1. Introduction
Nuclear factor I (NF-I) is a family of transcription factors comprising four members (NF-IA, NF-IB, NF-IC, and NF-IX), each with distinct functions depending on the cell type and target promoter context [
1]. Transcripts of each of the NF-I genes are differentially spliced, and NF-I recognition sequences are found in the sequences of many cellular and viral promoters, where they may act as activators or repressors of transcription [
2].
A growing number of studies have shown the importance of NF-I proteins in several cellular and viral mechanisms. NF-I recognition sites have been shown to be present in many cellular and viral promoters/enhancers as well as in genomes of several viruses including adenovirus [
3], BK virus (BKV) [
4], human JC virus (JCV) [
5], human papillomavirus (HPV) [
6], herpes simplex virus 1 (HSV-1) [
7], and cytomegalovirus (CMV) [
8]. Even though the functional importance of these NF-I sites in regulating gene transcription is well established, their role in regulation of other viruses has not been well understood.
HIV-1 proviral gene expression is tightly regulated by an interplay of host and viral proteins with the regulatory elements present in the LTR sequence of the viral genome [
2]. The HIV-1 LTR contains three discrete regions referred to as U3, R, and U5, and these contain four functional regions: the transactivation response (TAR) element, found within the R region (nt +1 to +60), the basal or core promoter (nt −78 to −1), a core enhancer region (nt −105 to −79), and a modulatory region (nt −454 to −104). The modulatory region contains the negative regulatory element (NRE) [
2,
9].
Key elements, such as the TATA, Sp1 and NF-kB region, that are present in the proximal part of the LTR (+1 to −112) have been extensively characterized [
2]. However, transcription control elements present in the −112 to −459 modulatory region of the LTR have been poorly characterized. This region includes regulatory elements that can mediate transcription in many cell types, under a variety of growth and differentiation conditions [
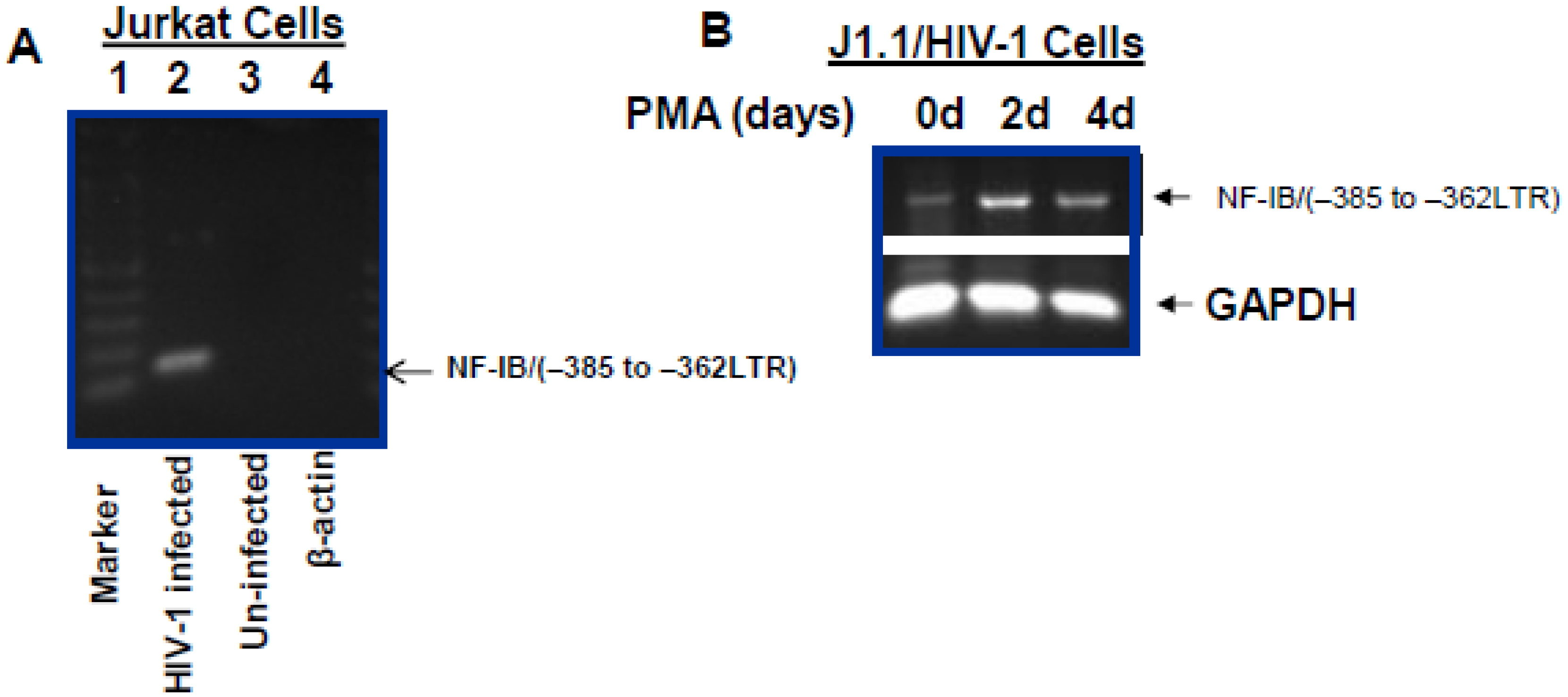
8]. One such regulatory element spanning the −385 to −362 region of the LTR, is directly adjacent to the NRE, has previously been foot printed using nuclear extracts from Jurkat cells and named as site A [
