. Parameters that determine virus adsorption kinetics: toward the design of better infection strategies for the insect cell-baculovirus expression system. Enzyme Microb. Technol. 2001, 29, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Voncken, J.W.; Vanlier, F.L.J.; Tramper, J.; Vlak, J.M. Detection and Analysis of Autographa-Californica Nuclear Polyhedrosis-Virus Mutants with Defective Interfering Properties. Virology 1991, 183, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Pijlman, G.P.; Dortmans, J.; Vermeesch, A.M.G.; Yang, K.; Martens, D.E.; Goldbach, R.W.; Vlak, J.M. Pivotal role of the non-hr origin of DNA replication in the genesis of defective interfering baculoviruses. J. Virol. 2002, 76, 5605–5611. [Google Scholar] [CrossRef] [PubMed]
- Giri, L.; Feiss, M.G.; Bonning, B.C.; Murhammer, D.W. Production of baculovirus defective interfering particles during serial passage is delayed by removing transposon target sites in fp25k. J. Gen. Virol. 2012, 93, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Rosinski, M.; Reid, S.; Nielsen, L.K. Kinetics of baculovirus replication and release using real-time quantitative polymerase chain reaction. Biotechnol. Bioeng. 2002, 77, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Matindoost, L.; Chan, L.C.L.; Qi, Y.M.; Nielsen, L.K.; Reid, S. Suspension culture titration: A simple method for measuring baculovirus titers. J. Virol. Methods 2012, 183, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Van Meter, K.E.; Stuart, M.K. A Monoclonal Antibody That Inhibits Translation in Sf2l Cell Lysates Is Specific for Glyceraldehyde-3-Phosphate Dehydrogenase. Arch. Insect Biochem. Physiol. 2008, 69, 107–117. [Google Scholar]
- Tie, F.; Banerjee, R.; Stratton, C.A.; Prasad-Sinha, J.; Stepanik, V.; Zlobin, A.; Diaz, M.O.; Scacheri, P.C.; Harte, P.J. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 2009, 136, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Ohkawa, T.; Volkman, L.E.; Welch, M.D. Actin-based motility drives baculovirus transit to the nucleus and cell surface. J. Cell Biol. 2010, 190, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Zhong, S.L.; Fei, Z.J.; Hashimoto, Y.; Xiang, J.Z.; Zhang, S.Y.; Blissard, G.W. The Transcriptome of the Baculovirus Autographa californica Multiple Nucleopolyhedrovirus in Trichoplusia ni Cells. J. Virol. 2013, 87, 6391–6405. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-R.; Zhong, S.; Fei, Z.; Gao, S.; Zhang, S.; Li, Z.; Wang, P.; Blissard, G.W. Transcriptome Responses of the Host Trichoplusia ni to Infection by the Baculovirus Autographa californica Multiple Nucleopolyhedrovirus. J. Virol. 2014, 88, 13781–13797. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.W.; Ijkel, W.F.J.; Tarchini, R.; Sun, X.L.; Sandbrink, H.; Wang, H.L.; Peters, S.; Zuidema, D.; Lankhorst, R.K.; Vlak, J.M.; et al. The sequence of the Helicoverpa armigera single nucleocapsid nucleopolyhedrovirus genome. J. Gen. Virol. 2001, 82, 241–257. [Google Scholar] [PubMed]
- Pedrini, M.R.S.; Nielsen, L.K.; Reid, S.; Chan, L.C.L. Properties of a unique mutant of Helicoverpa armigera single-nucleocapsid nucleopolyhedrovirus that exhibits a partial many polyhedra and few polyhedra phenotype on extended serial passaging in suspension cell cultures. Vitro Cell. Dev. Biol. Anim. 2005, 41, 289–297. [Google Scholar] [CrossRef]
- Westenberg, M.; Uijtdewilligent, P.; Vlak, J.M. Baculovirus envelope fusion proteins F and GP64 exploit distinct receptors to gain entry into cultured insect cells. J. Gen. Virol. 2007, 88, 3302–3306. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.T.; Song, J.H.; Liang, C.Y.; Yu, Z.H.; Chen, X.W. Construction of eukaryotic surface display based on the baculoviral F protein. Biotechniques 2006, 41, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Yin, F.F.; Shen, S.; Tan, Y.; Deng, F.; Vlak, J.M.; Hu, Z.H.; Wang, H.L. Partial functional rescue of Helicoverpa armigera Single Nucleocapsid Nucleopolyhedrovirus infectivity by replacement of F protein with GP64 from Autographa californica Multicapsid Nucleopolyhedrovirus. J. Virol. 2010, 84, 11505–11514. [Google Scholar] [CrossRef] [PubMed]
- Volkman, L.E. Baculovirus infectivity and the actin cytoskeleton. Curr. Drug Targets 2007, 8, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.F.; Blissard, G.W. Cellular VPS4 Is Required for Efficient Entry and Egress of Budded Virions of Autographa californica Multiple Nucleopolyhedrovirus. J. Virol. 2012, 86, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Au, S.; Pante, N. Nuclear transport of baculovirus: Revealing the nuclear pore complex passage. J. Struct. Biol. 2012, 177, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Stidwill, R.P.; Greber, U.F. Intracellular virus trafficking reveals physiological characteristics of the cytoskeleton. News Physiol. Sci. 2000, 15, 67–71. [Google Scholar] [PubMed]
- Fang, M.; Nie, Y.; Theilmann, D.A. Deletion of the AcMNPV core gene ac109 results in budded virions that are non-infectious. Virology 2009, 389, 66–74. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matindoost, L.; Nielsen, L.K.; Reid, S. Intracellular Trafficking of Baculovirus Particles: A Quantitative Study of the HearNPV/HzAM1 Cell and AcMNPV/Sf9 Cell Systems. Viruses 2015, 7, 2288-2307. https://doi.org/10.3390/v7052288
Matindoost L, Nielsen LK, Reid S. Intracellular Trafficking of Baculovirus Particles: A Quantitative Study of the HearNPV/HzAM1 Cell and AcMNPV/Sf9 Cell Systems. Viruses. 2015; 7(5):2288-2307. https://doi.org/10.3390/v7052288
Chicago/Turabian StyleMatindoost, Leila, Lars K. Nielsen, and Steve Reid. 2015. "Intracellular Trafficking of Baculovirus Particles: A Quantitative Study of the HearNPV/HzAM1 Cell and AcMNPV/Sf9 Cell Systems" Viruses 7, no. 5: 2288-2307. https://doi.org/10.3390/v7052288
APA StyleMatindoost, L., Nielsen, L. K., & Reid, S. (2015). Intracellular Trafficking of Baculovirus Particles: A Quantitative Study of the HearNPV/HzAM1 Cell and AcMNPV/Sf9 Cell Systems. Viruses, 7(5), 2288-2307. https://doi.org/10.3390/v7052288