9]. This core sequence, TGATTGGC, was shown to be the binding site for nuclear proteins present in a great variety of cell lines, such as Jurkat, HeLa, astrocytoma, oligodendroglioma and neuronal cells. Further, Schwartz
et al. [
9] have shown this region (−385 to −362) to be the binding site for nuclear protein and belonging to the nuclear factor I (NF-I) family. Interestingly, they observed an antagonistic role of this NF-I binding in the control of HIV-1 transcription in Jurkat cells.
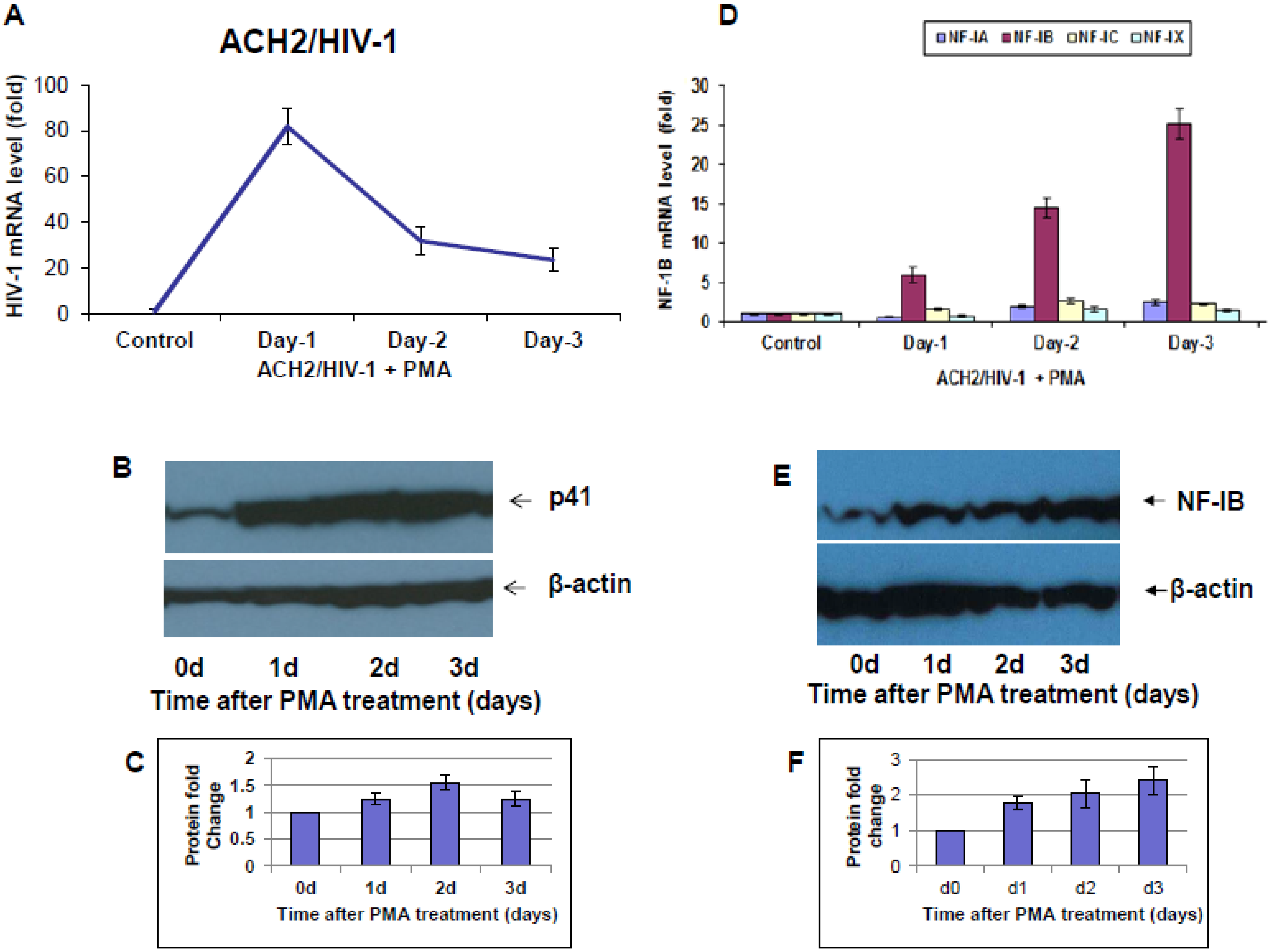
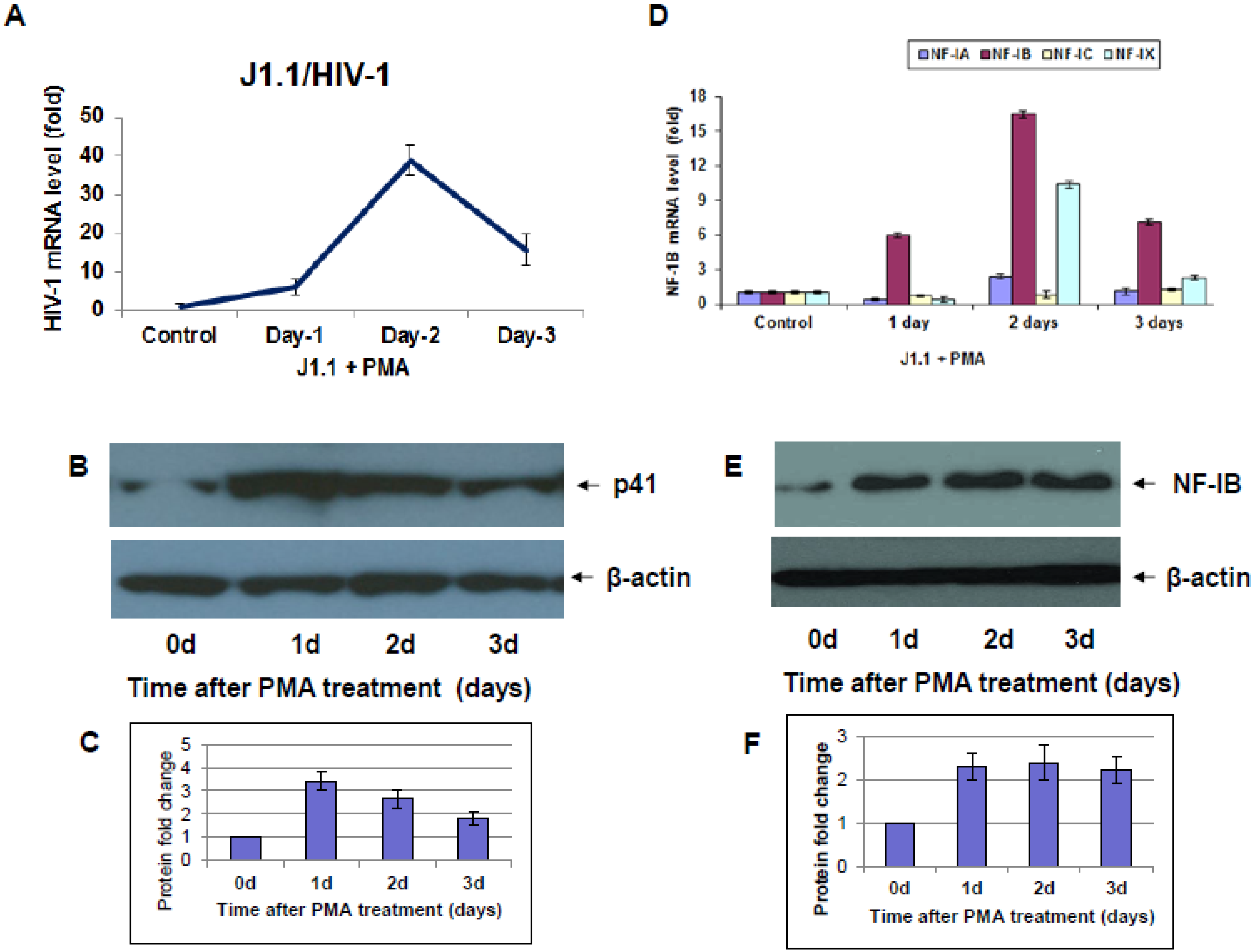
Following the emergence of HIV-1 from the latent state and during the subsequent completion of the infection cycle, the host cell exhibits ordered changes in the expression of a subset of its genes, which shadow the well-known, ordered changes in the pattern of viral gene expression characteristic of the HIV-1 replication cycle [
10]. Several studies have shown that treating host cells carrying HIV-1 provirus with different stimulants can re-activate the latent virus in these cells. We assumed that understanding of the differentially expressed NF-I protein levels during HIV-1 infection or reactivation of the latent virus into a replication cycle may provide potential new therapeutic approaches to eliminate viral infection.
2. Materials and Methods
2.1. Cells and Viruses
Jurkat, J-Lat-Tat-GFP-A1, ACH-2, and J1.1 cell lines were procured from the NIH AIDS Research and Reference Reagent Program (Rockville, MD, USA). PBMCs were isolated from three healthy HIV-seronegative donors obtained from the NIH blood bank by Ficoll-Hypaque separation (Sigma, St Louis, MO, USA). The cell lines and PBMCs were maintained in RPMI 1640 growth medium supplemented with 10% fetal bovine serum (FBS), 1 × penicillin/streptomycin, and 2 mM L-glutamine for 4 days in presence of 5 ug/mL phytohemagglutinin (PHA, Sigma) before the addition of 5 U/mL of human recombinant interleukin-2 (R&D systems, Minneapolis, MN, USA). The HIV-1 IIIB strain used in this study was obtained from the AIDS Research and Reference Reagent Program and propagated in Jurkat cells and PBMCs. Culture medium from HIV-1 infected cultures was collected every 3 days, clarified by centrifugation at 1500 RPM for 5 min, and quantitated for p24 antigen using an in-house HIV-1 europium nanoparticle P24 ELISA.
2.2. Infection of PBMCs and Jurkat Cells with HIV-1 Virus and Reactivation of Latently Infected Cells
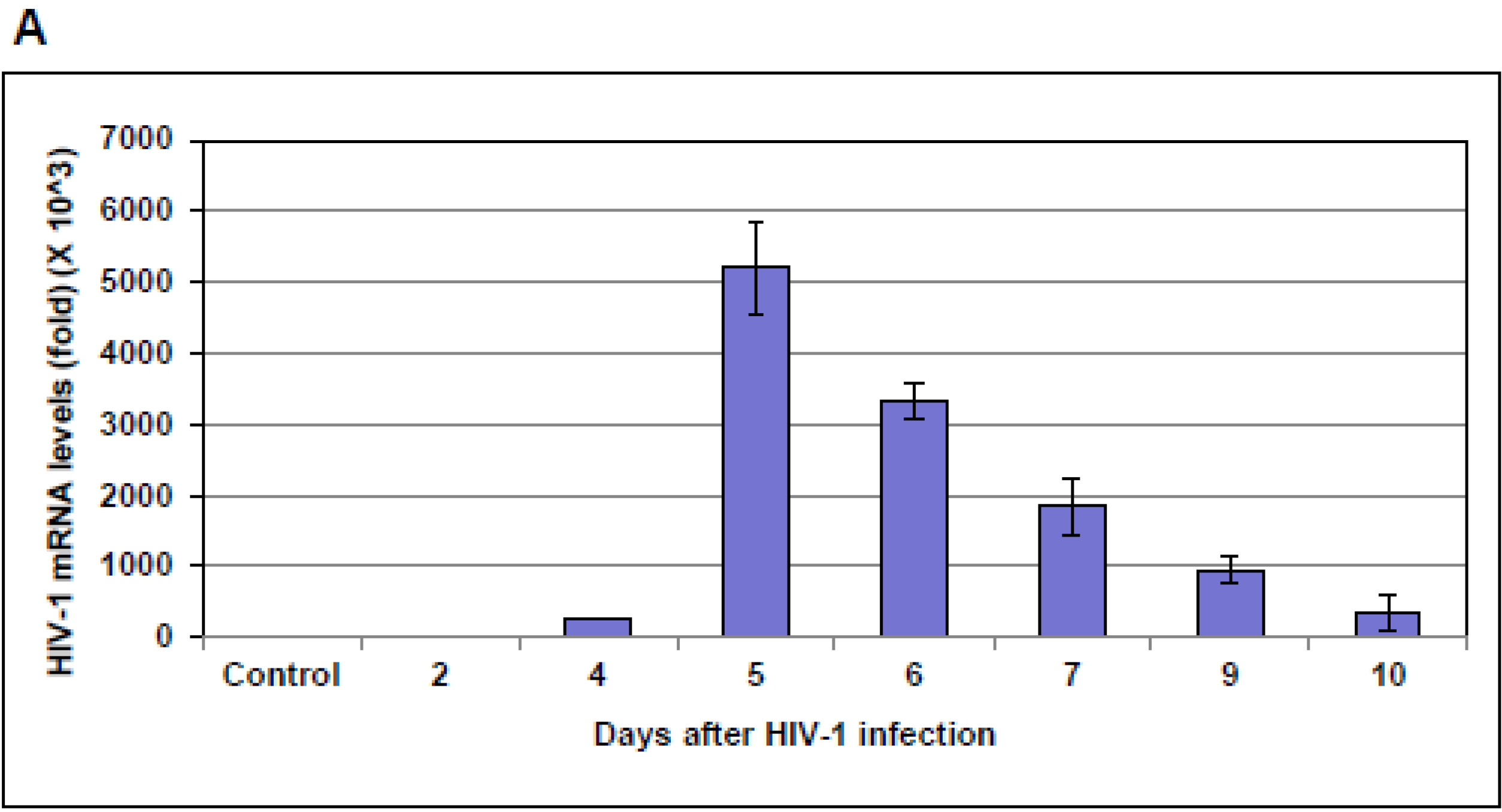
The expression of NF-IB during HIV-1 infection was tested in HIV-1 infected Jurkat cells and PBMCs. PBMCs and Jurkat cells were infected with HIV-1 III-B strain using 5 ng equivalent of p24/10
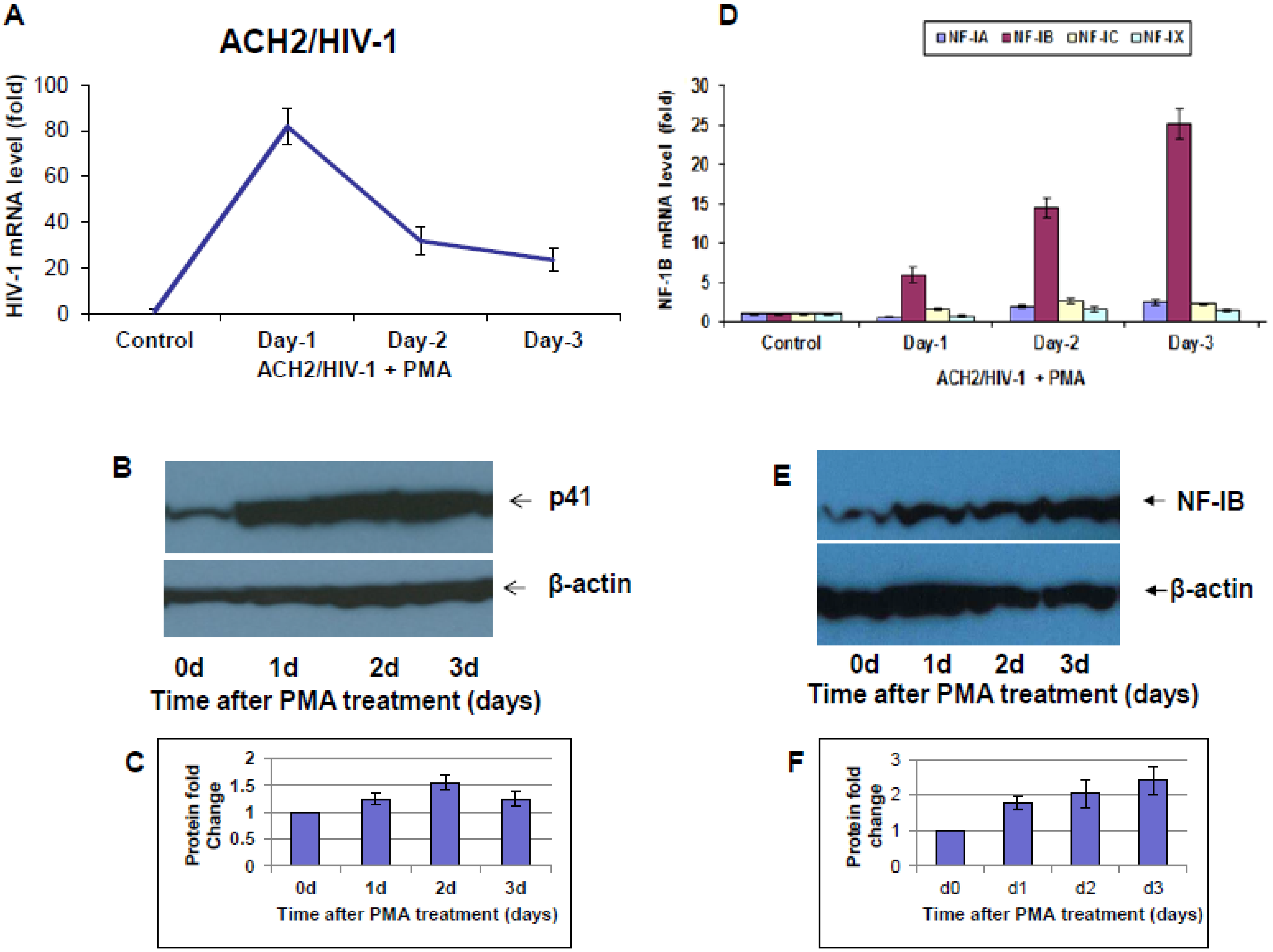
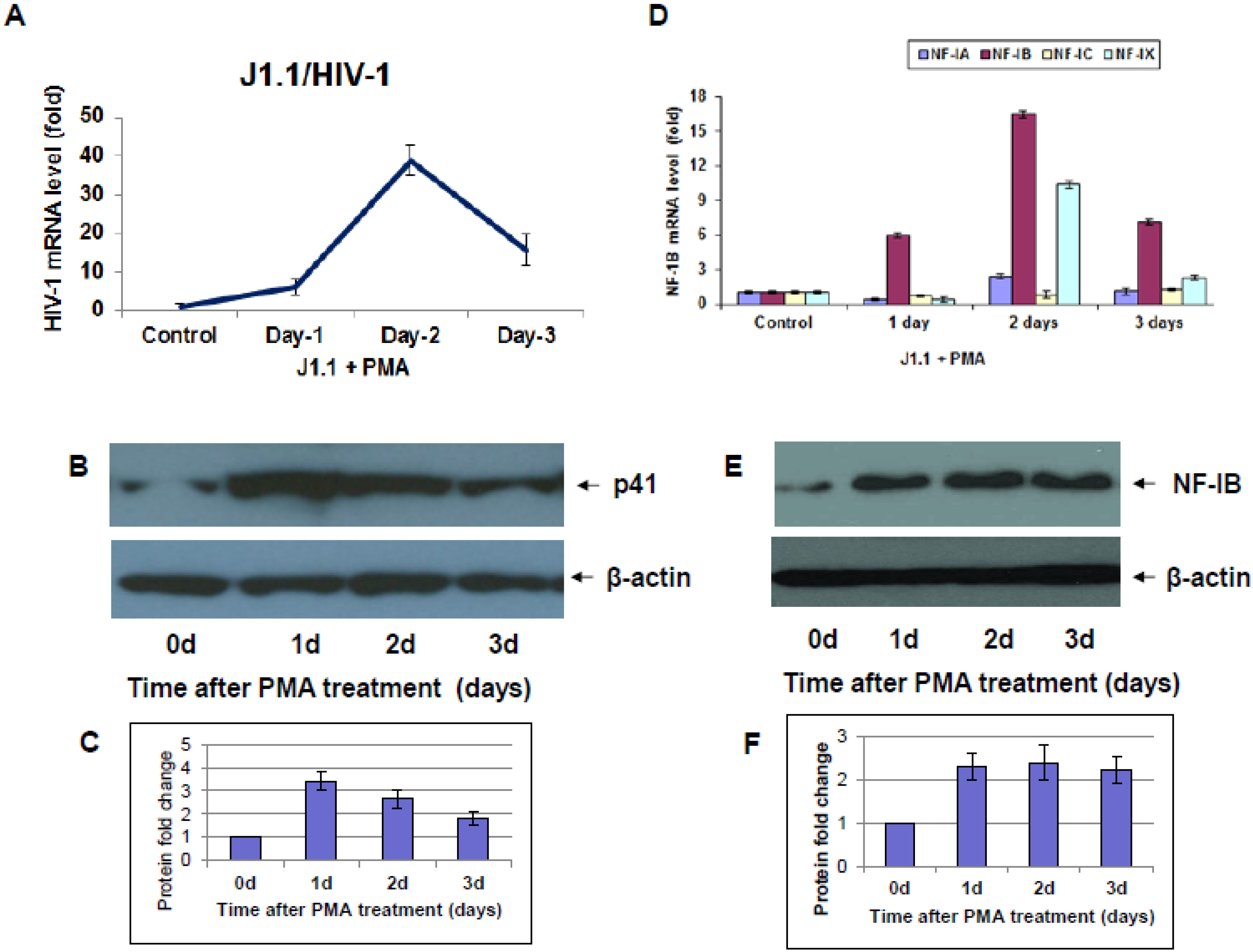
6 cells) in RPMI 1640 growth medium for 3 h at 37 °C. Infected cells were washed once with PBS and resuspended in RPMI 1640 growth media. At various time-pointsfollowing HIV-1 infection, cell pellets and supernatants were collected and stored at −80 °C to measure the expression of HIV-1 gag and NF-IB gene and proteins respectively. Reactivation of latency by PMA treatment: ACH-2, and J1.1 cells were reactivated by treatment with 10 ng/mL of phorbol myristyl acetate (PMA) for 30 min at 37 °C [
10]. Following PMA treatment cells were washed twice in PBS. On days 1, 2, and 3 post activation cell pellets and supernatants were collected and stored at −80 °C.
2.4. Western Blotting
Equal amounts of protein separated on a NuPAGE 4%–12% Bis-Tris gel (Invitrogen, Grand Island, NY, USA) were transferred on to a 0.45 µm pore size polyvinylidene fluoride (PVDF) membrane (Invitrogen). Membranes were blocked for 1 h with 5% non-fat dry milk. Blots were then probed using antibodies against NF-IA (rabbit polyclonal from Active Motif), NF-IB (rabbit polyclonal from Active Motif), β-actin (clone AC-74 from Sigma), gp41 (goat polyclonal from Thermo Fisher Scientific, Rockford, IL, USA).
The bound antibodies were detected using appropriate secondary antibodies conjugated with horseradish peroxidase (Santa Cruz biotechnology). Blots were developed and visualized by Western blotting Luminol reagent (Santa Cruz biotechnology) according to the manufacturer instructions. Quantification of signals from Western blots was done using Image J software, version.
2.5. LTR Pull-Down Assay
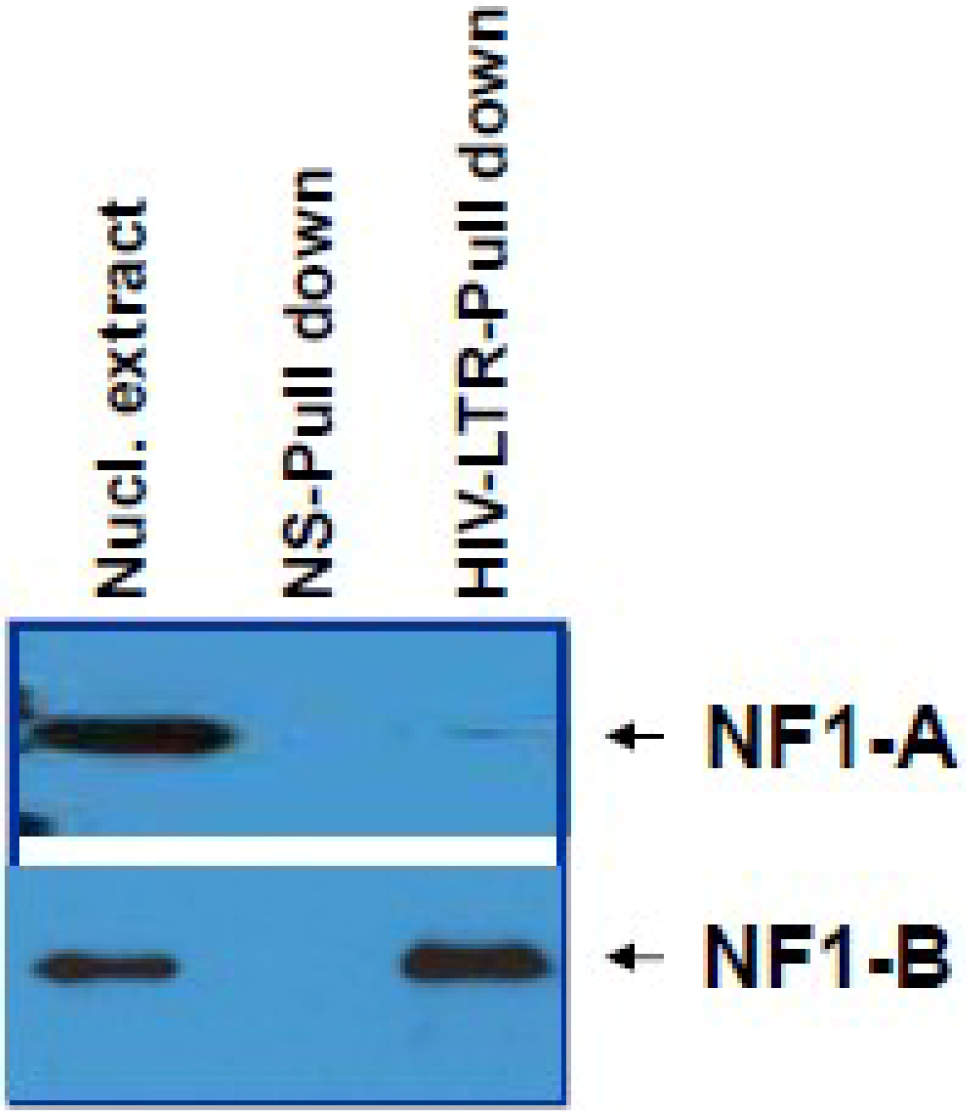
The HIV-1 LTR region was amplified using a full length HIV-1 IIIB clone as the template. A 456 bp fragment amplified from GAPDH coding region was used as a negative control. Both non-specific biotinylated DNA (bio-NS), and biotinylated LTR-probes (bio-LTR) were added with streptavidin-agarose in a binding buffer (10 mM HEPES, pH 7.8; supplemented with 1 mM EDTA, 0.5 mM PMSF Complete® mini protease inhibitor cocktail (Roche Diagnostics GmbH) and gently rotated for 2 h at 4 °C. Unbound biotinylated probes were removed by washing with binding buffer, and bound Biotin-streptavidin-agarose beads (agar-strep-bio-NS or agar-strep-bio-LTR) were made into 50% slurry using binding buffer.
Nuclear extracts were pre-cleared by gently rotating with 10% slurry of NS-Bio-streptavidin-agarose beads for 1 h at 4 °C. After centrifugation, the supernatants (pre-cleared nuclear extracts) were added to equal volumes of protein binding buffer containing agar-strep-bio-NS or agar-strep-bio-LTR. After 2 h at 4 °C with gentle rotation, the agar-strep-bio-NS or agar-strep-bio-LTR were pelleted by centrifugation, and washed three times with wash buffer (10 mM HEPES, pH 7.8, 0.1% NP-40, 0.5% glycerol, Complete® mini protease inhibitor cocktail (Roche Diagnostics). Proteins that bound to the agar-strep-bio-NS or agar-strep-bio-LTR were eluted by boiling for 3 min in Laemmli protein loading buffer and then resolved on a 4%–12% NuPAGE Bis-Tris gel. The immunoblotting was performed as described earlier.
2.6. Chromatin Immunoprecipitation (Chip) Assay
Chip assay was performed using the Chip-IT Express enzymatic kit (Active Motif) according to the manufacturer’s instructions. Briefly, the cells were fixed using 1% formaldehyde, lysed and then the cellular DNA was sheared using a Enzymatic shearing cocktail. One-tenth of the total cell lysate was used for total genomic DNA as “input DNA” control. Immunoprecipitation was performed overnight in cold room with 2 µg of NF-1B antibody (Active Motif) or β-actin (Sigma) and mouse IgG (Active Motif) antibodies as negative controls. Protein A/G was added and incubated at 4 °C for 1 h. Beads were then washed three times in Chip buffer, after which elution of the digested chromatin, reversion of cross-linking, and proteinase K treatment was performed. The eluted immunoprecipitated DNA and the sample of Chip input DNA were purified with the QIAquick PCR Purification Kit and then subjected to quantitative PCR (qPCR) by employing NF-1B site flanking primers: LTR Chip primer fwd (5'-TGGAAGGGCTAATTCACTCC-3'), LTR Chip primer rev (5'-GTAGTTCTGCCAATCAGGGAAGTAGC-3'). GAPDH was amplified using primers (FWD: 5’-GACAGTCAGCCGCATCTTCT; and REV: 5’- TTAAAAGCAGCCCTGGTGAC) and used to normalize the amount of input material.
2.7. Real-Time RT- PCR
Total RNA was extracted from the cells using the RNAzol-RT kit (Molecular Research Center, Inc., Cincinnati, OH, USA) according to the manufacturerss instructions. RNA concentration was measured using a Nanodrop spectrophotometer. First-strand cDNA was synthesized from 5 µg of total RNA using a SuperScript III first-strand synthesis kit (Invitrogen). Quantitation of NF-IA, NF-IB, NF-IC, and NF-IX, HIV-1 gag, and GAPDH were done using the following primers (NFI-A, Forward-CAGCCAAGTGACGCTGACA, Reverse-CCTCATTGCTCCTGGACTCAT; NF-1B, Forward-GCCACAATGATCCTGCCAAGAA, Reverse-GGTGGAGAAGACAGAGACCTCTGA; NFI-C, Forward-GGACAGGGATGGGCTCTG, Reverse-CGTTCTTCTGAGGCCAGTGC; NFI-X, Forward-CCACTGCCCAACGGACACTT, Reverse-CCGGGATAGAACACGTCATCA; HIV-1 gag Forward-CTAGGAACGATTCGCAGTTAATCCT, Reverse-CTCTATTGTGTGCATCAAAGGATAG; GAPDH Forward-GACAGTCAGCCGCATCTTCT, Reverse-TTAAAAGCAGCCCTGGTGAC).
Real-time PCR was performed on ABI Prism 7500 instrument (Applied Biosystems, Foster City, CA, USA) with a quantitect SYBR green kit (Qiagen, Valencia, CA, USA). The expression levels were normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
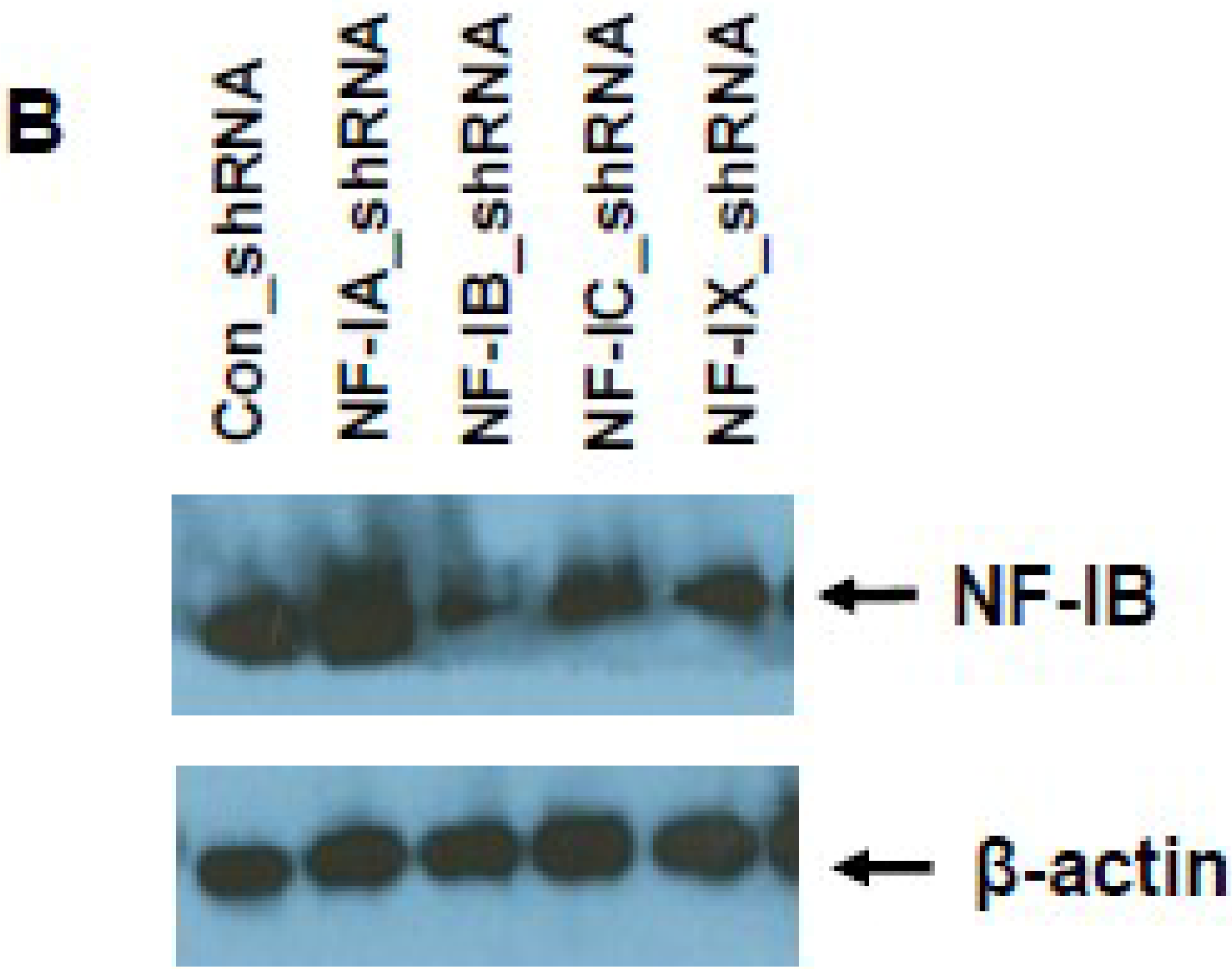
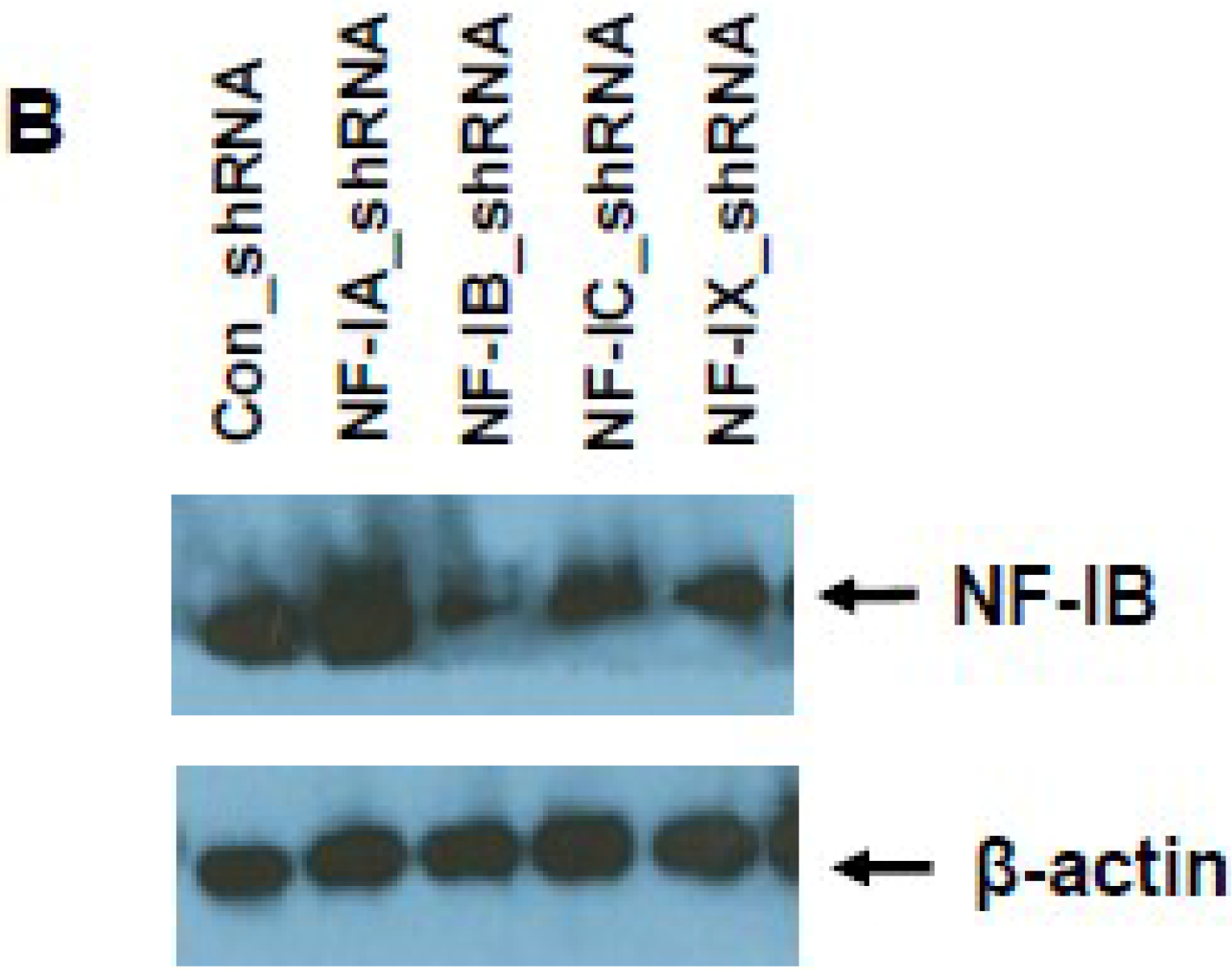
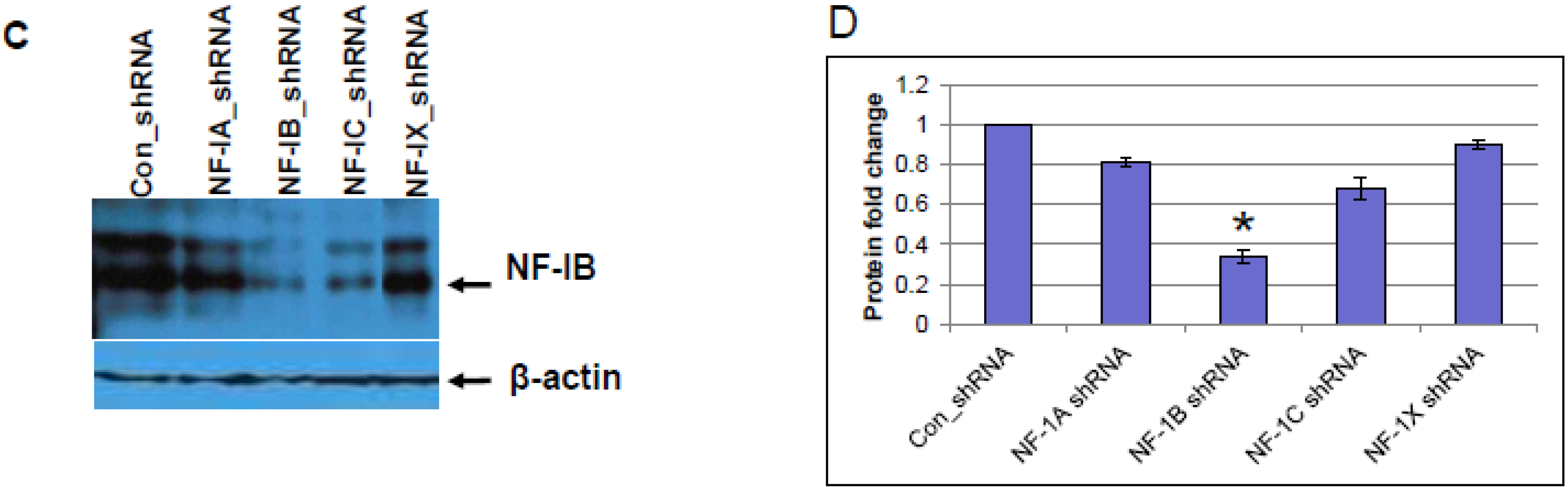
2.8. Knock-Down of NF-IB Expression Using Lentivirus-Based shRNA Transduction
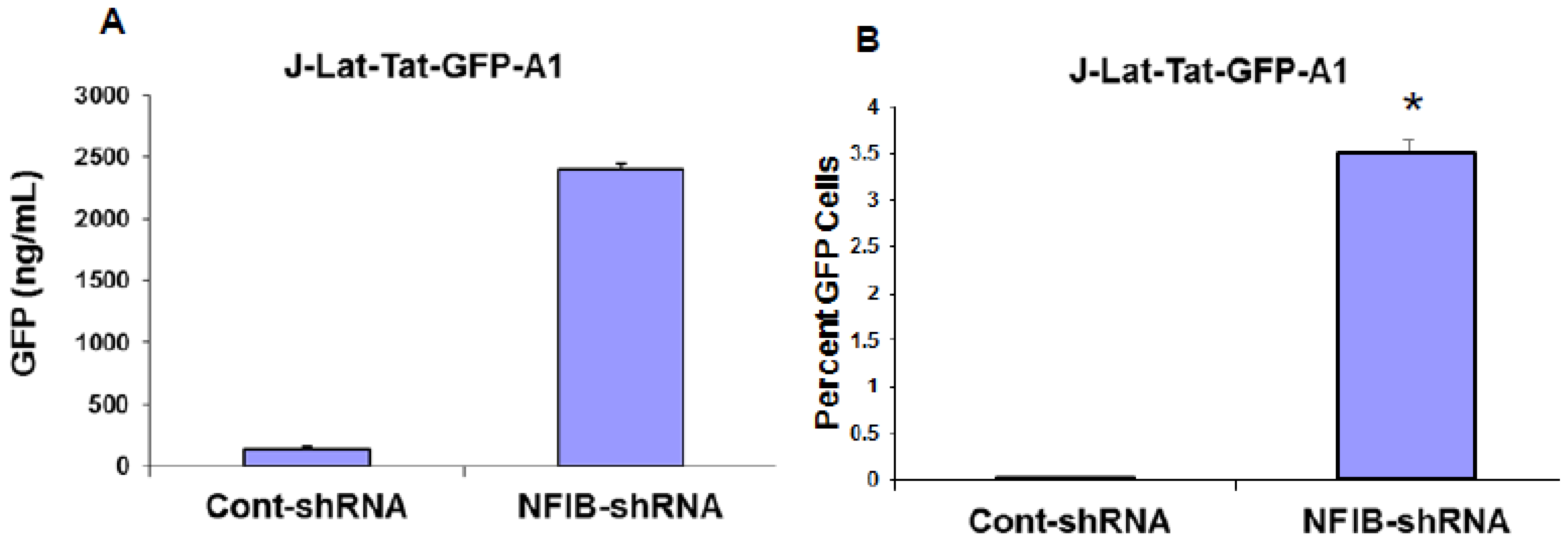
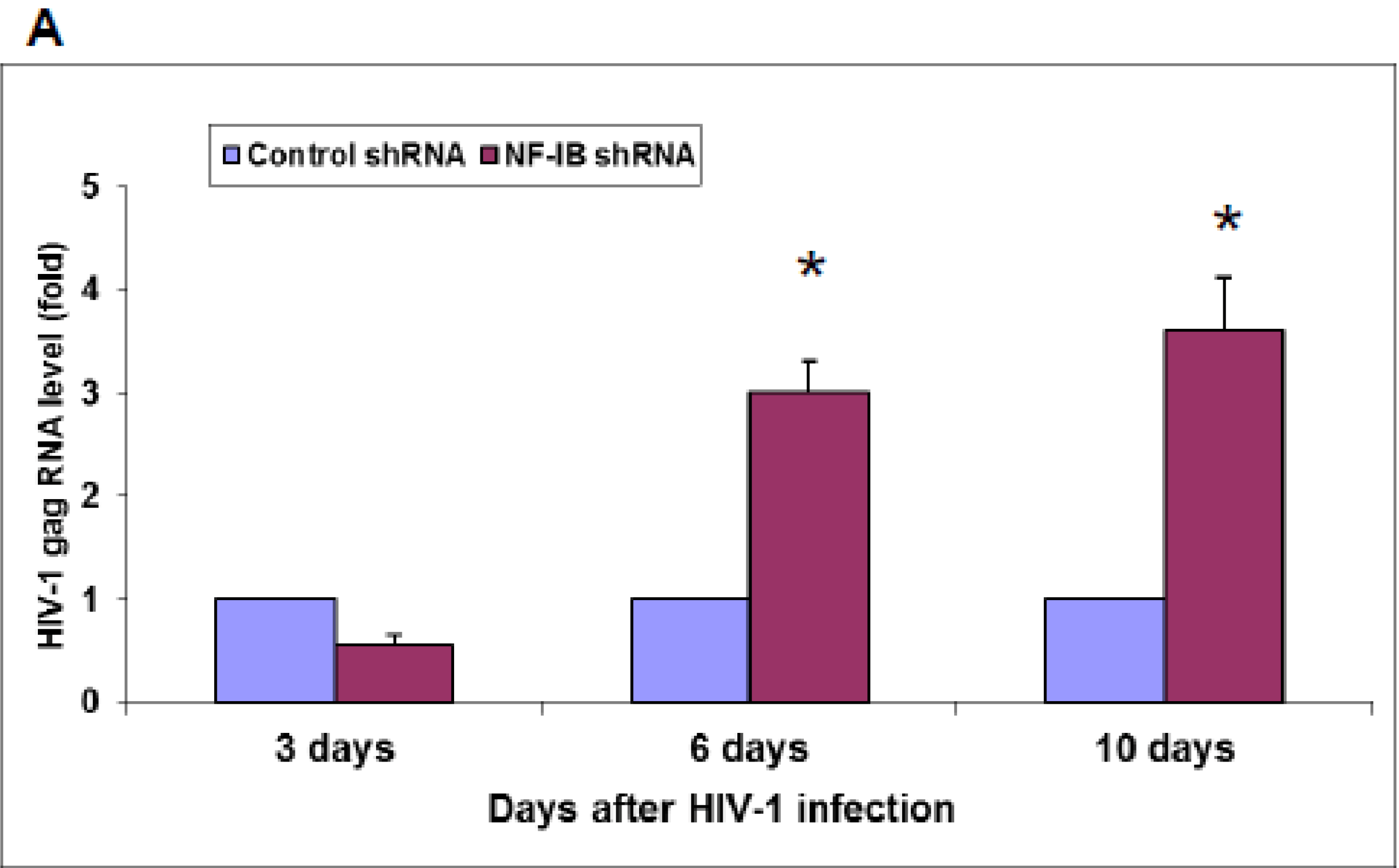
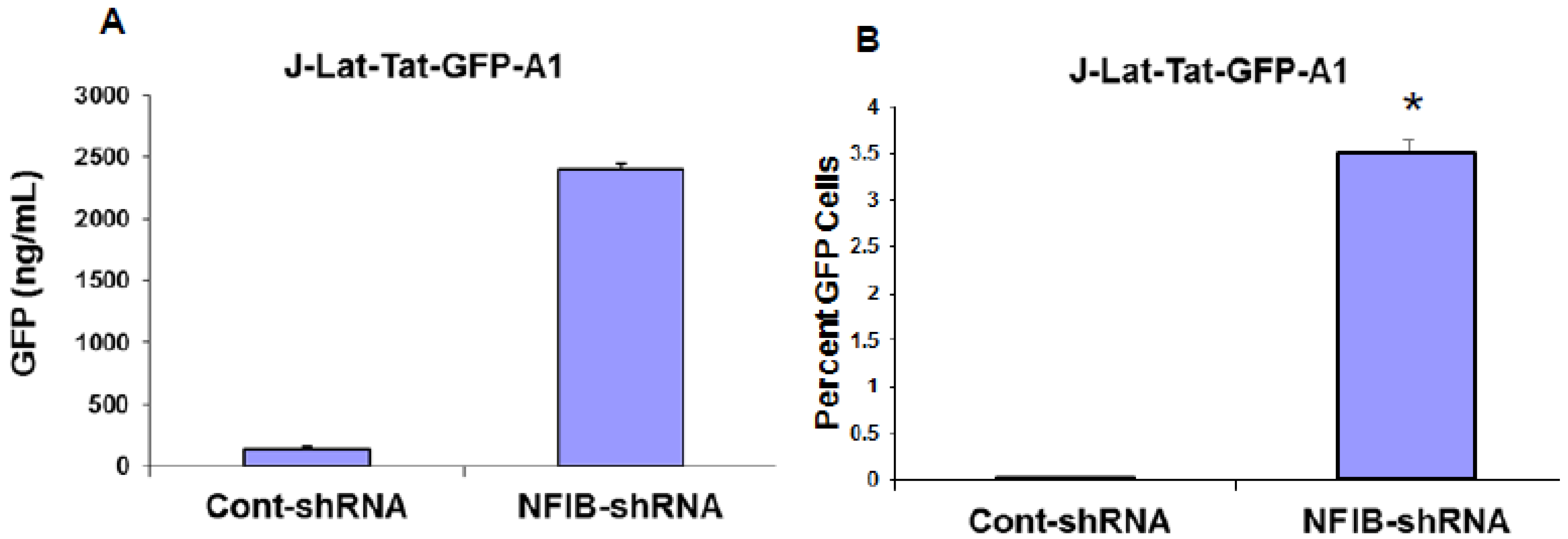
NF-IB level was knocked down in Jurkat, J1.1, and ACH-2 cells using MISSION shRNA lentiviral particles (Sigma). Briefly, cells (1.0 × 105) were transduced with 1.0 × 105 TU Lentiviral particles targeting human NF-1B (shNF-IB). As negative controls, cells were transduced with the MISSION pLKO.1-puro Non-Target shRNA Control Transduction Particles (sh-Control) that contain an shRNA insert that does not target any human gene. Three days post-infection, the cell media were supplemented with puromycin (10 ug/mL), and 2 weeks after selection, cells were used for the study. Knockdown of NF-1B expression in the resultant cell lines was confirmed by Western blot analysis. sh-Control or sh-NF-IB-selected Jurkat cells were infected with HIV-1 (III-B strain; 5 ng equivalent of p24/mL) and incubated for 10 days. J-Lat-Tat-GFP-A1 cells (1.0 × 105) were infected with 1.0 × 105 TU lentiviral particles (shNF-IB or sh-control). Three days post-infection, the media was supplemented with puromycin (10 µg/mL), and 2 weeks after selection, cells were used for the study. Jurkat cells were used to normalize cellular autofluorescence. Cells were washed in PBS and fixed in 1% paraformaldehyde. GFP fluorescence was measured using a FACScan flow cytometer (Becton Dickinson, Mountain View, CA, USA). In another set of experiments, the fluorescence from lysed cells was measured using GFP ELISA kit (Cell Biolabs. Inc., San Diego, CA, USA).
2.9. Statistical Analysis
The Students t-test was used for calculation of significance. The significance was set at p < 0.05.
4. Discussion
The persistence of latently infected viral reservoirs, despite prolonged highly active antiviral retroviral therapy (HAART), represents a major obstacle to the eradication of HIV-1 in infected patients [
11]. Latently infected cells contain replication-competent integrated HIV-1 genomes that are blocked at the transcriptional level, resulting in the absence of viral protein expression [
12,
13].
During
de novo HIV-1 infection, or reactivation of latently infected cells, various host cell genes are differentially expressed, most of which play an important role in regulating HIV-1 replication [
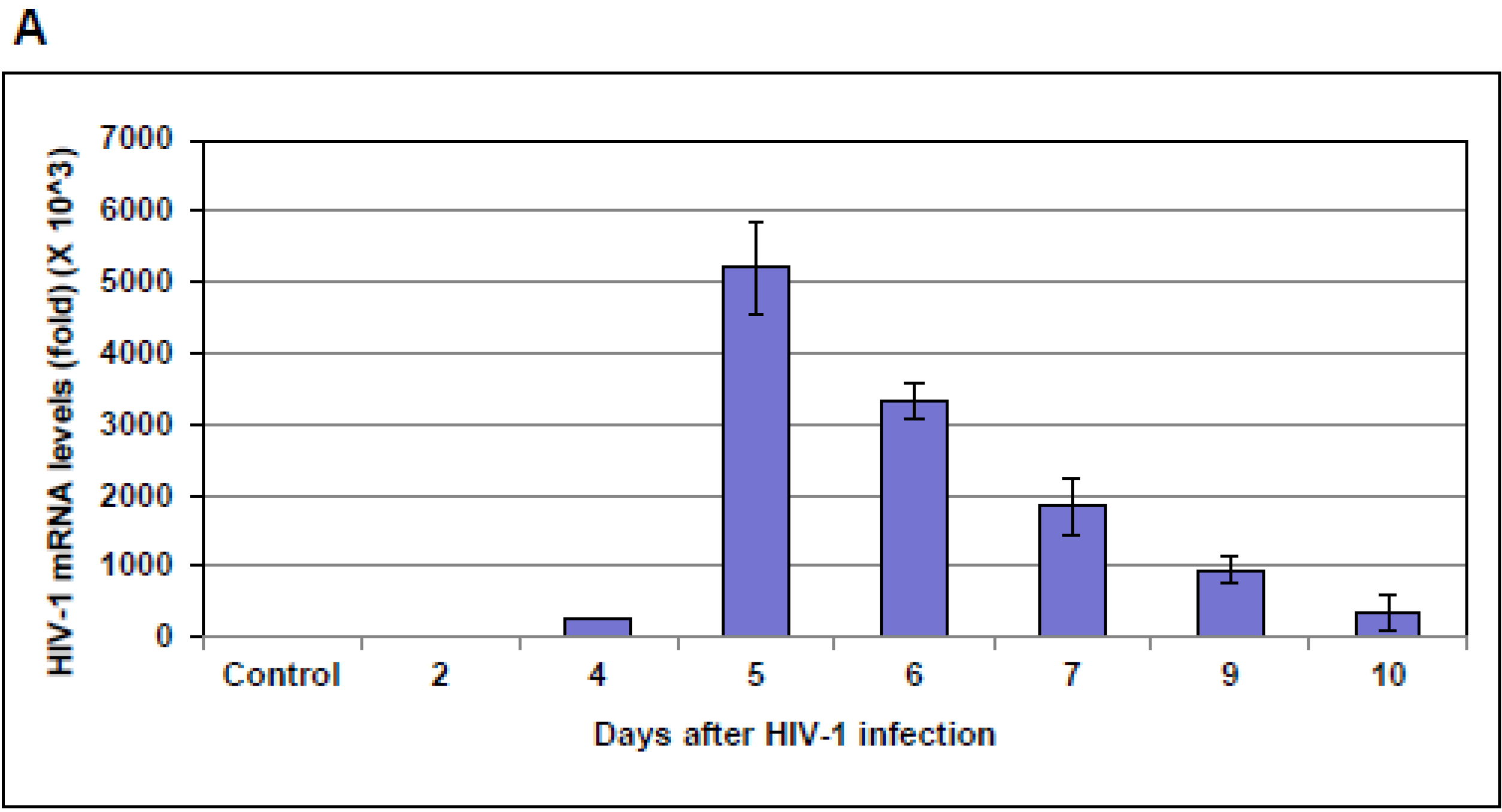
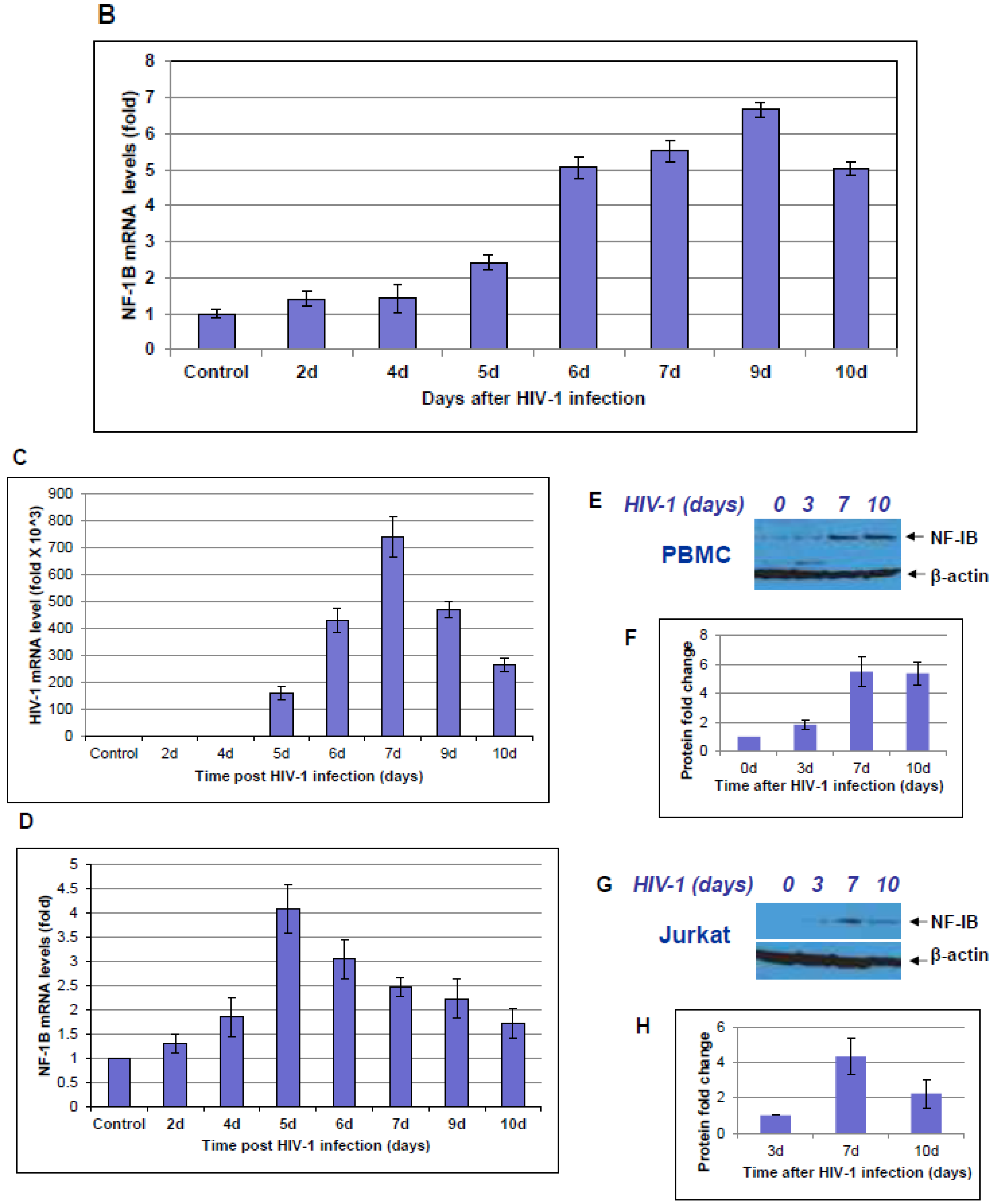
10]. Here, we observed a marked difference in NF-IB expression during HIV-1 infection, or reactivation of latently infected cells. Further understanding of NF1-B modulation may offer interesting and new insights into viral pathogenesis and latency. Because NFI family members are shown to either repress or activate cellular and viral genes, it was not evident whether the up-regulation of NF-IB during HIV-1 infection was favorable for the virus or cell. Hence we studied the role of NF-IB on HIV-1 transcription, and observed a novel mechanism of negative regulation of HIV-1 transcription by NF-IB, which is induced by HIV-1 infection.
The NRE region of the HIV-1 LTR has been reported to down-regulate LTR-directed HIV gene expression [
2,
9]. However, the mechanism for down regulation is not well understood. Since recent data have highlighted the importance of the U3 region of the LTR in determining the pathogenicity of HIV-1 [
9], studies concerning the nature and role of cellular transcription factors that interact with the LTR continue to be essential to our understanding of viral pathogenesis. Our results show induction of NF-IB protein in HIV-1 infection, and from our Chip experiment results it is apparent that HIV-1 induced NF-IB binds to the NRE region of the HIV-1 LTR (–385 to –362).
HIV-1 is known to manipulate host gene expression in favor of its replication and existence. For example, from our current study, the observed induction of NF-IB during HIV-1 infection and reactivation of latent infection, and its subsequent association with the NRE region of HIV-1 LTR suggests a negative feedback regulation of HIV-1 transcription through NF-IB. To support our hypothesis, in a lone available published result regarding the association of NF-I with LTR, it was demonstrated that the association of NF-I with TGATTGGC region of HIV-1 LTR resulted in antagonistic effect in the control of basal transcription of the HIV-1 genome in Jurkat cells [
9].
Several studies have described changes in certain cellular genes during HIV-1 infection. While many studies provide insights into the extensive effects of specific viral proteins on acute HIV-1 infection, changes in gene expression that may accompany HIV-1 infection and subsequent transition of the acute phase into a virologically latent phase have not been fully understood. It is believed that after the entry into the cell, HIV-1 establishes efficient control of viral replication by manipulating the cellular machinery. Based on our observation, we speculate that the induction of NF-IB during HIV-1 infection is to contain viral transcription through binding with the NRE region of the HIV-1 LTR.
Interestingly, pre-existence of the association of NF-IB with the NRE region of the HIV-1 LTR in latently infected (un-stimulated) cells, observed in our Chip experiment, further substantiates our notion that NF-IB acts as a negative regulator. As the reactivation of latently infected cells by external stimuli activates HIV-1 transcription, which in turn induces the expression of NF-IB resulting in increased association with the LTR. Subsequently HIV-1 transcription level is diminished to near normal levels; ironically, as HIV-1 transcription decreased, the NF-IB levels return to background, normal levels. Therefore, we observed a direct correlation between HIV-1 infection and NF-IB induction, and an inverse correlation between the NF-IB association with the LTR and HIV-1 transcription. Consistent with these results, we observed an increase in HIV-1 transcription when NF-IB was knocked-down by siRNA treatment in latent infected cells, confirming the negative regulatory role of NF-IB on HIV-1 transcription.
In a separate study depicting the relationship between HIV-1 infection and NF-IB expression, Sheeter
et al. [
14] proposed a negative role of NF-IB on cell surface CD4 expression. For optimal viral production, HIV-1 infection requires surface CD4 receptor down-modulation. On the basis of their findings, they speculated that NF-1B may be a cellular factor that HIV-1 induces to assist in the down-modulation of CD4, thereby restricting the new viral entry into the cell. Taken together, HIV-1-induced NF-IB expression may have a dual role in containing HIV-1 transcription: (1) by binding with the NRE region of the HIV-1 LTR to contain viral transcription; and (2) by down-modulating surface expression of CD4 to sustain viral infection. These findings suggest that NF-IB is a cellular antiviral response triggered by HIV-1 to contain its infection.
NFI consensus binding sites have been found on many cellular gene promoters, and association of NFI proteins with these promoters has been proposed to play a role in the determination of gene expression [
1]. Although further work will be required to clarify the correlation between NF-IB expression and HIV-1 transcription, data presented here conclusively demonstrates negative feedback regulation of HIV-1 transcription through the induction of NF-IB. In addition to binding to the HIV-1 LTR, HIV-1 induced NF-IB could also bind with the cellular gene promoters thereby altering the cellular mechanisms. The existence of the NF-I binding site on the various viral promoters raises the possibility that HIV-1 induced NF-IB could have effect on the transcription of other viruses, thereby regulating co-infections. The effect of HIV-1 induced NF-IB on the activation/inhibition of cellular and viral genes through binding on to the promoter remain open questions for future studies.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}